
Targeting MUC1-C Suppresses Chronic Activation of Cytosolic Nucleotide Receptors and STING in Triple-Negative Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
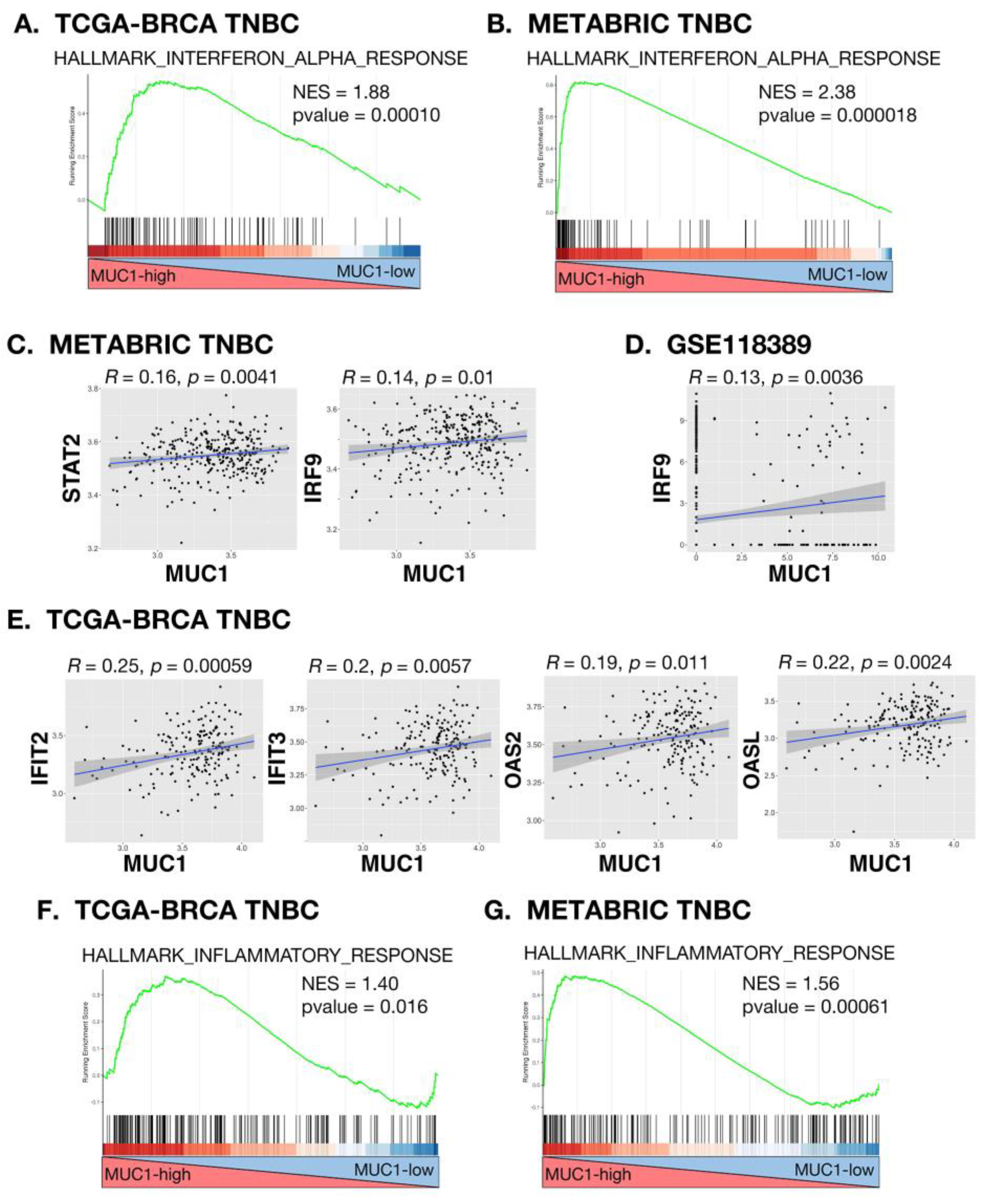
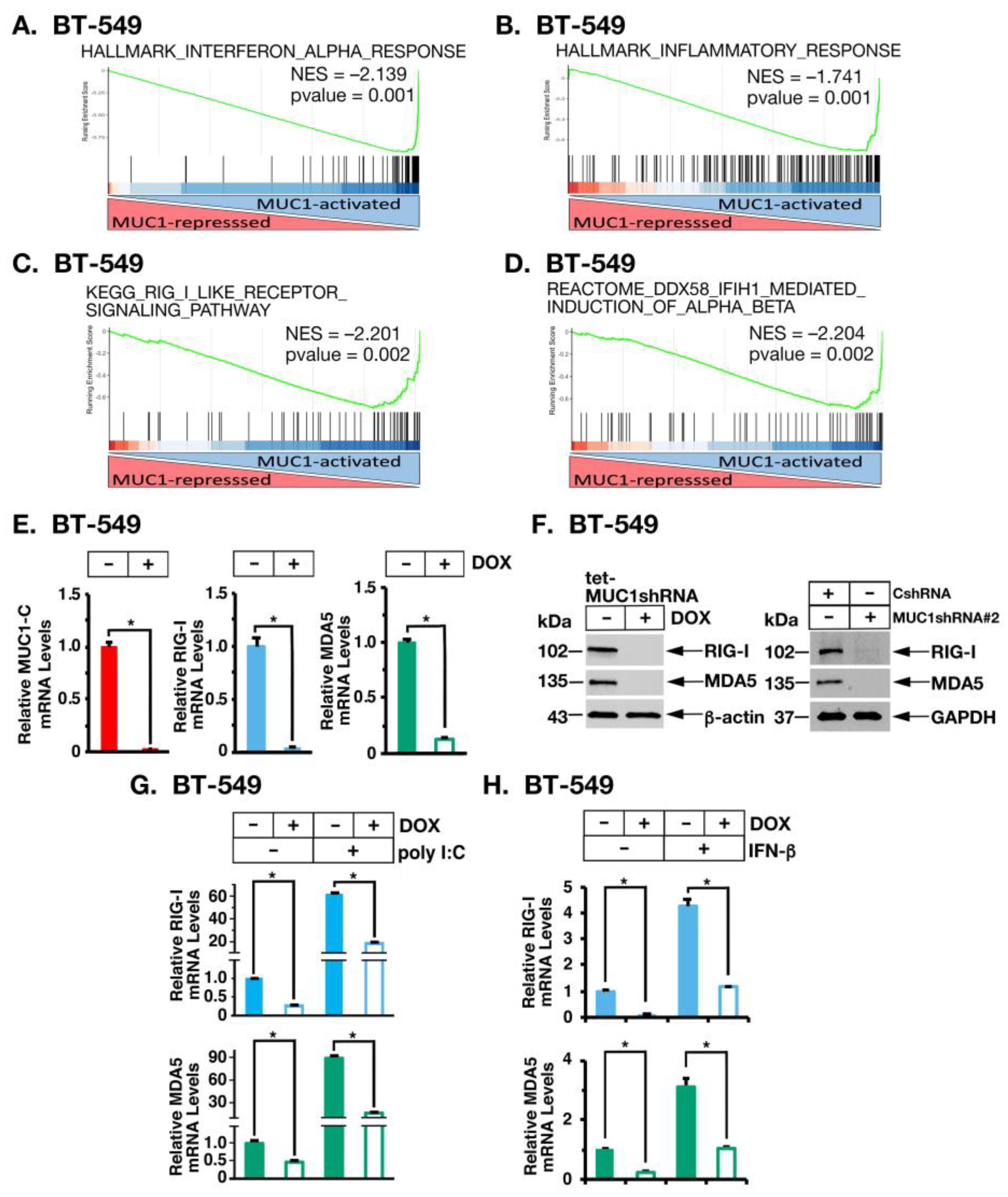
2.1. MUC1-C Induces RIG-I and MDA5 Cytosolic RNA PRRs in TNBC Cells
2.2. MUC1-C Activates the cGAS/STING Cytosolic DNA Sensing Pathway in TNBC Cells
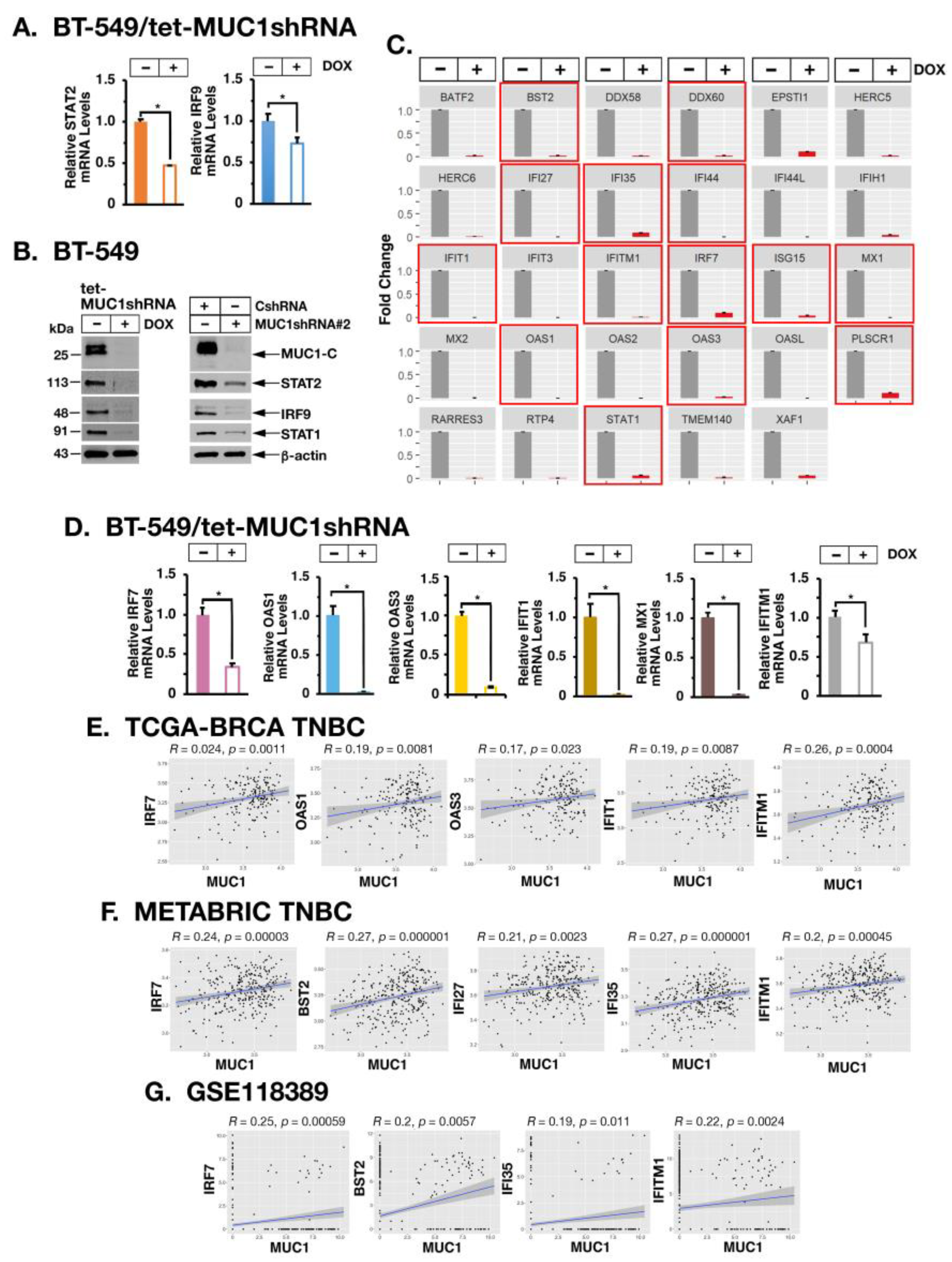
2.3. MUC1-C Drives U-ISGF3 Target DNA Damage Resistance Genes in TNBC Cells and Tumors
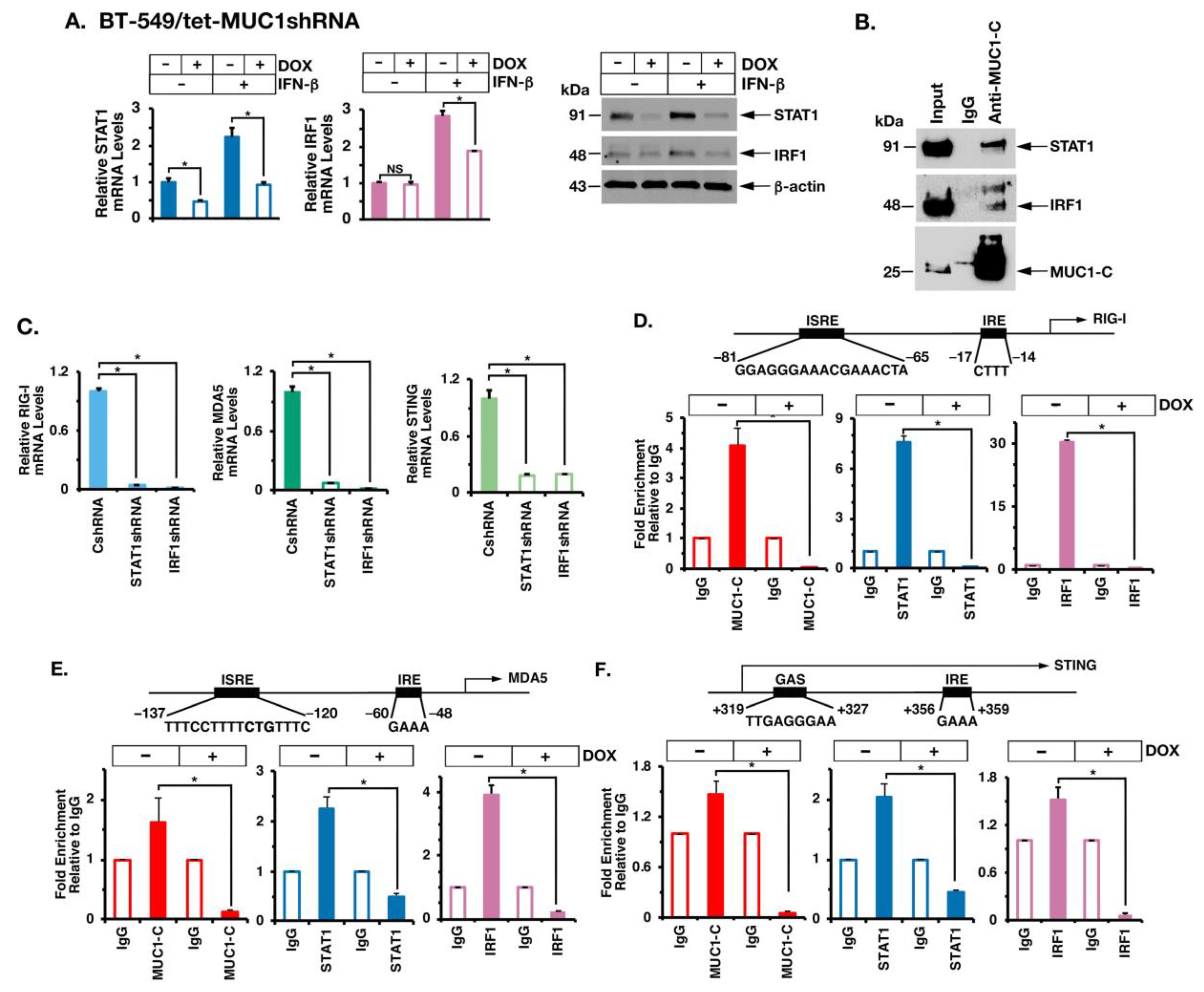
2.4. MUC1-C Interacts with STAT1 and IRF1 in Chronic IFN-β Stimulation of the Type I IFN Pathway
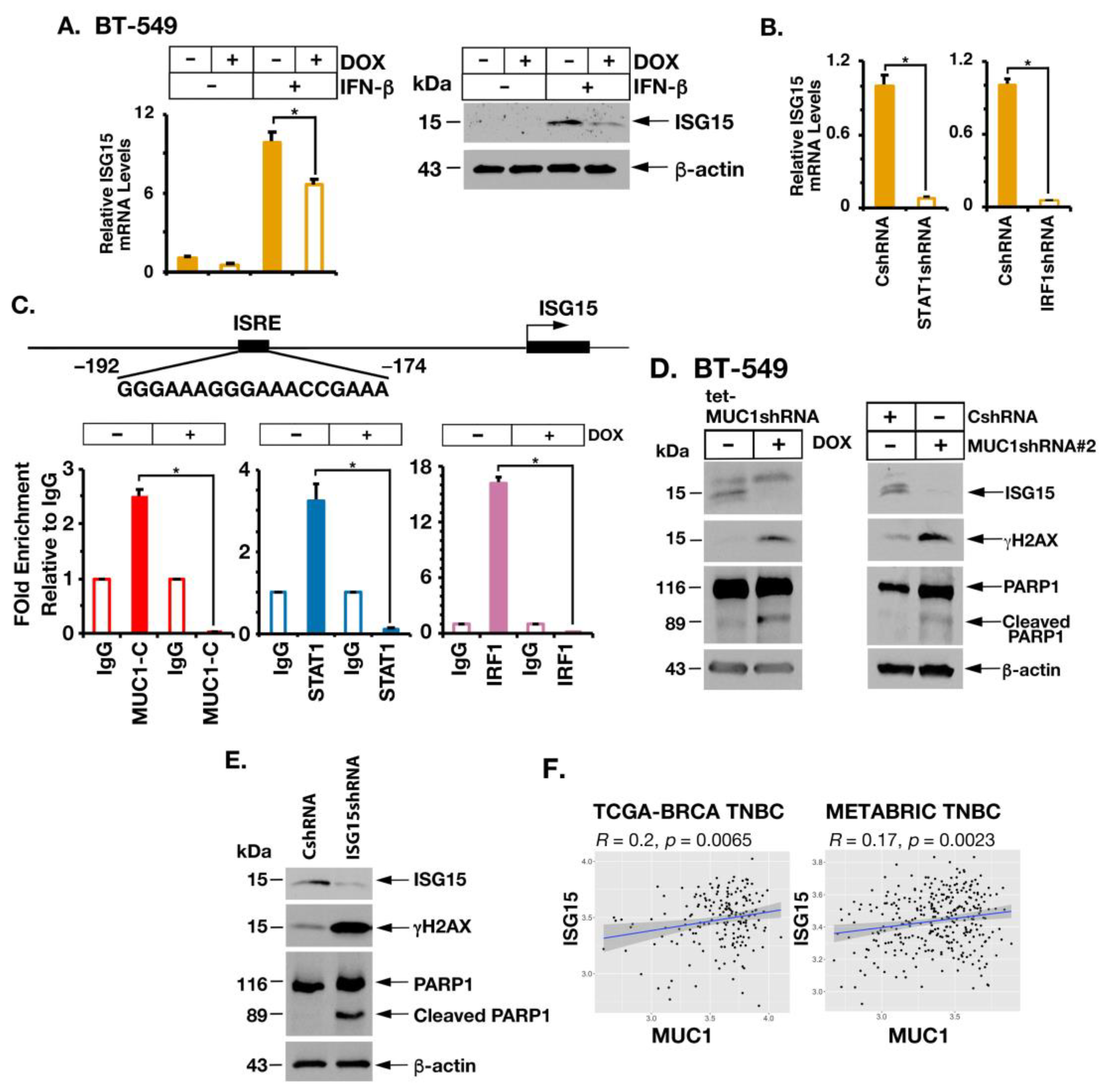
2.5. MUC1-C Is Necessary for Induction of ISG15 Expression
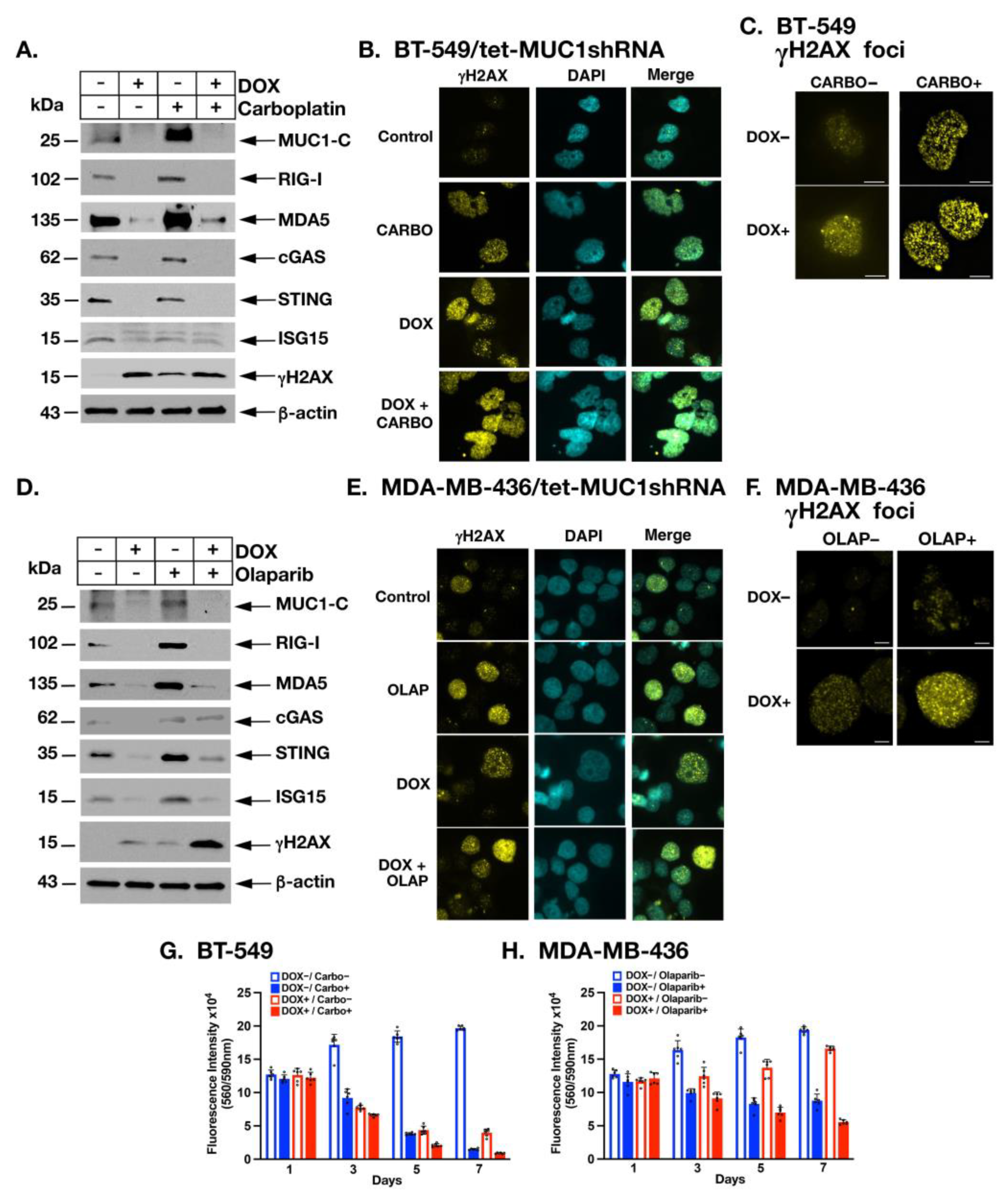
2.6. MUC1-C Is Necessary for Induction of PRRs, STING and ISG15 in the Response to Treatment with Carboplatin and Olaparib
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Gene Silencing and Rescue
4.3. Real-Time Quantitative Reverse-Transcription PCR (qRT-PCR)
4.4. Immunoblot Analysis
4.5. Coimmunoprecipitation of Nuclear Proteins
4.6. Chromatin Immunoprecipitation (ChIP)
4.7. ELISA for Measurement of IFN-β Levels
4.8. Cell Proliferation Assays
4.9. Apoptosis Assays
4.10. ICC Analysis of γH2AX Expression
4.11. Statistical Analysis
4.12. Analysis of Publicly Available TNBC Cohort and scRNA-Seq Datasets
4.13. Data and Software Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kufe, D. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis 2020, 41, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, H.; Hiraki, M.; Kufe, D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene 2018, 37, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Rajabi, H.; Takahashi, H.; Yasumizu, Y.; Li, W.; Jin, C.; Long, M.; Hu, Q.; Liu, S.; Fushimi, A.; et al. MUC1-C activates the NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res. 2019, 79, 5711–5722. [Google Scholar] [CrossRef] [Green Version]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C drives lineage plasticity in progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. JCI Insight 2020, 5, 137112. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Fushimi, A.; Yamashita, N.; Battacharya, A.; Rajabi, H.; Long, M.; Yasumizu, Y.; Oya, M.; Liu, S.; Kufe, D. MUC1-C activates the PBAF chromatin remodeling complex in integrating redox balance with progression of human prostate cancer stem cells. Oncogene 2021, 40, 4920–4940. [Google Scholar] [CrossRef]
- Hagiwara, M.; Yasumizu, Y.; Yamashita, N.; Rajabi, H.; Fushimi, A.; Long, M.D.; Li, W.; Bhattacharya, A.; Ahmad, R.; Oya, M.; et al. MUC1-C activates the BAF (mSWI/SNF) complex in prostate cancer stem cells. Cancer Res. 2021, 81, 1111–1122. [Google Scholar] [CrossRef]
- Yamashita, N.; Long, M.; Fushimi, A.; Yamamoto, M.; Hata, T.; Hagiwara, M.; Bhattacharya, A.; Hu, Q.; Wong, K.; Liu, S.; et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002115. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M.; Loo, Y.M., Jr. RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic sensing of immuno-stimulatory DNA.; the enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.; Wang, Z.; Ban, T.; Yanai, H.; Lu, Y.; Koshiba, R.; Nakaima, Y.; Hangai, S.; Savitsky, D.; Nakasato, M.; et al. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. USA 2009, 106, 17870–17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Cheon, H.; Wang, Y. Responses to cytokines and interferons that depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 2018, 10, a028555. [Google Scholar] [CrossRef] [Green Version]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [Green Version]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I interferon-mediated responses in cancer therapy: From anti-tumor immunity to resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [Green Version]
- Khodarev, N.; Ahmad, R.; Rajabi, H.; Pitroda, S.; Kufe, T.; McClary, C.; Joshi, M.D.; MacDermed, D.; Weichselbaum, R.; Kufe, D. Cooperativity of the MUC1 oncoprotein and STAT1 pathway in poor prognosis human breast cancer. Oncogene 2010, 29, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.D. ISG15, a double edged sword in cancer. Oncoimmunology 2015, 4, e1052935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandy, Z.; da Costa, I.C.; Schmidt, C.K. More than meets the ISG15, emerging roles in the DNA damage response and beyond. Biomolecules 2020, 10, 1557. [Google Scholar] [CrossRef] [PubMed]
- Raso, M.C.; Djoric, N.; Walser, F.; Hess, S.; Schmid, F.M.; Burger, S.; Knobeloch, K.P.; Penengo, L. Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J. Cell Biol. 2020, 219, e202002175. [Google Scholar] [CrossRef] [PubMed]
- Rengachari, S.; Groiss, S.; Devos, J.M.; Caron, E.; Grandvaux, N.; Panne, D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc. Natl. Acad. Sci. USA 2018, 115, E601–E609. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.M.; Liu, C.J.; Lee, T.C. Emerging functions of human IFIT proteins in cancer. Front Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.P. OAS proteins and cGAS: Unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 2014, 14, 521–528. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Khodarev, N.N. Intracellular RNA sensing in mammalian cells: Role in stress response and cancer therapies. Int. Rev. Cell Mol. Biol. 2019, 344, 31–89. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padariya, M.; Sznarkowska, A.; Kote, S.; Gomez-Herranz, M.; Mikac, S.; Pilch, M.; Alfaro, J.; Fahraeus, R.; Hupp, T.; Kalathiya, U. Functional interfaces, biological pathways, and regulations of interferon-related DNA damage resistance signature (IRDS) genes. Biomolecules 2021, 11, 622. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls Type I and Type II IFN responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef]
- Kondratova, A.A.; Cheon, H.; Dong, B.; Holvey-Bates, E.G.; Hasipek, M.; Taran, I.; Gaughan, C.; Jha, B.K.; Silverman, R.H.; Stark, G.R. Suppressing PARylation by 2′,5′-oligoadenylate synthetase 1 inhibits DNA damage-induced cell death. EMBO J. 2020, 39, e101573. [Google Scholar] [CrossRef]
- Jung, H.E.; Oh, J.E.; Lee, H.K. Cell-penetrating Mx1 enhances anti-viral resistance against mucosal influenza viral infection. Viruses 2019, 11, 109. [Google Scholar] [CrossRef]
- Liang, R.; Li, X.; Zhu, X. Deciphering the roles of IFITM1 in tumors. Mol. Diagn. Ther. 2020, 24, 433–441. [Google Scholar] [CrossRef]
- Forero, A.; Ozarkar, S.; Li, H.; Lee, C.H.; Hemann, E.A.; Nadjsombati, M.S.; Hendricks, M.R.; So, L.; Green, R.; Roy, C.N.; et al. Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by Type I and Type III interferons. Immunity 2019, 51, 451–464.E6. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.; Evans, B.; Levy, D.; Fahey, D.; Knight, E., Jr.; Darnell, J.E., Jr. Interferon-induced transcription of a gene encoding a 15-kDa protein depends on an upstream enhancer element. Proc. Natl. Acad. Sci. USA 1987, 84, 6394–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on triple-negative breast cancer: Current treatment strategies, unmet needs, and potential targets for future therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- Yamamoto, M.; Jin, C.; Hata, T.; Yasumizu, Y.; Zhang, Y.; Hong, D.; Maeda, T.; Miyo, M.; Hiraki, M.; Suzuki, Y.; et al. MUC1-C integrates chromatin remodeling and PARP1 activity in the DNA damage response of triple-negative breast cancer cells. Cancer Res. 2019, 79, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Solier, S.; Pommier, Y. The nuclear gamma-H2AX apoptotic ring: Implications for cancers and autoimmune diseases. Cell Mol. Life Sci. 2014, 71, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Francica, P.; Rottenberg, S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; de Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Liao, X.; Beckett, M.; Li, Y.; Khanna, K.K.; Wang, Z.; Kharbanda, S.; Weichselbaum, R.; Kufe, D. MUC1-C oncoprotein interacts directly with ATM and promotes the DNA damage response to ionizing radiation. Genes Cancer 2010, 1, 239–250. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerbrey, K.L.; Hu, G.; Kessler, J.D.; Roarty, K.; Li, M.Z.; Fang, J.E.; Herschkowitz, J.I.; Burrows, A.E.; Ciccia, A.; Sun, T.; et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 3665–3670. [Google Scholar] [CrossRef] [Green Version]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, N.; Fushimi, A.; Morimoto, Y.; Bhattacharya, A.; Hagiwara, M.; Yamamoto, M.; Hata, T.; Shapiro, G.I.; Long, M.D.; Liu, S.; et al. Targeting MUC1-C Suppresses Chronic Activation of Cytosolic Nucleotide Receptors and STING in Triple-Negative Breast Cancer. Cancers 2022, 14, 2580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112580
Yamashita N, Fushimi A, Morimoto Y, Bhattacharya A, Hagiwara M, Yamamoto M, Hata T, Shapiro GI, Long MD, Liu S, et al. Targeting MUC1-C Suppresses Chronic Activation of Cytosolic Nucleotide Receptors and STING in Triple-Negative Breast Cancer. Cancers. 2022; 14(11):2580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112580
Chicago/Turabian StyleYamashita, Nami, Atsushi Fushimi, Yoshihiro Morimoto, Atrayee Bhattacharya, Masayuki Hagiwara, Masaaki Yamamoto, Tsuyoshi Hata, Geoffrey I. Shapiro, Mark D. Long, Song Liu, and et al. 2022. "Targeting MUC1-C Suppresses Chronic Activation of Cytosolic Nucleotide Receptors and STING in Triple-Negative Breast Cancer" Cancers 14, no. 11: 2580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112580