
Deciphering Biomarkers for Leptomeningeal Metastasis in Malignant Hemopathies (Lymphoma/Leukemia) Patients by Comprehensive Multipronged Proteomics Characterization of Cerebrospinal Fluid

 , , , , , , ,
, , , , , , ,
Abstract
:Simple Summary
Abstract
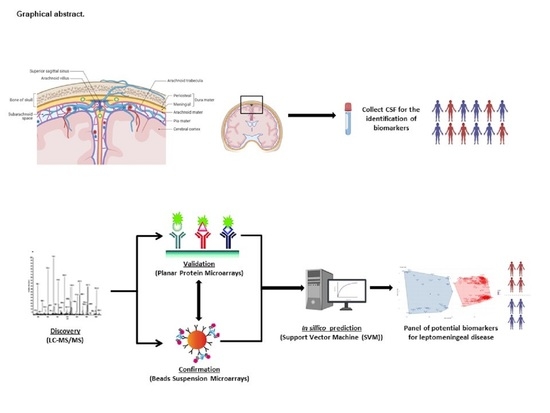
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.1.1. CSF Samples from LM Patients
2.1.2. CSF Samples from Control Subjects
2.2. LC-MS/MS Characterization
2.2.1. Sample Preparation for LC-MS/MS Characterization
2.2.2. Protein Depletion Strategies
2.2.3. SDS-PAGE Separation
2.2.4. In-Gel Digestion and Nano-UPLC-MS/MS Analysis
2.3. Multipronged Proteomic Screenings
2.3.1. CSF Biotinylation for Protein Microarrays Screening
2.3.2. Customized Protein Microarrays
2.3.3. Customized Beads Suspension Microarrays
2.4. Data Analysis and Biostatistics
2.4.1. Mass Spectrometry Datasets
2.4.2. LC-MS/MS Data Analysis
2.4.3. Functional Enrichment Analysis
2.4.4. Sharing of Data through PRIDE
2.4.5. Protein Microarray Datasets
2.4.6. Beads Suspension Microarrays Datasets
2.4.7. In Silico Prediction Datasets of Potential Biomarkers Candidates
3. Results
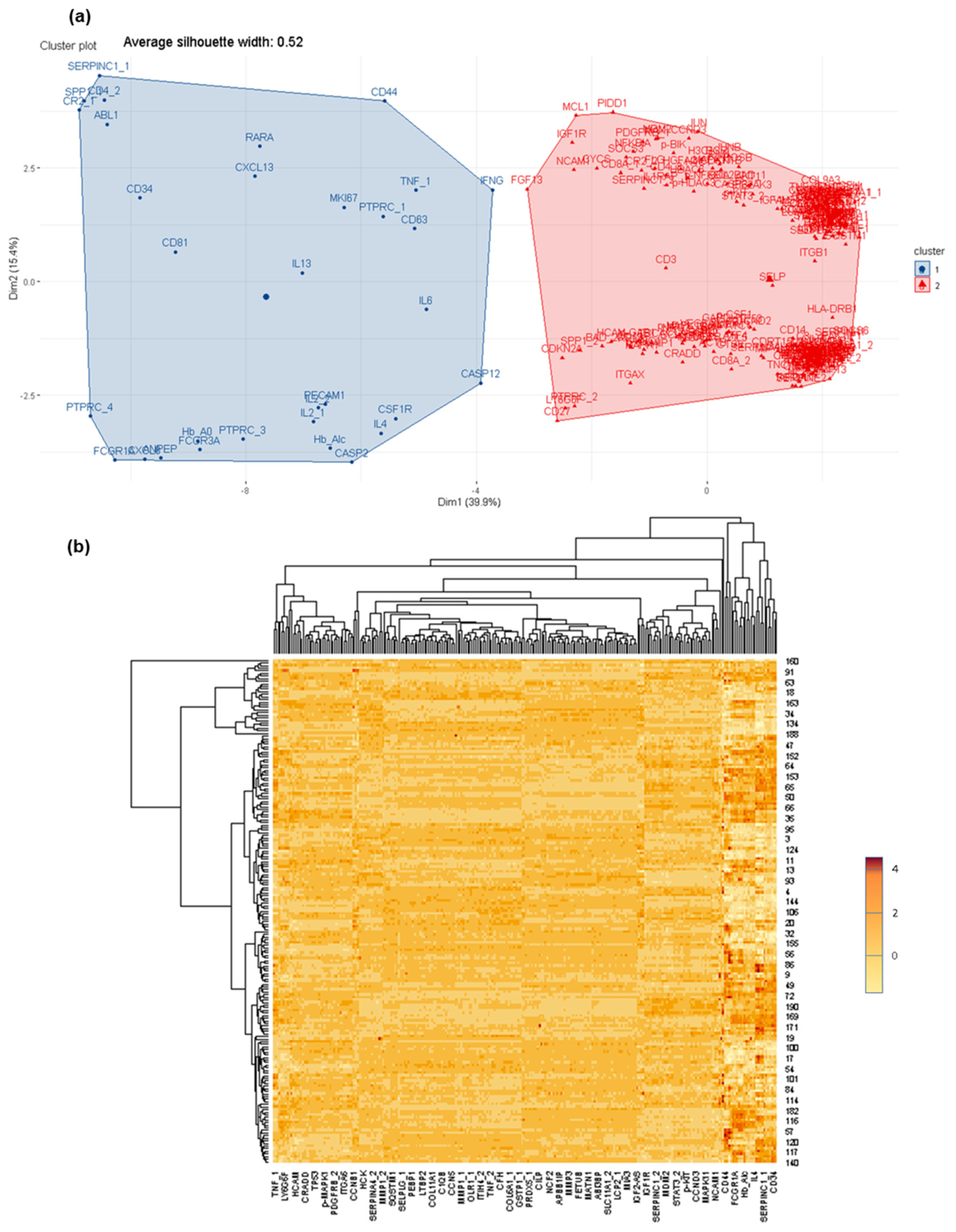
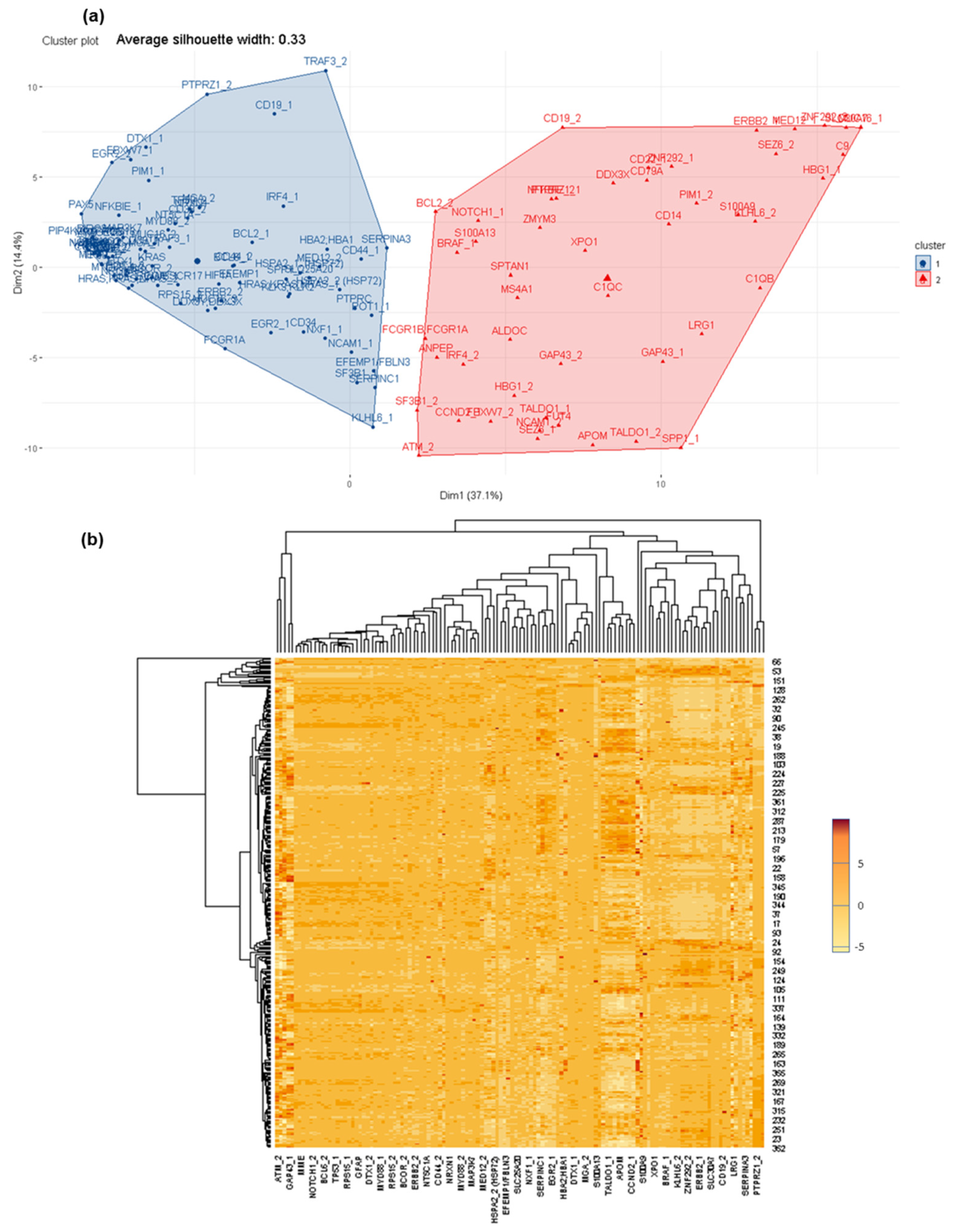
3.1. Determination of Global Differential Protein Profiles in CSF
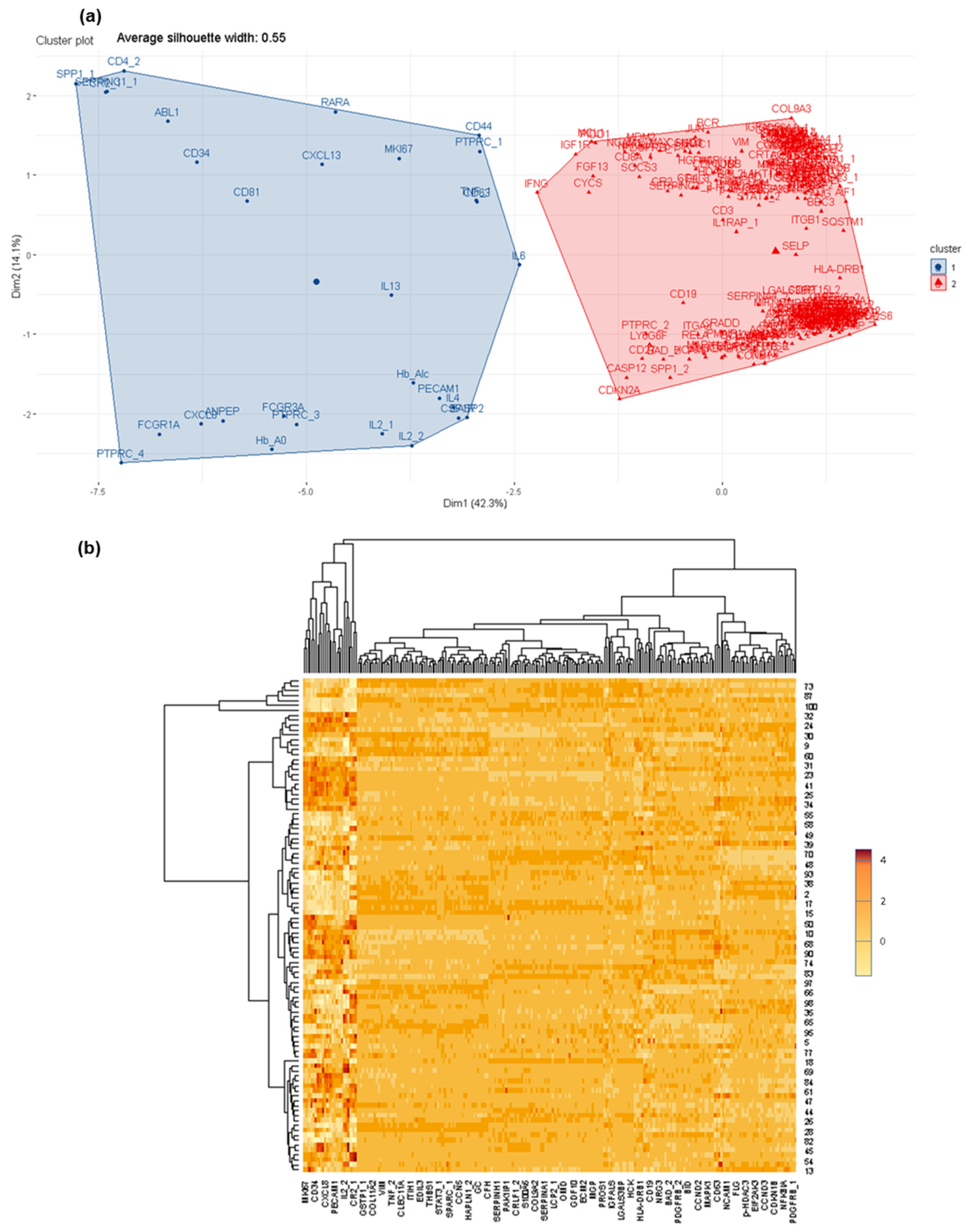
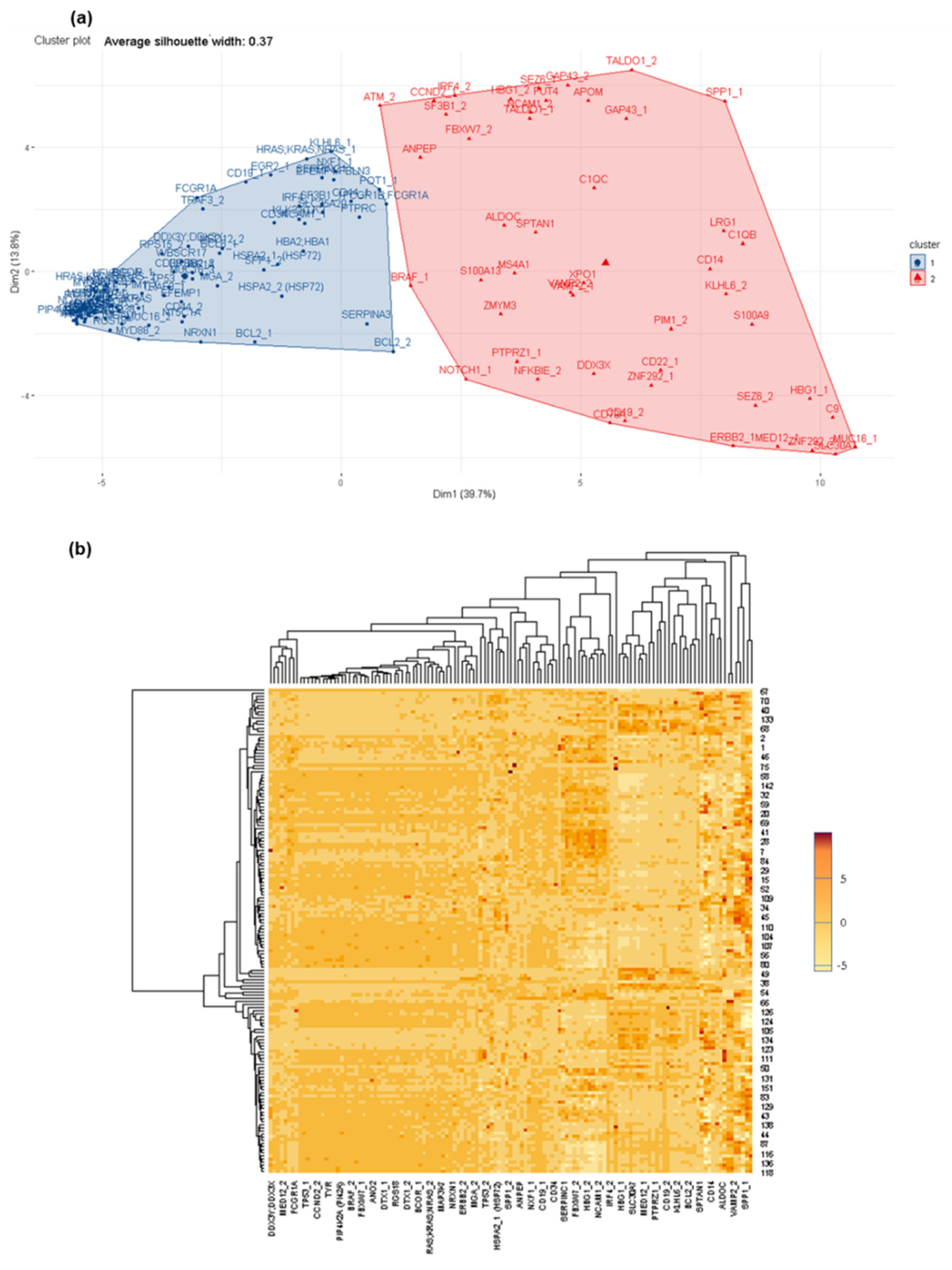
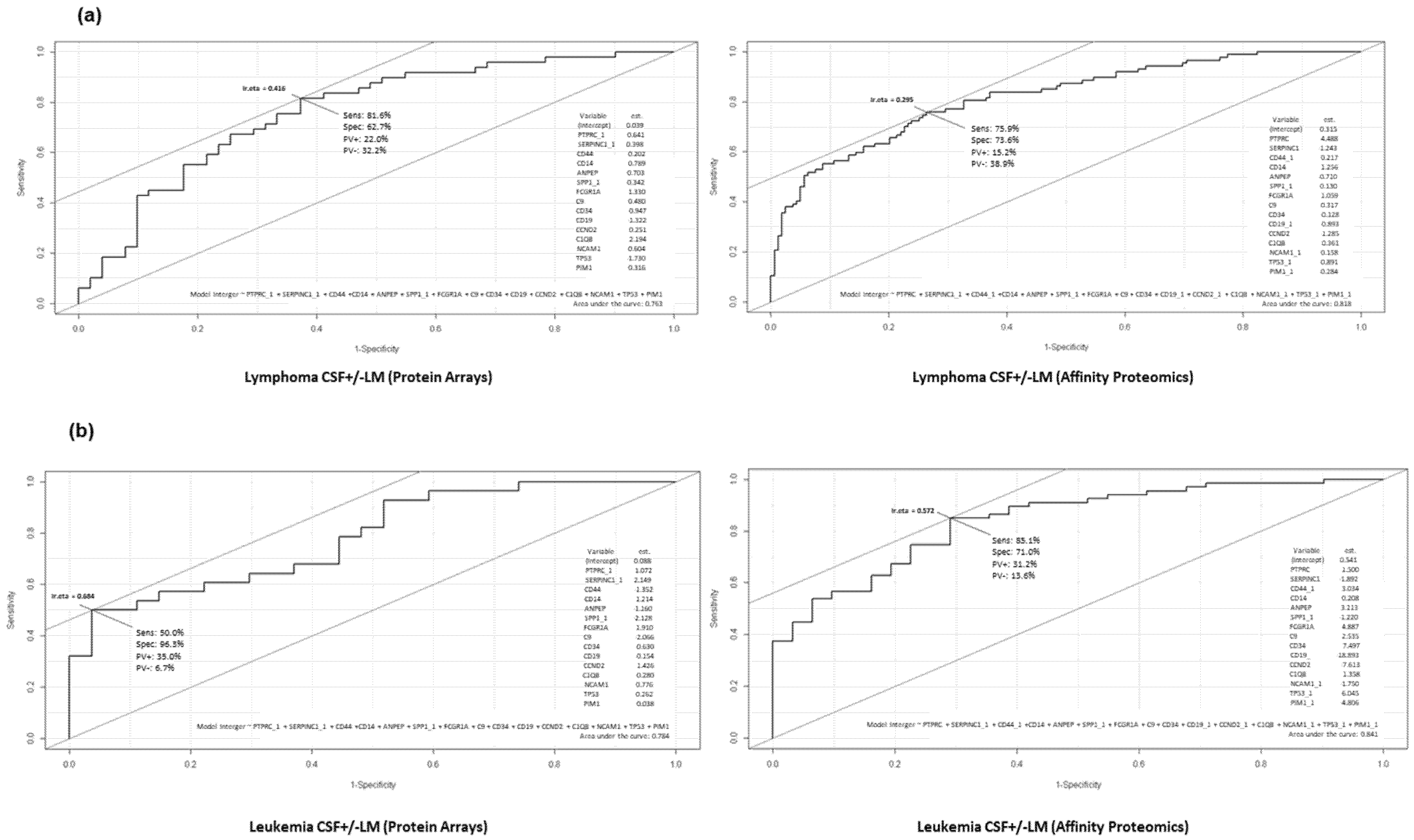
3.2. Deciphering Differential Protein Profiles in CSF by Customized Protein Arrays
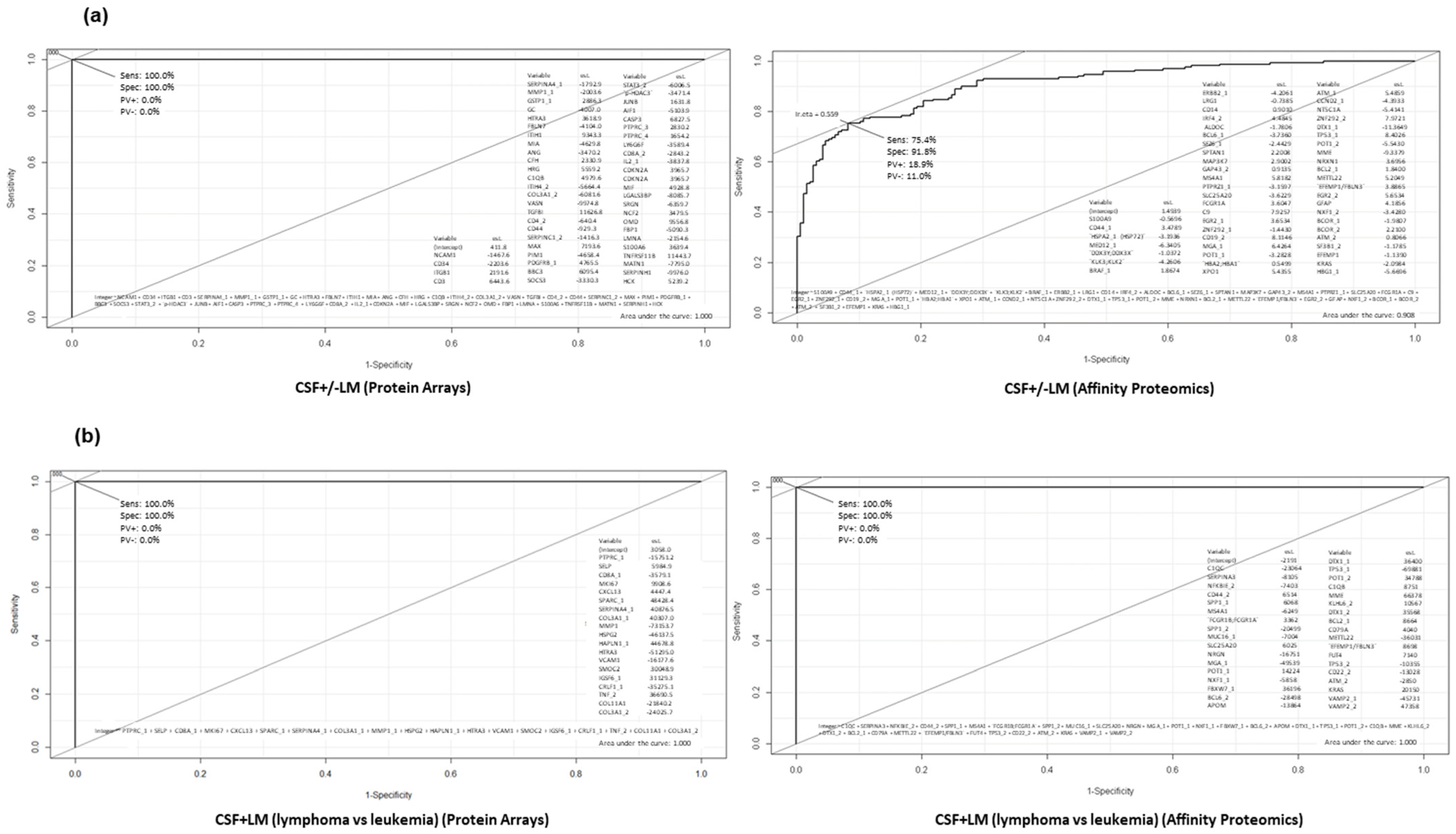
3.3. Confirmation of Differential Protein Profiles in CSF by Affinity Proteomics
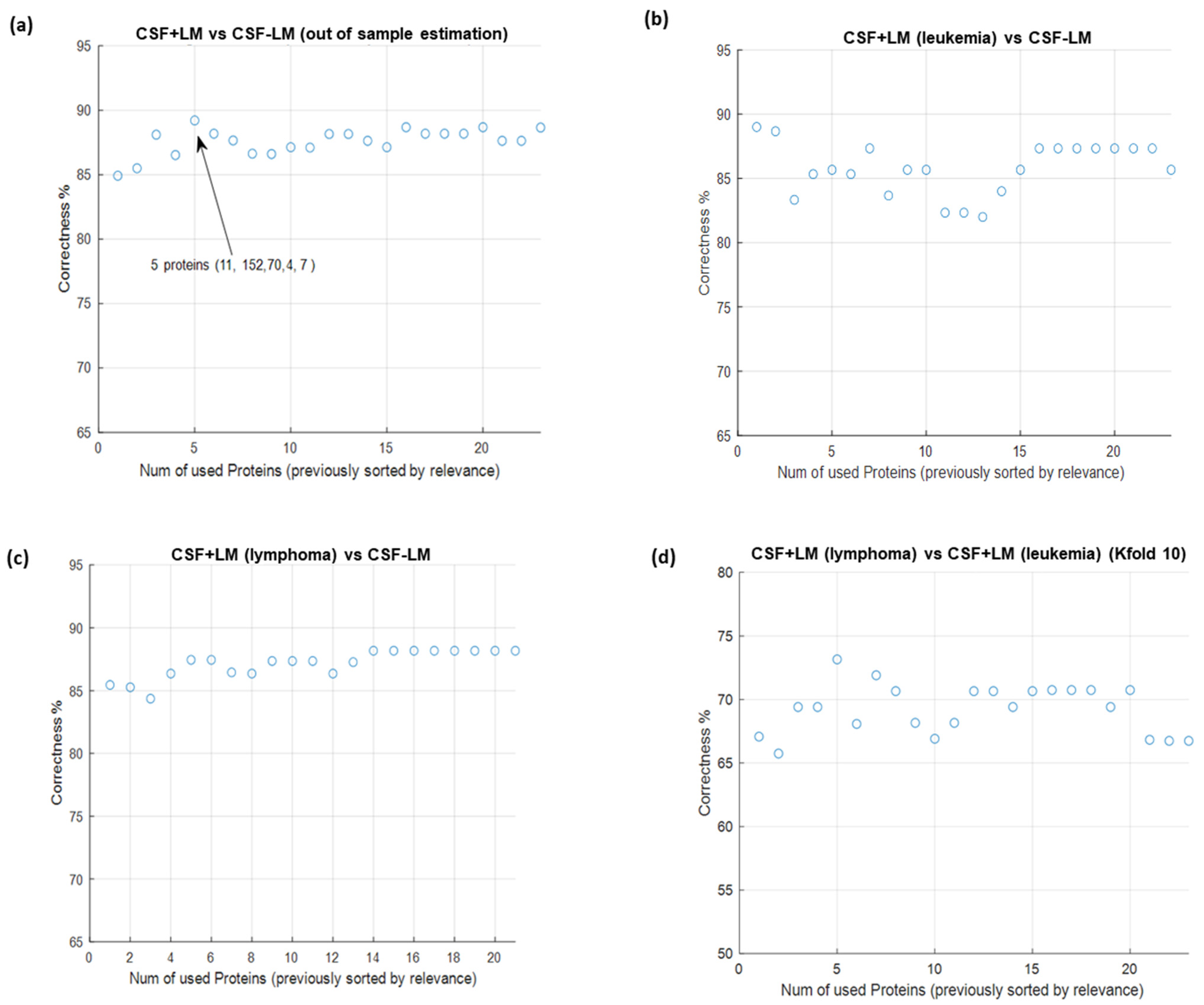
3.4. In Silico Prediction of Potential Diagnostic Biomarkers for LM in CSF
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corbin, Z.A.; Nagpal, S. Leptomeningeal Metastases. JAMA Oncol. 2016, 2, 839. [Google Scholar] [CrossRef]
- Sahebjam, S.; Forsyth, P.A.; Smalley, K.S.; Tran, N.D. Experimental Treatments for Leptomeningeal Metastases From Solid Malignancies. Cancer Control 2017, 24, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Rhun, E.; Galanis, E. Leptomeningeal metastases of solid cancer. Curr. Opin. Neurol. 2016, 29, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Mack, F.; Baumert, B.G.; Schäfer, N.; Hattingen, E.; Scheffler, B.; Herrlinger, U.; Glas, M. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat. Rev. 2016, 43, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P. Intrathecal chemotherapy for treatment of leptomeningeal dissemination of metastatic tumours. Lancet Oncol. 2010, 11, 871–879. [Google Scholar] [CrossRef]
- Joshi, A.; Ghosh, J.; Noronha, V.; Parikh, P.M.; Prabhash, K. Leptomeningeal metastasis in solid tumors with a special focus on lung cancer. Indian J. Cancer 2014, 51, 410–413. [Google Scholar]
- Galicia, N.; Dégano, R.; Díez, P.; González-González, M.; Góngora, R.; Ibarrola, N.; Fuentes, M. CSF analysis for protein biomarker identification in patients with leptomeningeal metastases from CNS lymphoma. Expert Rev. Proteom. 2017, 14, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Weller, M.; Brandsma, D.; Van den Bent, M.; De Azambuja, E.; Henriksson, R.; Preusser, M. EANO–ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up of patients with leptomeningeal metastasis from solid tumours. Ann. Oncol. 2017, 28, iv84–iv99. [Google Scholar] [CrossRef] [PubMed]
- Subirá, D.; Simó, M.; Illán, J.; Serrano, C.; Castañón, S.; Gonzalo, R.; Granizo, J.J.; Martínez-García, M.; Navarro, M.; Pardo, J.; et al. Diagnostic and prognostic significance of flow cytometry immunophenotyping in patients with leptomeningeal carcinomatosis. Clin. Exp. Metastasis 2015, 32, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Pellerino, A.; Brastianos, P.K.; Rudà, R.; Soffietti, R. Leptomeningeal Metastases from Solid Tumors: Recent Advances in Diagnosis and Molecular Approaches. Cancers 2021, 13, 2888. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ye, L.; Huang, L.; Zhou, L.; Chen, X.; Ye, M.; Song, Y. Serum Exosomal miRNA Might Be a Novel Liquid Biopsy to Identify Leptomeningeal Metastasis in Non-Small Cell Lung Cancer. OncoTargets Ther. 2021, 14, 2327. [Google Scholar] [CrossRef] [PubMed]
- van Bussel, M.T.; Pluim, D.; Bol, M.; Beijnen, J.H.; Schellens, J.H.; Brandsma, D. EpCAM-based assays for epithelial tumor cell detection in cerebrospinal fluid. J. Neuro-Oncol. 2018, 137, 1–10. [Google Scholar] [CrossRef]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Tu, Q.; Bittencourt, M.D.C.; Farre, I.; Mortier, L.; Cai, H.; Faure, G.C. Detection and quantification of CSF malignant cells by the CellSearch® technology in patients with melanoma leptomeningeal metastasis. Med. Oncol. 2013, 30, 538. [Google Scholar] [CrossRef]
- Patel, A.S.; Allen, J.E.; Dicker, D.T.; Peters, K.L.; Sheehan, J.M.; Glantz, M.J.; El-Deiry, W.S. Identification and enumeration of circulating tumor cells in the cerebrospinal fluid of breast cancer patients with central nervous system metastases. Oncotarget 2011, 2, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevel, K.S.; DiStefano, N.; Lin, X.; Skakodub, A.; Ogilvie, S.Q.; Reiner, A.S.; Boire, A. A retrospective, quantitative assessment of disease burden in patients with leptomeningeal metastases from non-small-cell lung cancer. Neuro-oncology 2020, 22, 675–683. [Google Scholar] [CrossRef]
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Berger, M.F. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J. Clin. Oncol. 2016, 34, 2404. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, X.; Xing, W.; Yu, H.; Si, T.; Guo, Z. Detection of circulating tumor DNA from non-small cell lung cancer brain metastasis in cerebrospinal fluid samples. Thorac. Cancer 2020, 11, 588–593. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xie, Y.; Lin, Y.; Yu, T.; Yin, Z. Different Gene Mutation Spectrum of the Paired CSF and Plasma Samples in Lung Adenocarcinoma with Leptomeningeal Metastases: The Liquid Biopsy Based on Circulating Tumor DNA. Chin. J. Lung Cancer 2020, 23, 646–654. [Google Scholar]
- Bastos, P.; Ferreira, R.; Manadas, B.; Moreira, P.I.; Vitorino, R. Insights into the human brain proteome: Disclosing the biological meaning of protein networks in cerebrospinal fluid. Crit. Rev. Clin. Lab. Sci. 2017, 54, 185–204. [Google Scholar] [CrossRef]
- Orešković, D.; Radoš, M.; Klarica, M. Role of choroid plexus in cerebrospinal fluid hydrodynamics. Neuroscience 2017, 354, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Josephson, S.A.; Fridlyand, J.; Karch, J.; Kadoch, C.; Karrim, J.; Damon, L.; Treseler, P.; Kunwar, S.; Shuman, M.A.; et al. Protein biomarker identification in the CSF of patients with CNS lymphoma. J. Clin. Oncol. 2008, 26, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Biomarkers: Hopes and challenges in the path from discovery to clinical practice. Transl. Res. 2012, 159, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantzi, M.; Bhat, A.; Latosinska, A. Clinical proteomic biomarkers: Relevant issues on study design & technical considerations in biomarker development. Clin. Transl. Med. 2014, 3, 7. [Google Scholar]
- Dasilva, N.; Díez, P.; Matarraz, S.; González-González, M.; Paradinas, S.; Orfao, A.; Fuentes, M. Biomarker discovery by novel sensors based on nanoproteomics approaches. Sensors 2012, 12, 2284–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acebes-Fernández, V.; Landeria-Viñuela, A.; Juanes-Velasco, P.; Hernández, A.P.; Otazo-Perez, A.; Manzano-Román, R.; Fuentes, M. Nanomedicine and Onco-Immunotherapy: From the Bench to Bedside to Biomarkers. Nanomaterials 2020, 10, 1274. [Google Scholar] [CrossRef]
- Matarraz, S.; González-González, M.; Jara, M.; Orfao, A.; Fuentes, M. New technologies in cancer. Protein microarrays for biomarker discovery. Clin. Transl. Oncol. 2011, 13, 156–161. [Google Scholar] [CrossRef]
- Liu, X.; Mo, F.; Zeng, H.; Zhu, S.; Ma, X. Quantitative proteomic analysis of cerebrospinal fluid from patients with diffuse large B-cell lymphoma with central nervous system involvement: A novel approach to diagnosis. Biomed. Rep. 2019, 11, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Fernández, C.; Santos, H.M.; Ruíz-Romero, C.; Blanco, F.J.; Capelo-Martínez, J.L. A comparison of depletion versus equalization for reducing high-abundance proteins in human serum. Electrophoresis 2011, 32, 2966–2974. [Google Scholar] [CrossRef]
- Larssen, E.; Brede, C.; Hjelle, A.B.; Øysæd, K.B.; Tjensvoll, A.B.; Omdal, R.; Ruoff, P. A rapid method for preparation of the cerebrospinal fluid proteome. Proteomics 2014, 15, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Olsen, J.V.; Godoy, L.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Pin, E.; Sjöberg, R.; Andersson, E.; Hellström, C.; Olofsson, J.; Falk, A.J.; Månberg, A. Array-based profiling of proteins and autoantibody repertoires in CSF. In Cerebrospinal Fluid (CSF) Proteomics; Humana: New York, NY, USA, 2019; pp. 303–318. [Google Scholar]
- González-González, M.; Bartolome, R.; Jara-Acevedo, R.; Casado-Vela, J.; Dasilva, N.; Matarraz, S.; García, J.; Alcazar, J.A.; Sayagues, J.M.; Orfao, A.; et al. Evaluation of homo-and hetero-functionally activated glass surfaces for optimized antibody arrays. Anal. Biochem. 2014, 450, 37–45. [Google Scholar] [CrossRef]
- Sierra-Sánchez, Á.; Garrido-Martín, D.; Lourido, L.; González-González, M.; Díez, P.; Ruiz-Romero, C.; SjÖber, R.; Droste, C.; De Las Rivas, J.; Nilsson, P.; et al. Screening and Validation of Novel Biomarkers in Osteoarticular Pathologies by Comprehensive Combination of Protein Array Technologies. J. Proteome Res. 2017, 16, 1890–1899. [Google Scholar] [CrossRef]
- Schwenk, J.M.; Igel, U.; Neiman, M.; Langen, H.; Becker, C.; Bjartell, A.; Ponten, F.; Wiklund, F.; Grönberg, H.; Nilsson, P.; et al. Toward next generation plasma profiling via heat-induced epitope retrieval and array-based assays. Mol. Cell. Proteom. 2010, 9, 2497–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Valiente, R.; Fernández-García, J.; Carabias-Sánchez, J.; Landeira-Viñuela, A.; Góngora, R.; Gonzalez-Gonzalez, M.; Fuentes, M.A. Systematic Analysis Workflow for High-Density Customized Protein Microarrays in Biomarker Screening. Methods Mol. Biol. 2019, 1871, 107–122. [Google Scholar] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 10 October 2019).
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef]
- Gaudet, P.; Michel, P.A.; Zahn-Zabal, M.; Cusin, I.; Duek, P.D.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Teixeira, D. The neXtProt knowledgebase on human proteins: Current status. Nucleic Acids Res. 2015, 43, D764–D770. [Google Scholar] [CrossRef] [Green Version]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; Milacic, M. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Ser. B Stat. Methodol. 1974, 36, 111–133. [Google Scholar] [CrossRef]
- Galicia, N.; Díez, P.; Dégano, R.M.; Guest, P.C.; Ibarrola, N.; Fuentes, M. Proteomic biomarker identification in cerebrospinal fluid for leptomeningeal metastases with neurological complications. Proteom. Methods Neuropsychiatr. Res. 2017, 974, 85–96. [Google Scholar]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massagué, J. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 2017, 168, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Dorzweiler, K.; Miller, M.A.; Lauffenburger, D.A.; Strik, H.; Bartsch, J.W. Profiling of metalloprotease activities in cerebrospinal fluids of patients with neoplastic meningitis. Fluids Barriers CNS 2017, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Smalley, I.; Law, V.; Wyatt, C.; Evernden, B.; Fang, B.; Koomen, J.M.; Smalley, K.S. Proteomic analysis of CSF from patients with leptomeningeal melanoma metastases identifies signatures associated with disease progression and therapeutic resistance. Clin. Cancer Res. 2020, 26, 2163–2175. [Google Scholar] [CrossRef] [Green Version]
- Bader, J.M.; Geyer, P.E.; Müller, J.B.; Strauss, M.T.; Koch, M.; Leypoldt, F.; Mann, M. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Mol. Syst. Biol. 2020, 16, e9356. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Remsik, J.; Kiseliovas, V.; Derderian, C.; Sener, U.; Alghader, M.; Boire, A. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020, 369, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Cordone, I.; Masi, S.; Giannarelli, D.; Pasquale, A.; Conti, L.; Telera, S.; Pace, A.; Papa, E.; Marino, M.; de Fabritiis, P.; et al. Major Differences in Lymphocyte Subpopulations Between Cerebrospinal Fluid and Peripheral Blood in Non-Hodgkin Lymphoma Without Leptomeningeal Involvement: Flow Cytometry Evidence of a Cerebral Lymphatic System. Front. Oncol. 2021, 11, 1668. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juanes-Velasco, P.; Galicia, N.; Pin, E.; Jara-Acevedo, R.; Carabias-Sánchez, J.; García-Valiente, R.; Lecrevisse, Q.; Pedreira, C.E.; Gongora, R.; Sanchez-Santos, J.M.; et al. Deciphering Biomarkers for Leptomeningeal Metastasis in Malignant Hemopathies (Lymphoma/Leukemia) Patients by Comprehensive Multipronged Proteomics Characterization of Cerebrospinal Fluid. Cancers 2022, 14, 449. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020449
Juanes-Velasco P, Galicia N, Pin E, Jara-Acevedo R, Carabias-Sánchez J, García-Valiente R, Lecrevisse Q, Pedreira CE, Gongora R, Sanchez-Santos JM, et al. Deciphering Biomarkers for Leptomeningeal Metastasis in Malignant Hemopathies (Lymphoma/Leukemia) Patients by Comprehensive Multipronged Proteomics Characterization of Cerebrospinal Fluid. Cancers. 2022; 14(2):449. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020449
Chicago/Turabian StyleJuanes-Velasco, Pablo, Norma Galicia, Elisa Pin, Ricardo Jara-Acevedo, Javier Carabias-Sánchez, Rodrigo García-Valiente, Quentin Lecrevisse, Carlos Eduardo Pedreira, Rafael Gongora, Jose Manuel Sanchez-Santos, and et al. 2022. "Deciphering Biomarkers for Leptomeningeal Metastasis in Malignant Hemopathies (Lymphoma/Leukemia) Patients by Comprehensive Multipronged Proteomics Characterization of Cerebrospinal Fluid" Cancers 14, no. 2: 449. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020449