
Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
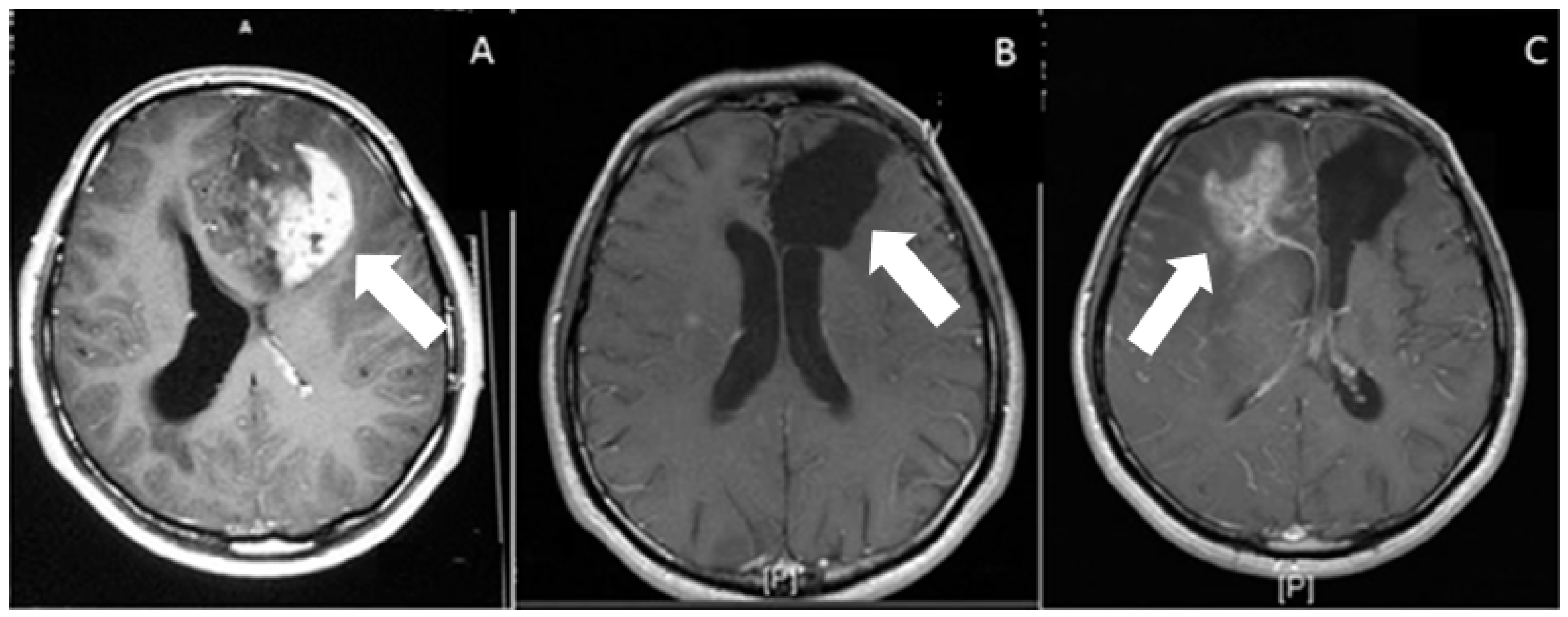
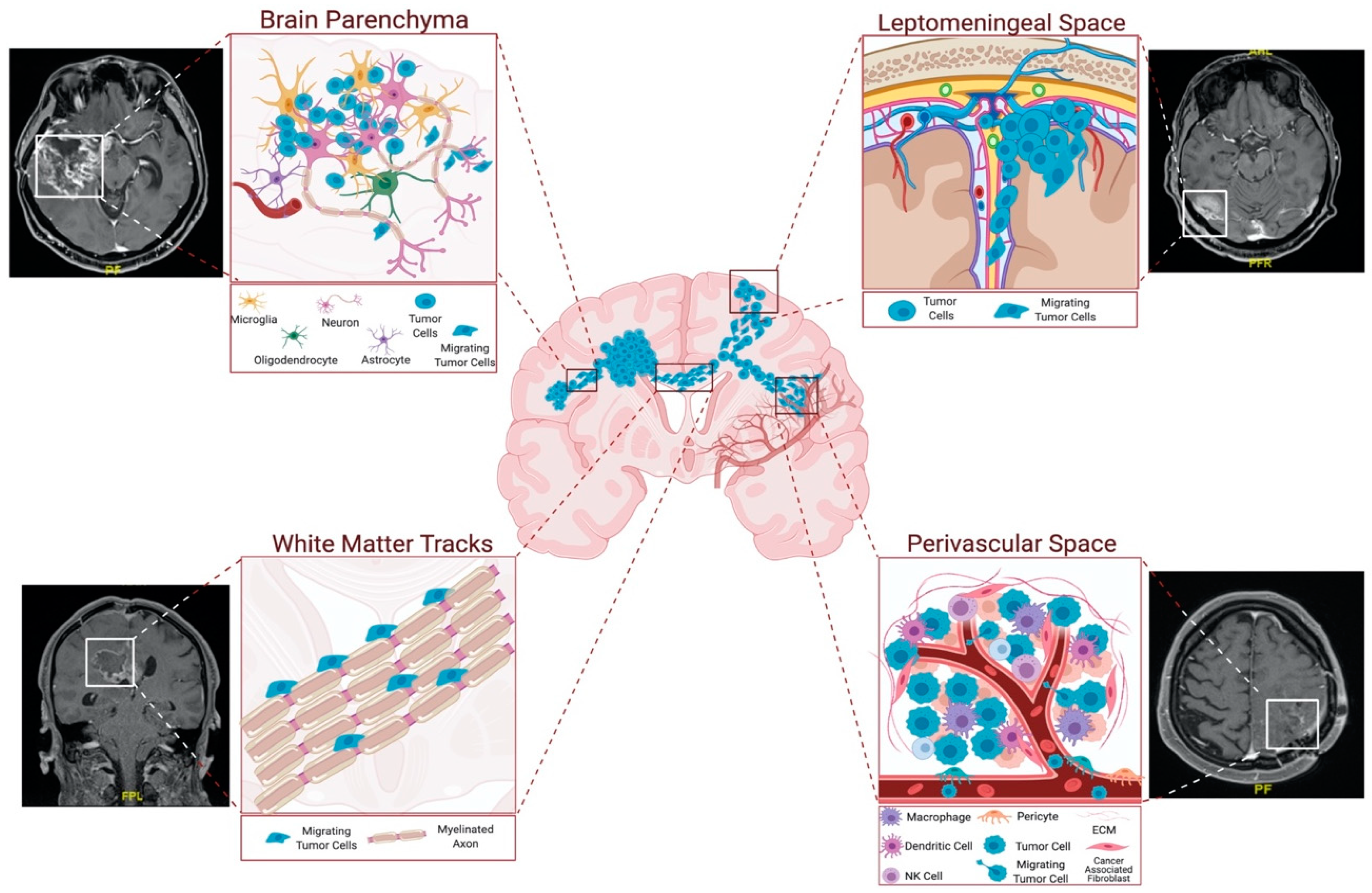
2. Routes of Glioblastoma Cell Invasion
3. Modes of Glioblastoma Invasion
4. Molecular Mechanisms of Glioblastoma Cell Invasion
4.1. Role of Extracellular Matrix in Invasion
4.2. Role of Adhesion Proteins in Invasion: Attachment to and Detachment from ECM
4.3. Role of Proteinases in Invasion: Remodeling of ECM
4.4. Cytoskeletal Changes during Invasion
4.5. Role of Other Motility Factors in Invasion
5. Role of Ion Channels in Glioma Invasion
6. Epithelial-to-Mesenchymal Transition and Invasion
7. Unbiased, High-Throughput Experimental Approaches to Study Glioblastoma Invasion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Gene/Protein Identified | Reference |
|---|---|---|
| Differential expression analysis of tumor cells from tumor core and the invasive rim by laser-capture microdissection | P311 | [144] |
| Microarray analysis of the cells from tumor cores and the cells that invaded White matter using laser-capture microdissection | ATX and BCLW | [145] |
| Microarray analysis of core and rim cells using cell-line spheroids invading collagen | MKK3 and p38 | [146] |
| Microarray analysis of cell lines and primary cultures with radial migration assay | CTGF | [147] |
| RNA sequencing of motile and nonmotile cells using a spheroid dispersal model | SERPINE1 | [149] |
| Differential expression analysis of long noncoding RNAs in glioma tissues, compared to normal brain tissues | NEAT1 | [150] |
| miRNA profiling of slow-growing, diffusely infiltrating glioma and noninvasive primitive neural tumors | miRNA-449a | [151] |
| Functional screen with monoclonal antibody library generated against primary glioblastoma cells | Itga7 | [152] |
| Analysis of enriched proteins on the cell membranes with different invasive capacities | Itga5, CD97 and Anxa1 | [153] |
| Analysis of proteins in cell lines with different invasive capacities | Cathepsin D | [154] |
| Functional proteomics approach with fluorophore-assisted light inactivation | Neuropilin-1 and Semaphorin3A | [155] |
| Functional proteomics approach with fluorophore-assisted light inactivation | CD155/PVR | [156] |
| Analysis of proteins from glioblastoma sections by microdissecting cells from invasive border and proliferative core | ELMO1 and Dock180 | [157] |
| Proteomics analysis of xenograft models generated by serial transplantation of human glioblastoma specimens into rat brains | PDI | [158] |
8. Clinical Targeting of Glioblastoma: Is There an Anti-Invasion/Anti-Migration Treatment?
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Cha, J.; Kang, S.G.; Kim, P. Strategies of Mesenchymal Invasion of Patient-derived Brain Tumors: Microenvironmental Adaptation. Sci. Rep. 2016, 6, 24912. [Google Scholar] [CrossRef] [Green Version]
- Keime-Guibert, F.; Chinot, O.; Taillandier, L.; Cartalat-Carel, S.; Frenay, M.; Kantor, G.; Guillamo, J.S.; Jadaud, E.; Colin, P.; Bondiau, P.Y.; et al. Radiotherapy for glioblastoma in the elderly. N. Engl. J. Med. 2007, 356, 1527–1535. [Google Scholar] [CrossRef]
- Lang, F.F.; Gilbert, M.R. Diffusely infiltrative low-grade gliomas in adults. J. Clin. Oncol. 2006, 24, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Laperriere, N.; Zuraw, L.; Cairncross, G. Radiotherapy for newly diagnosed malignant glioma in adults: A systematic review. Radiother. Oncol. 2002, 64, 259–273. [Google Scholar] [CrossRef]
- Yaşargil, M.G.; von Ammon, K.; Cavazos, E.; Doczi, T.; Reeves, J.D.; Roth, P. Tumours of the limbic and paralimbic systems. Acta Neurochir. 1992, 118, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Schramm, J.; Aliashkevich, A.F. Surgery for temporal mediobasal tumors: Experience based on a series of 235 patients. Neurosurgery 2007, 60, 285–294. [Google Scholar] [CrossRef]
- Scherer, H.J. Structural Development in Gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar]
- De Gooijer, M.C.; Guillén Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Montana, V.; Sontheimer, H. Bradykinin promotes the Chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [Green Version]
- Diksin, M.; Smith, S.J.; Rahman, R. The molecular and phenotypic basis of the glioma invasive perivascular niche. Int. J. Mol. Sci. 2017, 18, 2342. [Google Scholar] [CrossRef] [Green Version]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [PubMed] [Green Version]
- Mughal, A.A.; Zhang, L.; Fayzullin, A.; Server, A.; Li, Y.; Wu, Y.; Glass, R.; Meling, T.; Langmoen, I.A.; Leergaard, T.B.; et al. Patterns of Invasive Growth in Malignant Gliomas—The Hippocampus Emerges as an Invasion-Spared Brain Region. Neoplasia 2018, 20, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yi, L.; Kang, Q.; Zhou, J.; Chen, T.; Hugnot, J.; Yu, S. Glioma invasion along white matter tracts: A dilemma for neurosurgeons. Cancer Lett. 2022, 526, 103–111. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chédotal, A.; Kerjan, G.; Moreau-Fauvarque, C. The brain within the tumor: New roles for axon guidance molecules in cancers. Cell Death Differ. 2005, 12, 1044–1056. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Kunkel, P.; Müller, S.; Schirmacher, P.; Stavrou, D.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro. Oncol. 2004, 3, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro. Oncol. 2014, 16, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, T.A.; De Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, K.M.; Monsefi, N.; Dawson, J.C.; Degasperi, A.; Bukowski-Wills, J.C.; Volinsky, N.; Dobrzyński, M.; Birtwistle, M.R.; Tsyganov, M.A.; Kiyatkin, A.; et al. Bistability in the Rac1, PAK, and RhoA Signaling Network Drives Actin Cytoskeleton Dynamics and Cell Motility Switches. Cell Syst. 2016, 2, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Paňková, K.; Rösel, D.; Novotný, M.; Brábek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2010, 67, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, G.M.; Zhong, J.; Paul, A.; Kellie, S.J. Mesenchymal migration as a therapeutic target in glioblastoma. J. Oncol. 2010, 2010, 430142. [Google Scholar]
- Maaser, K.; Wolf, K.; Klein, C.E.; Niggemann, B.; Zänker, K.S.; Bröcker, E.B.; Friedl, P. Functional hierarchy of simultaneously expressed adhesion receptors: Integrin α2β1 but not CD44 mediates MV3 melanoma cell migration and matrix reorganization within three-dimensional hyaluronan-containing collagen matrices. Mol. Biol. Cell 1999, 10, 3067–3079. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef] [PubMed]
- Alieva, M.; Leidgens, V.; Riemenschneider, M.J.; Klein, C.A.; Hau, P.; van Rheenen, J. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 2019, 9, 2054. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virga, J.; Szivos, L.; Hortobágyi, T.; Chalsaraei, M.K.; Zahuczky, G.; Steiner, L.; Tóth, J.; Reményi-Puskár, J.; Bognár, L.; Klekner, A. Extracellular matrix differences in glioblastoma patients with different prognoses. Oncol. Lett. 2019, 17, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, U.; Kaye, A.H. Extracellular matrix and the brain: Components and function. J. Clin. Neurosci. 2000, 7, 280–290. [Google Scholar] [CrossRef]
- Goldbrunner, R.H.; Bernstein, J.J.; Tonn, J.C. Cell-extracellular matrix interaction in glioma invasion. Acta Neurochir. 1999, 141, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, B.B.; Mahesparan, R.; Thorsen, F.; Haugland, H.K.; Porwol, T.; Enger, P.O.; Lund-Johansen, M.; Bjerkvig, R. Laminin expression by glial fibrillary acidic protein positive cells in human gliomas. Int. J. Dev. Neurosci. 1999, 17, 531–539. [Google Scholar] [CrossRef]
- Read, R.M.T.; Kai, M.L.; Skaftnesmo, O.; Bjerkvig, R.; Engebraaten, O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003, 195, 49–57. [Google Scholar]
- Bjerkvig, R.; Laerum, O.D.; Rucklidge, G.J. Immunocytochemical characterization of extracellular matrix proteins expressed by cultured glioma cells. Cancer Res. 1989, 49, 5424–5428. [Google Scholar]
- Baldwin, J.R.; McKeever, P.E.; Booker, T.R. Products of cultured neuroglial cells: II. The production of fibronectin by C6 glioma cells. Neurochem. Res. 1985, 10, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Savaraj, N.; Wu, C.; Landy, H.; Wangpaijit, M.; Wei, Y.; Kuo, M.T.; Robles, C.; Furst, A.J.; Lampidis, T.; Feun, L. Procollagen alpha 1 type I: A potential aide in histopathological grading of glioma. Cancer Investig. 2005, 23, 577–581. [Google Scholar] [CrossRef]
- Paulus, W.; Roggendorf, W.; Schuppan, D. Immunohistochemical investigation of collagen subtypes in human glioblastomas. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 413, 325–332. [Google Scholar] [CrossRef]
- Han, J.; Daniel, J.C.; Pappas, G.D. Invasion in Brain Tissue Cultures. Cancer 1995, 88, 127–132. [Google Scholar]
- De Clerck, Y.A.; Shimada, H.; Gonzalez-Gomez, I.; Raffel, C. Tumoral invasion in the central nervous system. J. Neurooncol. 1993, 18, 111–121. [Google Scholar] [CrossRef]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, A.; Westphal, M. Glioma invasion in the central nervous system. Neurosurgery 1996, 39, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Mechanism, A. Glioblastoma Expression of Vitronectin and the av/83 Integrin. Adhesion mechanism for transformed glial cells. J. Clin. Investig. 1991, 88, 1924–1932. [Google Scholar]
- Uhm, J.H.; Dooley, N.P.; Kyritsis, A.P.; Rao, J.S.; Gladson, C.L. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin. Cancer Res. 1999, 5, 1587–1594. [Google Scholar]
- Higuchi, M.; Ohnishi, T.; Arita, N.; Hiraga, S.; Hayakawa, T. Expression of tenascin in human gliomas: Its relation to histological malignancy, tumor dedifferentiation and angiogenesis. Acta Neuropathol. 1993, 85, 481–487. [Google Scholar] [CrossRef]
- Bourdon, M.A.; Wikstrand, C.J.; Furthmayr, H.; Matthews, T.J.; Bigner, D.D. Human Glioma-Mesenchymal Extracellular Matrix Antigen Defined by Monoclonal Antibody Human Glioma-Mesenchymal Monoclonal Antibody Extracellular Matrix Antigen Defined by. Cancer Res. 1983, 43, 2796–2805. [Google Scholar]
- McComb, R.D.; Bigner, D.D. Immunolocalization of Laminin in Neoplasms of the Central and Peripheral Nervous Systems. J. Neuropathol. Exp. Neurol. 1985, 44, 242–253. [Google Scholar] [CrossRef]
- Marienhagen, K.; Bjerkvig, R. Migratory Pattern of Fetal Rat Brain Cells and Human Glioma Cells in the Adult Rat Brain. Cancer Res. 1993, 53, 5158–5165. [Google Scholar]
- Gkretsi, V.; Stylianopoulos, T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 930–935. [Google Scholar] [CrossRef]
- Ranuncolo, S.M.; Ladeda, V.; Specterman, S. CD44 Expression in Human Gliomas. J. Surg. Oncol. 2002, 79, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Kawano, Y.; Matsumoto, M.; Suga, M.; Kaibuchi, K.; Ando, M.; Saya, H. Regulated CD44 Cleavage under the Control of Protein Kinase C, Calcium Influx, and the Rho Family of Small G Proteins. J. Biol. Chem. 1999, 274, 25525–25534. [Google Scholar] [CrossRef] [Green Version]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef] [PubMed]
- Edvardsen, K.; Chent, W.; Rucklidgef, G.; Walsh, F.S.; Obrinkt, B.; Bock, E. Transmembrane neural cell-adhesion molecule (NCAM), but not secretion of matrix metalloproteinases. Proc. Natl. Acad. Sci. USA 2000, 90, 11463–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prag, S.; Lepekhin, E.A.; Kolkova, K.; Hartmann-petersen, R.; Kawa, A.; Walmod, P.S.; Belman, V.; Gallagher, H.C.; Berezin, V.; Bock, E.; et al. NCAM regulates cell motility. J. Cell Sci. 2002, 115, 283–292. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Vanoni, C.; Massari, S.; Raimondi, A.; Pola, S.; Cattaneo, M.G.; Francolini, M.; Vicentini, L.M.; Pietrini, G. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin- catenin adhesion system. J. Cell Sci. 2002, 115, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.B.; Midwood, K.S.; Schwarzbauer, J.E. Tenascin-C Suppresses Rho Activation. J. Cell Biol. 2000, 150, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin signaling in glioma pathogenesis: From biology to therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, A.F. Integrins and health. Sci. Am. 1997, 276, 68–75. [Google Scholar] [CrossRef]
- Biology, C.; Hospital, H. Stimulation of extracellular matrix components in the normal. Int. J. Cancer 1998, 872, 864–872. [Google Scholar]
- Paulus, W.; Baur, I.; Schuppan, D.; Roggendorf, W. Characterization of integrin receptors in normal and neoplastic human brain. Am. J. Pathol. 1993, 143, 154–163. [Google Scholar]
- Gingras, M.C.; Roussel, E.; Bruner, J.M.; Branch, C.D.; Moser, R.P. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J. Neuroimmunol. 1995, 57, 143–153. [Google Scholar] [CrossRef]
- Cary, L.A.; Guan, J.L. Focal adhesion kinase in integrin-mediated signaling. Front. Biosci. 1999, 4, 102–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, E.A.; Huttenlocher, A. Regulation of Integrin-Mediated Adhesion During Cell Migration. Microsc. Res. Tech. 1998, 419, 412–419. [Google Scholar] [CrossRef]
- Munson, J.; Bonner, M.; Fried, L.; Hofmekler, J.; Arbiser, J.; Bellamkonda, R. Identifying new small molecule anti-invasive compounds for glioma treatment. Cell Cycle 2013, 12, 2200–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauck, C.R.; Hsia, D.A.; Schlaepfer, D.D. The Focal Adhesion Kinase—A Regulator of Cell Migration and Invasion Structural Characteristics of FAK-like Protein Tyrosine Kinases Focal adhesion kinase (FAK) 1 together with Pyk2 (1) form a subfamily of FAK-like protein-tyrosine kinases (PTKs). FAK. IUBMB Life 2002, 53, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Owens, L.V.; Xu, L.H.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the Focal Adhesion Kinase (p125FAK) in Invasive Human Tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar]
- Zagzag, D.; Friedlander, D.R.; Margolis, B.; Grumet, M.; Semenza, G.L.; Zhong, H.; Simons, J.W.; Holash, J.; Wiegand, S.J.; Yancopoulos, G.D. Molecular events implicated in brain tumor angiogenesis and invasion. Pediatr. Neurosurg. 2000, 33, 49–55. [Google Scholar] [CrossRef]
- Wilkins-port, C.E.; Freytag, J.; Higgins, S.P.; Higgins, P.J. PAI-1: A Multifunctional SERPIN with Complex Roles in Cell Signaling and Migration. Cell Commun. Insights 2010, 3, 562481. [Google Scholar]
- Deryugina, E.I.; Ratnikov, B.; Monosov, E.; Postnova, T.I.; Discipio, R.; Smith, J.W.; Strongin, A.Y. MT1-MMP Initiates Activation of pro-MMP-2 and Integrin αvβ3 Promotes Maturation of MMP-2 in Breast Carcinoma Cells. Exp. Cell Res. 2001, 223, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Steck, P.A.; Tofilon, P.; Boyd, D.; Aliosman, F.; Stetlerstevenson, W.G.; Liotta, L.A.; Sawaya, R. Role of Plasminogen-Activator and of 92-Kda Type-Iv Collagenase in Glioblastoma Invasion Using an in-Vitro Matrigel Model. J. Neurooncol. 1994, 18, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene 2013, 32, 327–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raithatha, S.A.; Muzik, H.; Muzik, H.; Rnewcastle, N.B.; Johnston, R.N.; Edwards, D.R.; Forsyth, P.A. Localization of gelatinase-A and gelatinase-B mRNA and protein in human gliomas. Neuro-Oncology 2000, 2, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Miyamori, H.; Kita, D. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005, 4, 239–246. [Google Scholar] [CrossRef]
- Held-feindt, J.; Paredes, E.B.; Bl, U.; Seidenbecher, C.; Stark, A.M.; Mehdorn, H.M.; Mentlein, R. Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int. J. Cancer 2006, 61, 55–61. [Google Scholar] [CrossRef]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.J.; Wang, S.Y.; Xie, G.F.; Zeng, Q.; Chen, C.; Dong, A.N.; Huang, Z.M. Subdivision of M category for nasopharyngeal carcinoma with synchronous metastasis: Time to expand the M categorization system. Chin. J. Cancer 2015, 34, 40. [Google Scholar] [CrossRef] [Green Version]
- Geis, T.; Döring, C.; Popp, R.; Grossmann, N.; Fleming, I.; Hansmann, M.; Dehne, N.; Brüne, B. HIF-2alpha-dependent PAI-1 induction contributes to angiogenesis in hepatocellular carcinoma. Exp. Cell Res. 2014, 331, 46–57. [Google Scholar] [CrossRef]
- Colin, C.; Voutsinos-Porche, B.; Nanni, I.; Fina, F.; Metellus, P.; Intagliata, D.; Baeza, N.; Bouvier, C.; Delfino, C.; Loundou, A.; et al. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol. 2009, 118, 745. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Hayama, M.; Adachi, A.; Matsutani, T. High Serum Level of Plasminogen Activator Inhibitor-1 Predicts Histological Grade of Intracerebral Gliomas. Anticancer Res. 2008, 418, 415–418. [Google Scholar]
- Ford, H.; Hospital, H.F.; State, W. Immunolocalization of cathepsin B in human glioma: Implications for tumor invasion and angiogenesis. J. Neurosurg. 1995, 83, 285–290. [Google Scholar]
- Gondi, C.S.; Lakka, S.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Tung, C.H.; Weissleder, R.; Rao, J.S. Advances in Brief Adenovirus-Mediated Expression of Antisense Urokinase Plasminogen Activator Receptor and Antisense Cathepsin B Inhibits Tumor Growth, Invasion, and Angiogenesis in Gliomas. Cancer Res. 2004, 4069–4077. [Google Scholar] [CrossRef] [Green Version]
- Demchik, L.L.; Sameni, M.; Nelson, K.; Mikkelsen, T.; Sloane, B.F. Cathepsin B and glioma invasion. Int. J. Dev. Neurosci. 1999, 17, 483–494. [Google Scholar] [CrossRef]
- Sivaparvathi, M.; Sawaya, R.; Wu Wang, S.; Rayford, A.; Yamamoto, M.; Liottat, L.A.; Nicolson, G.L.; Rao, J.S. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin. Exp. Metastasis 1995, 13, 49–56. [Google Scholar] [CrossRef]
- Tysnes, B.B.; Mahesparan, R. Biological mechanisms of glioma invasion and potential therapeutic targets. J. Neurooncol. 2001, 53, 129–147. [Google Scholar] [CrossRef]
- Nicholson-Dykstra, S.; Higgs, H.N.; Harris, E.S. Actin dynamics: Growth from dendritic branches. Curr. Biol. 2005, 15, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Whale, A.; Hashim, F.N.; Fram, S.; Jones, G.E.; Wells, C.M. Signalling to cancer cell invasion through PAK family kinases. Front. Biosci. 2011, 16, 849–864. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Oikawa, T. Membrane lipids in invadopodia and podosomes: Key structures for cancer invasion and metastasis. Oncotarget 2010, 1, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta-Mol. Cell Res. 2007, 1773, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Han, M.; Whetsell, W.; Wang, J.; Rich, J.; Hallahan, D.; Han, Z. Tax-interacting protein 1 coordinates the spatiotemporal activation of Rho GTPases and regulates the infiltrative growth of human glioblastoma. Oncogene 2014, 33, 1558–1569. [Google Scholar] [CrossRef] [Green Version]
- Lamszus, K.; Schmidt, N.O.; Jin, L.; Laterra, J.; Zagzag, D.; Way, D.; Witte, M.; Weinand, M.; Goldberg, I.D.; Westphal, M.; et al. Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int. J. Cancer 1998, 75, 19–28. [Google Scholar] [CrossRef]
- Watanabe, K.; Tachibana, O.; Sato, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Lund-johansen, M.; Bjcrkvig, R.; Humphrey, P.A.; Bigner, S.H.; Bigner, D.D.; Laerum, O. Effect of Epidermal Growth Factor on Glioma Cell Growth, Migration, and Invasion in Vitro. Cancer Res. 1990, 50, 6039–6044. [Google Scholar]
- Coniglio, S.J.; Segall, J.E. Microglial-stimulation of glioma invasion involves the EGFR ligand amphiregulin. PLoS ONE 2021, 16, e0260252. [Google Scholar] [CrossRef]
- Joseph, J.V.; Magaut, C.R.; Storevik, S.; Geraldo, L.H.; Mathivet, T.; Latif, M.A.; Rudewicz, J.; Guyon, J.; Gambaretti, M.; Haukas, F.; et al. TGF-β promotes microtube formation in glioblastoma through Thrombospondin 1. Neuro. Oncol. 2021, 1–10. [Google Scholar] [CrossRef]
- Hemler, M.E. Integrin associated proteins. Curr. Opin. Cell Biol. 1998, 10, 578–585. [Google Scholar] [CrossRef]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukierman, E.; Cukierman, E.; Pankov, R.; Stevens, D.R. Taking Cell-Matrix Adhesions to the Third Dimension. Science 2013, 294, 1708–1712. [Google Scholar] [CrossRef]
- Rabinovitz, I.; Mercurio, A.M. The Integrin α6β4 Functions in Carcinoma Cell Migration on Laminin-1 by Mediating the Formation and Stabilization of Actin-containing Motility Structures. J. Cell Biol. 1997, 139, 1873–1884. [Google Scholar] [CrossRef]
- De Semir, D.; Bezrookove, V.; Nosrati, M.; Scanlon, K.R.; Singer, E.; Judkins, J.; Rieken, C.; Wu, C.; Shen, J.; Schmudermayer, C.; et al. PHIP drives glioblastoma motility and invasion by regulating the focal adhesion complex. Proc. Natl. Acad. Sci. USA 2020, 117, 9064–9073. [Google Scholar] [CrossRef]
- Porčnik, A.; Novak, M.; Breznik, B.; Majc, B.; Hrastar, B.; Šamec, N.; Zottel, A.; Jovčevska, I. TRIM28 Selective Nanobody Reduces Glioblastoma Stem Cell Invasion. Molecules 2021, 26, 5141. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, M.; Yu, S.; Hai, L.; Zhang, L.; Zhang, C.; Zhao, P.; Zhou, H.; Wang, S.; Yang, X. Arg kinase mediates CXCL12/CXCR4-induced invadopodia formation and invasion of glioma cells. Exp. Cell Res. 2020, 389, 111893. [Google Scholar] [CrossRef]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef] [Green Version]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [Green Version]
- Caramia, M.; Sforna, L.; Franciolini, F.; Catacuzzeno, L. The volume-regulated anion channel in glioblastoma. Cancers 2019, 11, 307. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Long, S.; Wu, B.; Liu, J.; Li, G. NKCC1 involvement in the epithelial-to-mesenchymal transition is a prognostic biomarker in gliomas. PeerJ 2020, 8, e8787. [Google Scholar] [CrossRef]
- Zou, W. Potassium Channel and Glioma. Biomed. J. Sci. Tech. Res. 2019, 16, 12179–12184. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 channels in modulating Ca2+ oscillations during glioblastoma cell migration and invasion. Int. J. Mol. Sci. 2018, 19, 2970. [Google Scholar] [CrossRef] [Green Version]
- Thuringer, D.; Chanteloup, G.; Boucher, J.; Pernet, N.; Boudesco, C.; Jego, G.; Chatelier, A.; Bois, P.; Gobbo, J.; Cronier, L.; et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017, 8, 37681–37693. [Google Scholar] [CrossRef] [Green Version]
- Brandalise, F.; Ratto, D.; Leone, R.; Olivero, F.; Roda, E.; Locatelli, C.A.; Grazia Bottone, M.; Rossi, P. Deeper and Deeper on the Role of BK and Kir4.1 Channels in Glioblastoma Invasiveness: A Novel Summative Mechanism? Front. Neurosci. 2020, 14, 595664. [Google Scholar] [CrossRef]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z.-G. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell. Signal. 2014, 26, 2773–2781. [Google Scholar] [CrossRef] [Green Version]
- Bao, M.H.; Lv, Q.L.; Szeto, V.; Wong, R.; Zhu, S.Z.; Zhang, Y.Y.; Feng, Z.P.; Sun, H.S. TRPM2-AS inhibits the growth, migration, and invasion of gliomas through JNK, c-Jun, and RGS4. J. Cell. Physiol. 2020, 235, 4594–4604. [Google Scholar] [CrossRef]
- Wong, R.; Gong, H.; Alanazi, R.; Bondoc, A.; Luck, A.; Sabha, N.; Horgen, F.D.; Fleig, A.; Rutka, J.T.; Feng, Z.P.; et al. Inhibition of TRPM7 with waixenicin A reduces glioblastoma cellular functions. Cell Calcium 2020, 92, 102307. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Chu, Y.; Cegielski, V.; Chu, X.P. Acid-Sensing Ion Channel 1 Contributes to Weak Acid-Induced Migration of Human Malignant Glioma Cells. Front. Physiol. 2021, 12, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Thompson, R.; Hardy, K.; DeCamp, L.; Berghuis, B.; Sigler, R.; Knudsen, B.; Cottingham, S.; Zhao, P.; Dykema, K.; et al. A highly invasive human glioblastoma pre-clinical model for testing therapeutics. J. Transl. Med. 2008, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Jia, Y.; Feng, Q.; Tang, B.; Luo, X.; Yang, Q.; Yang, H.; Li, Q. Decorin Suppresses Invasion and EMT Phenotype of Glioma by Inducing Autophagy via c-Met/Akt/mTOR Axis. Front. Oncol. 2021, 11, 659353. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2014, 13, 963–972. [Google Scholar] [CrossRef]
- Myung, J.K.; Choi, S.A.; Kim, S.K.; Wang, K.C.; Park, S.H. Snail plays an oncogenic role in glioblastoma by promoting epithelial mesenchymal transition. Int. J. Clin. Exp. Pathol. 2014, 7, 1977–1987. [Google Scholar]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Jiang, L.; Wang, X.; Wei, W.; Song, C.; Cui, Y.; Wu, X.; Qiu, G. P4HA2 Promotes Epithelial-to-Mesenchymal Transition and Glioma Malignancy through the Collagen-Dependent PI3K/AKT Pathway. J. Oncol. 2021, 2021, 1406853. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, X.; Wang, J.; Ji, R.; Zhang, L.; Qin, J.; Tian, M.; Jin, G.; Zhang, X. P4HA2 promotes cell proliferation and migration in glioblastoma. Oncol. Lett. 2021, 22, 601. [Google Scholar] [CrossRef]
- Storci, G.; Sansone, P.; Mari, S.; D’Uva, G.; Tavolari, S.; Guarnieri, T.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Chieco, P.; et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010, 225, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kong, J.; Chang, H.; Kim, H.; Kim, A. EGF induces epithelial-mesenchymal transition through phospho-Smad2/3-Snail signaling pathway in breast cancer cells. Oncotarget 2016, 7, 85021–85032. [Google Scholar] [CrossRef] [Green Version]
- Mariani, L.; McDonough, W.S.; Hoelzinger, D.B.; Beaudry, C.; Kacsmarek, E.; Coons, S.W.; Giese, A.; Moghaddam, M.; Seiler, R.W.; Berens, M.E. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 2001, 61, 4190–4196. [Google Scholar]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene Expression Profile of Glioblastoma Multiforme Invasive Phenotype Points to New Therapeutic Targets. Neoplasia 2006, 7, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Demuth, T.; Reavie, L.B.; Rennert, J.L.; Nakada, M.; Nakada, S.; Hoelzinger, D.B.; Beaudry, C.E.; Henrichs, A.N.; Anderson, E.M.; Berens, M.E. MAP-ing glioma invasion: Mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol. Cancer Ther. 2007, 6, 1212–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuth, T.; Rennert, J.L.; Hoelzinger, D.B.; Reavie, L.B.; Nakada, M.; Beaudry, C.; Nakada, S.; Anderson, E.M.; Henrichs, A.N.; McDonough, W.S.; et al. Glioma cells on the run—The migratory transcriptome of 10 human glioma cell lines. BMC Genom. 2008, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro. Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seker, F.; Cingoz, A.; Sur-Erdem, İ.; Erguder, N.; Erkent, A.; Uyulur, F.; Selvan, M.E.; Gümüş, Z.H.; Gönen, M.; Bayraktar, H.; et al. Identification of SERPINE1 as a regulator of glioblastoma cell dispersal with transcriptome profiling. Cancers 2019, 11, 1651. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Zhang, C.; Yao, H.; Zhang, X.; Zhou, Y.; Che, Y.; Huang, Y. Knockdown of long non-coding RNA NEAT1 inhibits glioma cell migration and invasion via modulation of SOX2 targeted by miR-132. Mol. Cancer 2018, 17, 1651. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, Y.; Sidlauskas, K.; Ellis, M.; Evans, I.; Frankel, P.; Lau, J.; El-Hassan, T.; Guglielmi, L.; Broni, J.; et al. Inhibition of GPR158 by microRNA-449a suppresses neural lineage of glioma stem/progenitor cells and correlates with higher glioma grades. Oncogene 2018, 37, 4313–4333. [Google Scholar] [CrossRef] [Green Version]
- Haas, T.L.; Sciuto, M.R.; Brunetto, L.; Valvo, C.; Signore, M.; Fiori, M.E.; di Martino, S.; Giannetti, S.; Morgante, L.; Boe, A.; et al. Integrin α7 Is a Functional Marker and Potential Therapeutic Target in Glioblastoma. Cell Stem Cell 2017, 21, 35–50.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallawaaratchy, D.M.; Buckland, M.E.; McDonald, K.L.; Li, C.C.Y.; Ly, L.; Sykes, E.K.; Christopherson, R.I.; Kaufman, K.L. Membrane Proteome Analysis of Glioblastoma Cell Invasion. J. Neuropathol. Exp. Neurol. 2015, 74, 425–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Moon, K.-S.; Pan, S.; Lee, K.-H.; Ryu, H.-H.; Jung, T.-Y.; Kim, I.-Y.; Jang, W.-Y.; Jung, C.-H.; Jung, S. Proteomic Analysis between U87MG and U343MG-A Cell Lines: Searching for Candidate Proteins for Glioma Invasion. Brain Tumor Res. Treat. 2014, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Bagci, T.; Wu, J.K.; Pfannl, R.; Ilag, L.L.; Jay, D.G. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 2009, 28, 3537–3550. [Google Scholar] [CrossRef] [Green Version]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarzynka, M.J.; Hu, B.; Hui, K.M.; Bar-Joseph, I.; Gu, W.; Hirose, T.; Haney, L.B.; Ravichandran, K.S.; Nishikawa, R.; Cheng, S.Y. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res. 2007, 67, 7203–7211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goplen, D.; Wang, J.; Enger, P.; Tysnes, B.B.; Terzis, A.J.A.; Laerum, O.D.; Bjerkvig, R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006, 66, 9895–9902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Yang, H.; Zhu, X.-L.; Zhang, Y.; Kun, L. Knockdown of long non-coding RNA SLC8A1-AS1 attenuates cell invasion and migration in glioma via suppression of Wnt/β-catenin signaling pathways. Brain Res. Bull. 2021, 176, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Jin, B.; Huang, L.; Zhou, W. MiR-129-5p inhibits glioma cell progression in vitro and in vivo by targeting TGIF2. J. Cell. Mol. Med. 2018, 22, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, Q.; Zeng, X.; Li, M.; Liao, J. Lnc-NLC1-C inhibits migration, invasion and autophagy of glioma cells by targeting miR-383 and regulating PRDX-3 expression. Oncol. Lett. 2021, 22, 640. [Google Scholar] [CrossRef]
- Lin, Y.; Wei, L.; Hu, B.; Zhang, J.; Wei, J.; Qian, Z.; Zou, D. RBM8A Promotes Glioblastoma Growth and Invasion Through the Notch/STAT3 Pathway. Front. Oncol. 2021, 11, 736941. [Google Scholar] [CrossRef]
- Formolo, C.A.; Williams, R.; Gordish-Dressman, H.; MacDonald, T.J.; Lee, N.H.; Hathout, Y. Secretome Signature of Invasive Glioblastoma Multiforme. J. Proteome Res. 2011, 10, 3149–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzikowski, L.; Mirzaei, R.; Sarkar, S.; Kumar, M.; Bose, P.; Bellail, A.; Hao, C.; Yong, V.W. Fibrinogen in the glioblastoma microenvironment contributes to the invasiveness of brain tumor-initiating cells. Brain Pathol. 2021, 31, e12947. [Google Scholar] [CrossRef]
- Gritsenko, P.; Leenders, W.; Friedl, P. Recapitulating in vivo-like plasticity of glioma cell invasion along blood vessels and in astrocyte-rich stroma. Histochem. Cell Biol. 2017, 148, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Cofano, F.; Salvati, L.F.; Monticelli, M.; Zeppa, P.; Di Perna, G.; Melcarne, A.; Altieri, R.; La Rocca, G.; Sabatino, G.; et al. Fluorescence-Guided Surgery for High-Grade Gliomas: State of the Art and New Perspectives. Technol. Cancer Res. Treat. 2021, 20, 1–11. [Google Scholar] [CrossRef]
- Pekmezci, M.; Morshed, R.A.; Chunduru, P.; Pandian, B.; Young, J.; Villanueva-Meyer, J.E.; Tihan, T.; Sloan, E.A.; Aghi, M.K.; Molinaro, A.M.; et al. Detection of glioma infiltration at the tumor margin using quantitative stimulated Raman scattering histology. Sci. Rep. 2021, 11, 12162. [Google Scholar] [CrossRef]
- Castro, L.N.G.; Tirosh, I.; Suvà, M.L. Decoding cancer biology one cell at a time. Cancer Discov. 2021, 11, 960–970. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2011, 5, 102–114. [Google Scholar] [CrossRef]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Hu, J.A.; Zheng, J.S. Mechanism of temozolomide-induced antitumour effects on glioma cells. J. Int. Med. Res. 2014, 42, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Somasundaram, K. Glioblastoma vs temozolomide: Can the red queen race be won? Cancer Biol. Ther. 2019, 20, 1083–1090. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, J.; Wang, T.; Wang, K.; Wu, Z.; Sun, T. Efficacy and safety of bevacizumab in the treatment of adult gliomas: A systematic review and meta-analysis. BMJ Open 2021, 11, e048975. [Google Scholar] [CrossRef]
- Dhermain, F. Radiotherapy of high-grade gliomas: Current standards and new concepts, innovations in imaging and radiotherapy, and new therapeutic approaches. Chin. J. Cancer 2014, 33, 16–24. [Google Scholar] [CrossRef]
- Brem, S.; Abdullah, K.G. Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Singapore, 2017; ISBN 9780994438126. [Google Scholar]
- Mooney, J.; Bernstock, J.D.; Ilyas, A.; Ibrahim, A.; Yamashita, D.; Markert, J.M.; Nakano, I. Current Approaches and Challenges in the Molecular Therapeutic Targeting of Glioblastoma. World Neurosurg. 2019, 129, 90–100. [Google Scholar] [CrossRef]
- Lu, C.; Shervington, A. Chemoresistance in gliomas. Mol. Cell. Biochem. 2008, 312, 71–80. [Google Scholar] [CrossRef]
- Ghia, A.J. Fractionated Radiotherapy of Intracranial Gliomas. Prog. Neurol. Surg. 2018, 31, 38–47. [Google Scholar] [PubMed]
- Larson, E.W.; Peterson, H.E.; Lamoreaux, W.T.; MacKay, A.R.; Fairbanks, R.K.; Call, J.A.; Carlson, J.D.; Ling, B.C.; Demakas, J.J.; Cooke, B.S.; et al. Clinical outcomes following salvage Gamma Knife radiosurgery for recurrent glioblastoma. World J. Clin. Oncol. 2014, 5, 142–148. [Google Scholar] [CrossRef]
- Moghaddasi, L.; Bezak, E. Development of an integrated Monte Carlo model for glioblastoma multiforme treated with boron neutron capture therapy. Sci. Rep. 2017, 7, 7069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schijns, V.E.J.C.; Pretto, C.; Devillers, L.; Pierre, D.; Hofman, F.M.; Chen, T.C.; Mespouille, P.; Hantos, P.; Glorieux, P.; Bota, D.A.; et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine 2015, 33, 2690–2696. [Google Scholar] [CrossRef] [Green Version]
- Martikainen, M.; Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagci-Onder, T.; Wakimoto, H.; Anderegg, M.; Cameron, C.; Shah, K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2011, 71, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbalý, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer 2012, 48, 2192–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma a randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A state of the science review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirson, E.D.; Dbalý, V.; Tovaryš, F.; Vymazal, J.; Soustiel, J.F.; Itzhaki, A.; Mordechovich, D.; Steinberg-Shapira, S.; Gurvich, Z.; Schneiderman, R.; et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 10152–10157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanner, A.A.; Wong, E.T.; Villano, J.L.; Ram, Z. Post hoc analyses of intention-to-treat population in phase III comparison of NovoTTF-100ATM system versus best physician’s choice chemotherapy. Semin. Oncol. 2014, 41, S25–S34. [Google Scholar] [CrossRef] [Green Version]
- Michelhaugh, S.K.; Kiousis, S.; Michelhaugh, S.A.; Klinger, N.V.; Mittal, S. Abstract 4398: In vitro Tumor Treating Fields (TTFields) alter proliferation and morphology of patient-derived high-grade meningioma cell lines. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; p. 4398. [Google Scholar]
- Giladi, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Munster, M.; Blat, R.; Sherbo, S.; Bomzon, Z.; Urman, N.; Itzhaki, A.; et al. Mitotic Spindle Disruption by Alternating Electric Fields Leads to Improper Chromosome Segregation and Mitotic Catastrophe in Cancer Cells. Sci. Rep. 2015, 5, 18046. [Google Scholar] [CrossRef] [PubMed]
- Ornelas, A.S.; Porter, A.B.; Sharma, A.; Knox, M.G.; Marks, L.A.; Wingerchuk, D.M.; O’Carroll, C.B. What is the role of tumor-Treating fields in newly diagnosed glioblastoma? Neurologist 2019, 24, 71–73. [Google Scholar] [CrossRef]
- Guzauskas, G.F.; Salzberg, M.; Wang, B.C. Estimated lifetime survival benefit of tumor treating fields and temozolomide for newly diagnosed glioblastoma patients. CNS Oncol. 2018, 7, CNS23. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Klinger, N.V.; Michelhaugh, S.K.; Barger, G.R.; Pannullo, S.C.; Juhász, C. Alternating electric tumor treating fields for treatment of glioblastoma: Rationale, preclinical, and clinical studies. J. Neurosurg. 2018, 128, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; De Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro. Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliemroth, J.; Feyerabend, T.; Gerlach, C.; Arnold, H.; Terzis, A.J.A. Proliferation, migration, and invasion of human glioma cells exposed to fractionated radiotherapy in vitro. Neurosurg. Rev. 2003, 26, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Wild-bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal Irradiation Promotes Migration and Invasiveness of Glioma Cells: Implications for Radiotherapy of Human Glioblastoma Sublethal Irradiation Promotes Migration and Invasiveness of Glioma Cells: Implications for Radiotherapy of Human Glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Giannelli, G.; Villa, E.; Lahn, M. Transforming Growth Factor-β as a Therapeutic Target in Hepatocellular Carcinoma. Cancer Res. 2014, 74, 1890–1895. [Google Scholar] [CrossRef] [Green Version]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, F.; Le, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Levin, V.A.; Phuphanich, S.; Yung, W.K.A.; Forsyth, P.A.; Del Maestro, R.; Perry, J.R.; Fuller, G.N.; Baillet, M. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J. Neurooncol. 2006, 78, 295–302. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.Y.; et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seker-Polat, F.; Pinarbasi Degirmenci, N.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020443
Seker-Polat F, Pinarbasi Degirmenci N, Solaroglu I, Bagci-Onder T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers. 2022; 14(2):443. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020443
Chicago/Turabian StyleSeker-Polat, Fidan, Nareg Pinarbasi Degirmenci, Ihsan Solaroglu, and Tugba Bagci-Onder. 2022. "Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives" Cancers 14, no. 2: 443. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020443