
Multiple ETS Factors Participate in the Transcriptional Control of TERT Mutant Promoter in Thyroid Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Analysis of Public Expression Datasets
2.2. Cell Culture
2.3. Gene Silencing
2.4. Real-Time Quantitative PCR
2.5. MAPK Inhibition Experiments
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
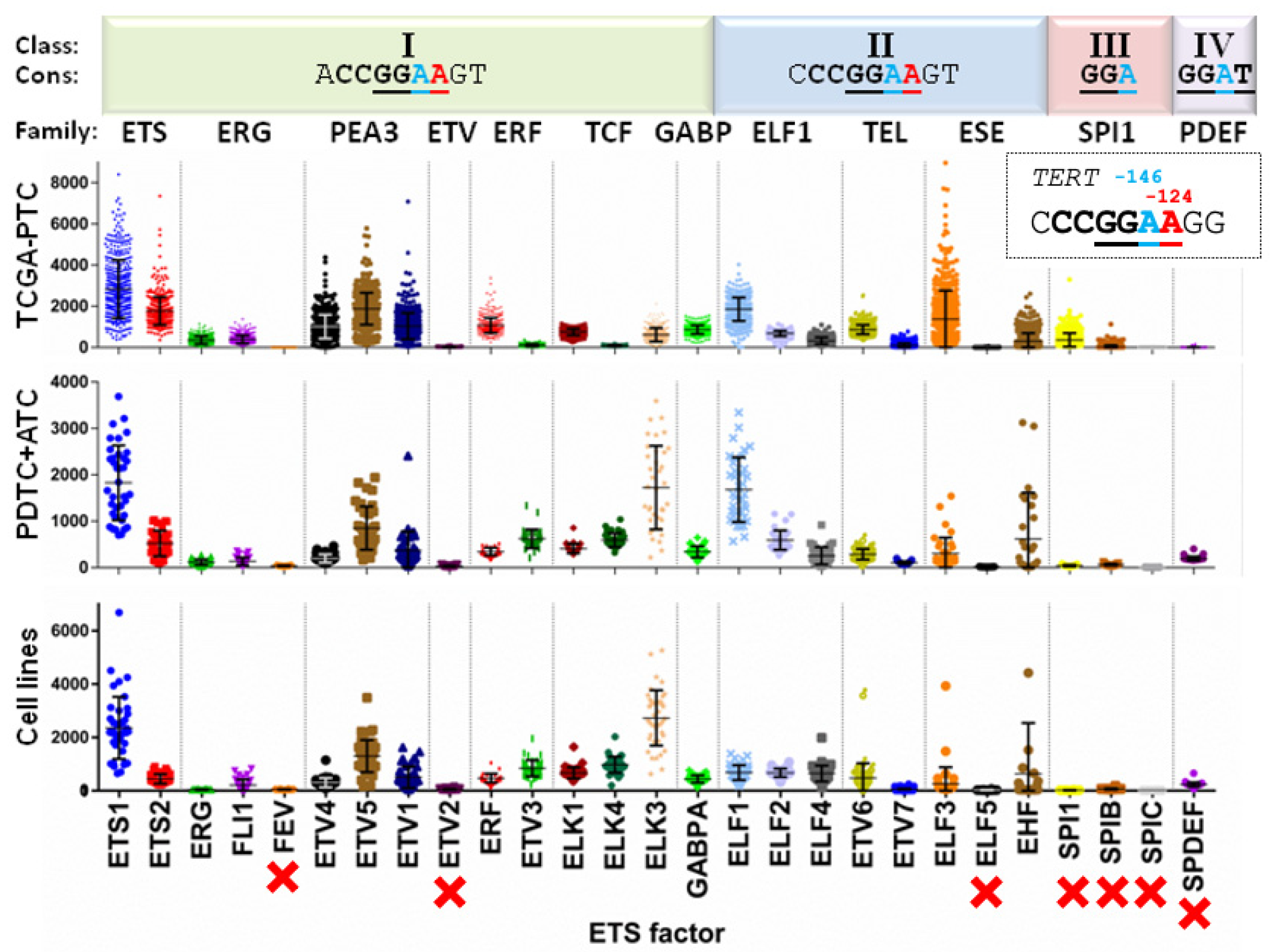
3.1. ETS Factors Show a Wide Range of Expression in Thyroid Cancer Specimens but Remain Comparable across Thyroid Cancer Types
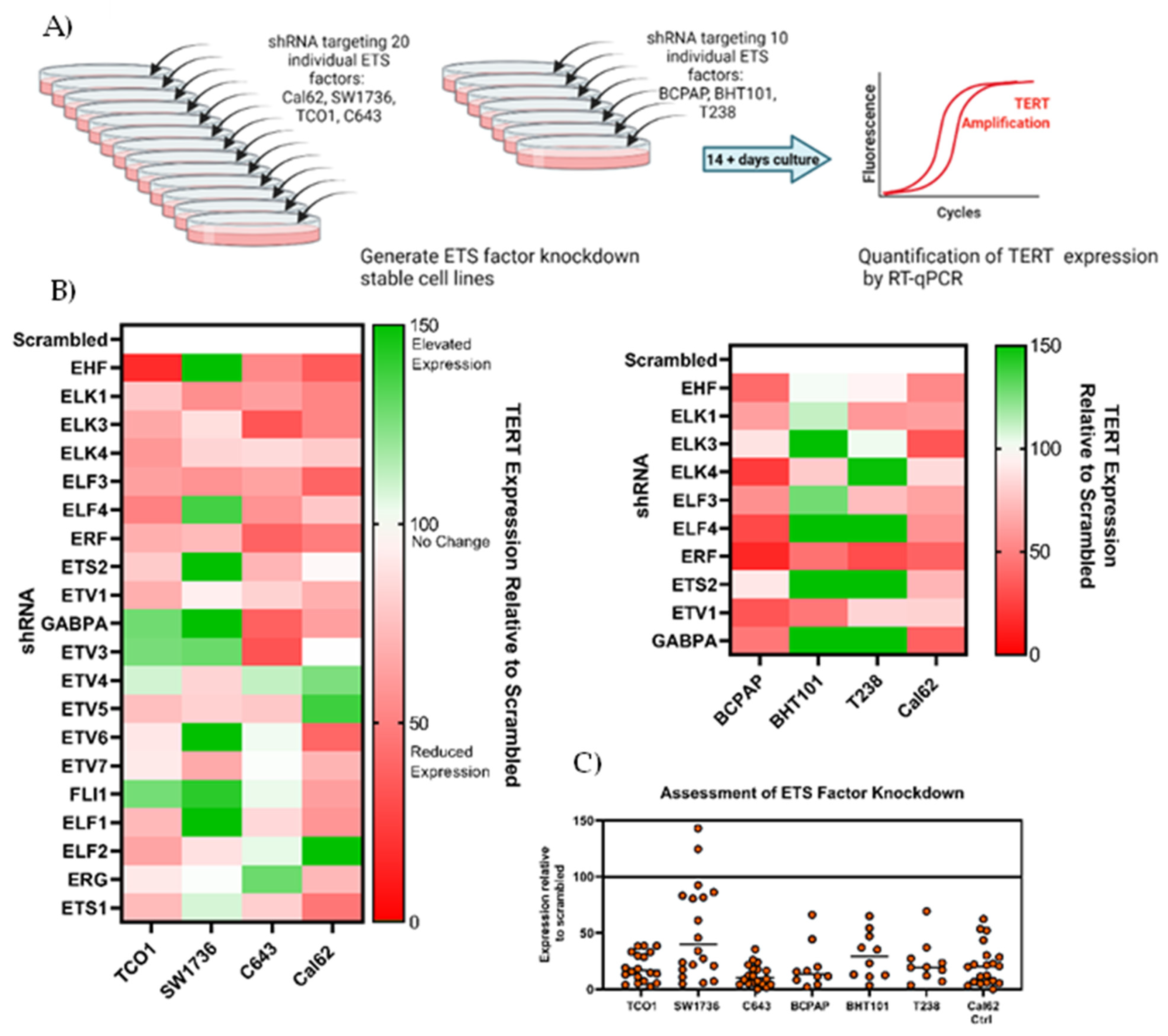
3.2. Screening of 20 ETS in Cell Lines Shows Vvariable Effects on TERT Expression
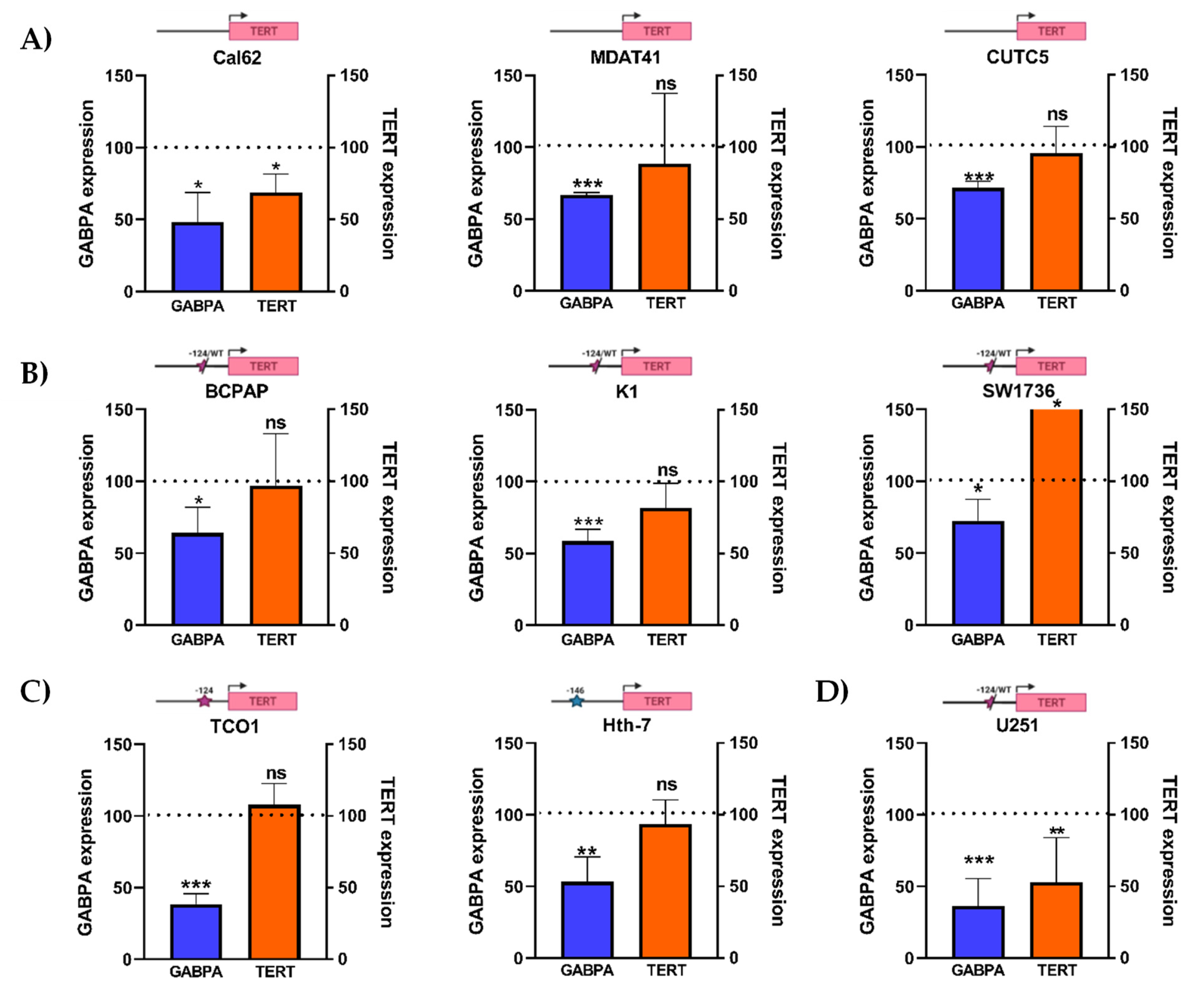
3.3. GABPA Knockdown Does Not Affect TERT Expression across Thyroid Cancer Cell Lines Carrying TPMs
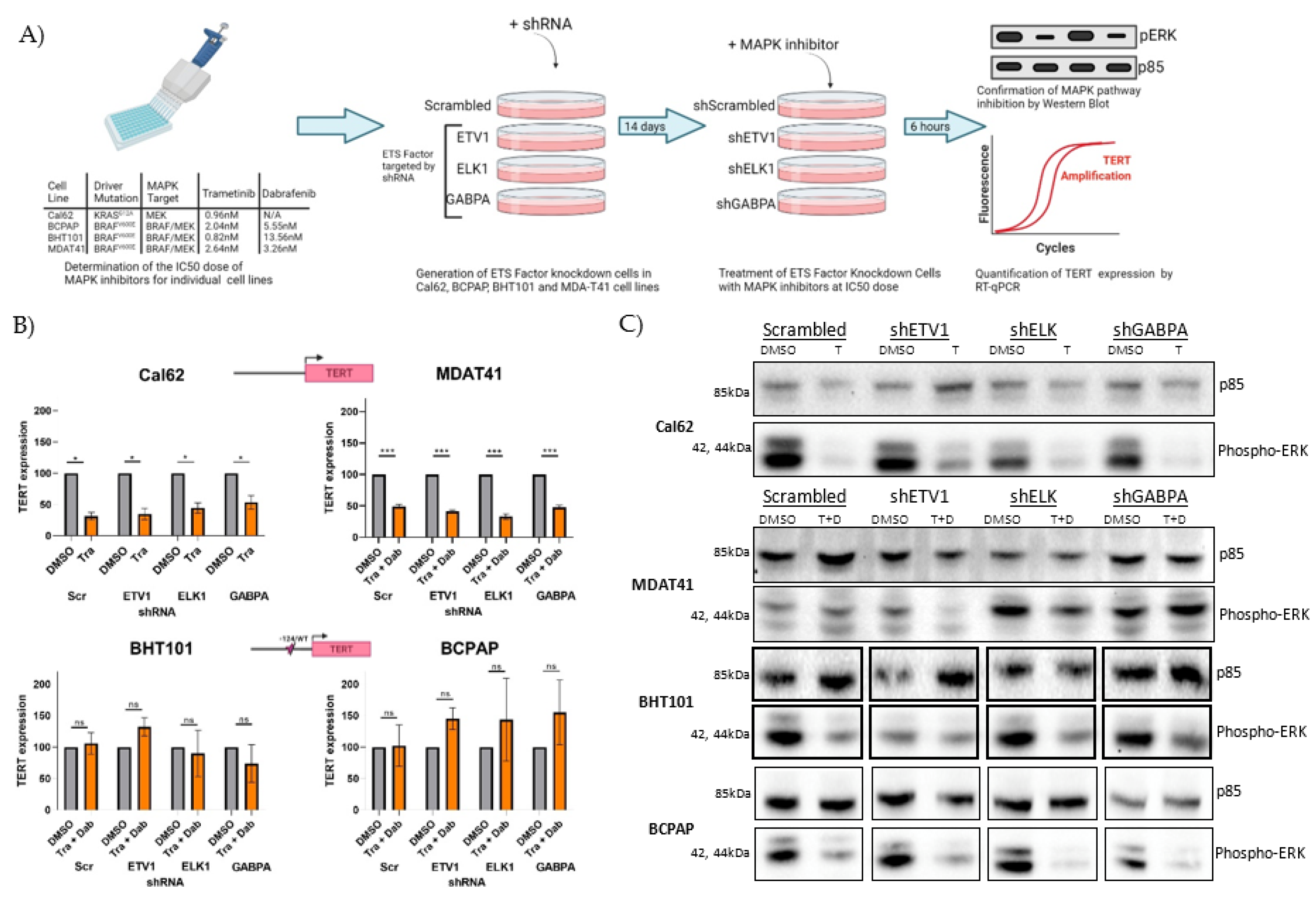
3.4. Inhibiting MAPK Signaling Primarily Affects Expression of TERT in Cells with Wild-Type TERT Promoter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metab. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr.-Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Bishop, J.; Zhu, G.; Zhang, T.; Ladenson, P.W.; Xing, M. Mortality Risk Stratification by Combining BRAF V600E and TERT Promoter Mutations in Papillary Thyroid Cancer: Genetic Duet of BRAF and TERT Promoter Mutations in Thyroid Cancer Mortality. JAMA Oncol. 2017, 3, 202–208. [Google Scholar] [CrossRef]
- Song, Y.S.; Yoo, S.K.; Kim, H.H.; Jung, G.; Oh, A.R.; Cha, J.Y.; Kim, S.J.; Cho, S.W.; Lee, K.E.; Seo, J.S.; et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr.-Relat. Cancer 2019, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [Green Version]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [Green Version]
- Mancini, A.; Xavier-Magalhaes, A.; Woods, W.S.; Nguyen, K.T.; Amen, A.M.; Hayes, J.L.; Fellmann, C.; Gapinske, M.; McKinney, A.M.; Hong, C.; et al. Disruption of the beta1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell 2018, 34, 513–528.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Bullock, M.; Lim, G.; Li, C.; Ho Choi, I.; Kochhar, S.; Liddle, C.; Zhang, L.; Clifton-Bligh, R.J. Thyroid transcription factor FOXE1 interacts with ETS factor ELK1 to co-regulate TERT. Oncotarget 2016, 7, 85948. [Google Scholar] [CrossRef] [Green Version]
- Bullock, M.; Lim, G.; Zhu, Y.; Aberg, H.; Kurdyukov, S.; Clifton-Bligh, R. ETS Factor ETV5 Activates the Mutant Telomerase Reverse Transcriptase Promoter in Thyroid Cancer. Thyroid 2019, 29, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Mu, N.; Wang, N.; Straat, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2018, 38, 965–979. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar]
- Greenberg, R.A.; O’Hagan, R.C.; Deng, H.; Xiao, Q.; Hann, S.R.; Adams, R.R.; Lichtsteiner, S.; Chin, L.; Morin, G.B.; DePinho, R.A. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene 1999, 18, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Maida, Y.; Kyo, S.; Kanaya, T.; Wang, Z.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Ohmichi, M.; Gotoh, N.; Murakami, S.; et al. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene 2002, 21, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N.; et al. Comprehensive genetic characterization of human thyroid cancer cell lines: A validated panel for preclinical studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef]
- Wei, G.H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef]
- Panebianco, F.; Nikitski, A.V.; Nikiforova, M.N.; Nikiforov, Y.E. Spectrum of TERT promoter mutations and mechanisms of activation in thyroid cancer. Cancer Med. 2019, 8, 5831–5839. [Google Scholar] [CrossRef] [Green Version]
- Stenman, A.; Yang, M.; Paulsson, J.O.; Zedenius, J.; Paulsson, K.; Juhlin, C.C. Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma. Cancers 2021, 13, 6340. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Shore, P.; Willingham, N.; Lakey, J.H.; Sharrocks, A.D. The mechanism of phosphorylation-inducible activation of the ETS-domain transcription factor Elk-1. EMBO J. 1999, 18, 5666–5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, S.; Loukinov, D.; Abdullaev, Z.; Guilleret, I.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007, 35, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avin, B.A.; Wang, Y.; Gilpatrick, T.; Workman, R.E.; Lee, I.; Timp, W.; Umbricht, C.B.; Zeiger, M.A. Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosomes Cancer 2019, 58, 530–540. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, B.A.; Gilpatrick, T.; Wang, Y.; Timp, W.; Umbricht, C.B.; Zeiger, M.A. Characterization of Allele-Specific Regulation of Telomerase Reverse Transcriptase in Promoter Mutant Thyroid Cancer Cell Lines. Thyroid 2020, 30, 1470–1481. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tissue Derivation | MAPK Driver | TERT Promoter | Use |
|---|---|---|---|---|
| TCO1 | Thyroid, ATC | BRAF V600E | c.-124C>T, HOM | 1st screen, 20 ETS |
| SW1736 | Thyroid, ATC | BRAF V600E | c.-124C>T, HET | 1st screen, 20 ETS |
| C643 | Thyroid, ATC | HRAS G13R | c.-124C>T, HET | 1st screen, 20 ETS |
| Cal62 | Thyroid, ATC | KRAS G12R | Wild type | 1st screen, 20 ETS + MAPKi experiments |
| BCPAP | Thyroid, PTC | BRAF V600E | c.-124C>T, HET | 2nd screen, 10 ETS + MAPKi experiments |
| BHT101 | Thyroid, ATC | BRAF V600E | c.-124C>T, HET | 2nd screen, 10 ETS + MAPKi experiments |
| T238 | Thyroid, ATC | BRAF V600E | c.-124C>T, HET | 2nd screen, 10 ETS |
| MDAT41 | Thyroid, PTC | BRAF V600E | Wild type | Extended screen, GABPA + MAPKi experiments |
| CUTC5 | Thyroid, PTC | BRAF V600E | Wild type | Extended screen, GABPA |
| K1 | Thyroid, PTC | BRAF V600E | c.-124C>T, HET | Extended screen, GABPA |
| Hth7 | Thyroid, ATC | NRAS Q61L | c.-146C>T, HOM | Extended screen, GABPA |
| U251 | Glioblastoma | Unknown | c.-124C>T, HOM | Extended screen, GABPA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thornton, C.E.M.; Hao, J.; Tamarapu, P.P.; Landa, I. Multiple ETS Factors Participate in the Transcriptional Control of TERT Mutant Promoter in Thyroid Cancers. Cancers 2022, 14, 357. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020357
Thornton CEM, Hao J, Tamarapu PP, Landa I. Multiple ETS Factors Participate in the Transcriptional Control of TERT Mutant Promoter in Thyroid Cancers. Cancers. 2022; 14(2):357. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020357
Chicago/Turabian StyleThornton, Caitlin E. M., Jingzhu Hao, Prasanna P. Tamarapu, and Iñigo Landa. 2022. "Multiple ETS Factors Participate in the Transcriptional Control of TERT Mutant Promoter in Thyroid Cancers" Cancers 14, no. 2: 357. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020357