
Autophagy Targeting and Hematological Mobilization in FLT3-ITD Acute Myeloid Leukemia Decrease Repopulating Capacity and Relapse by Inducing Apoptosis of Committed Leukemic Cells

 , , , , , , and
, , , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
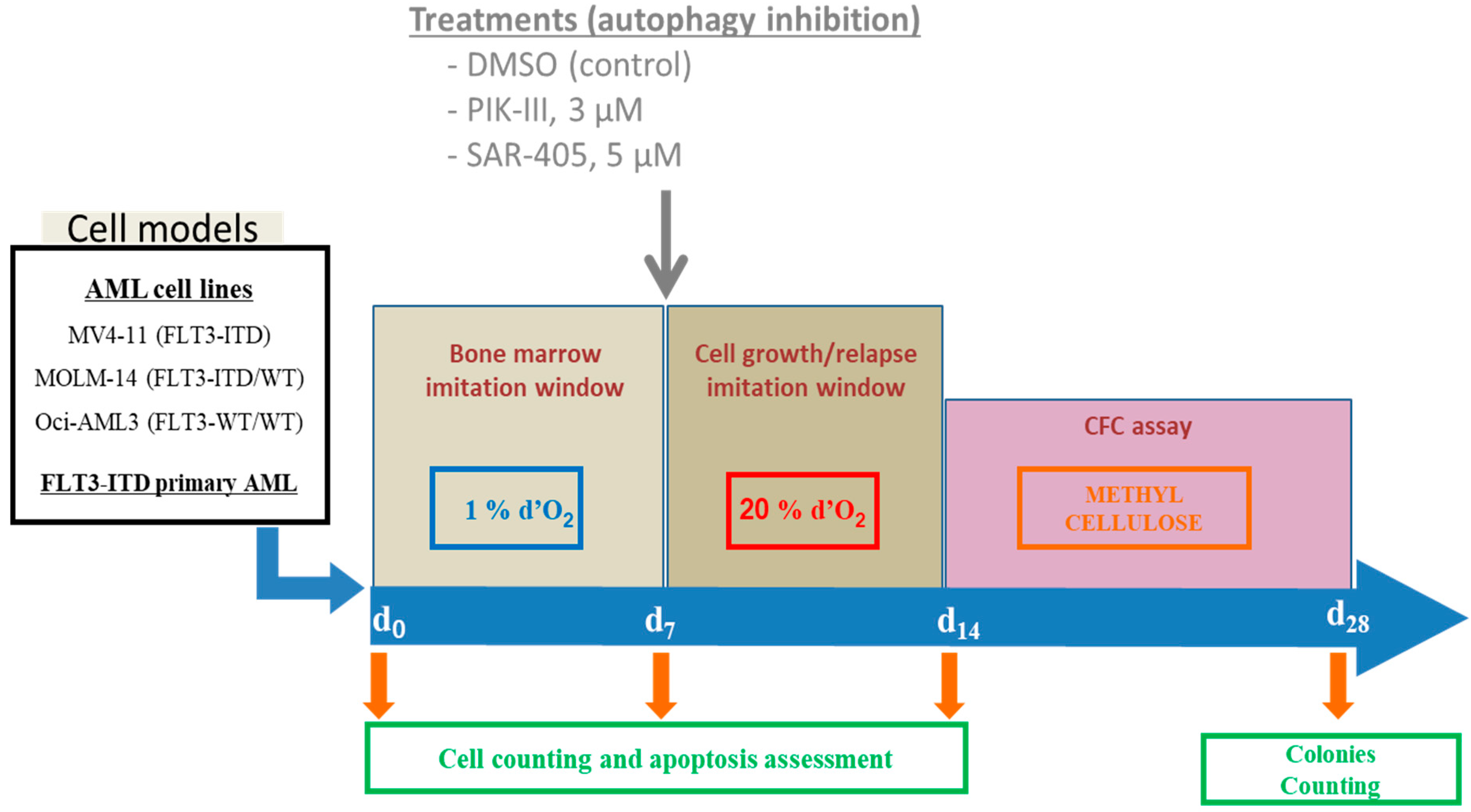
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. AML Patient Biological Samples
2.4. Culture at Low or Atmospheric Oxygen Concentration
2.5. Flow Cytometry
2.6. Autophagy Inhibition by PIK-III and SAR-405
2.7. Western Blotting
2.8. Animal Models for In Vivo Studies
2.9. Statistical Analysis
3. Results
3.1. Vps34 Inhibition Using PIK-III and SAR-405 Block Autophagy in AML Cells
3.2. Inhibition of Autophagy Reduces Expansion and Enhances Apoptosis of FLT3-ITD AML Cells Following Transfer to Growth-Permissive Cultures at Atmosphere O2 Concentration
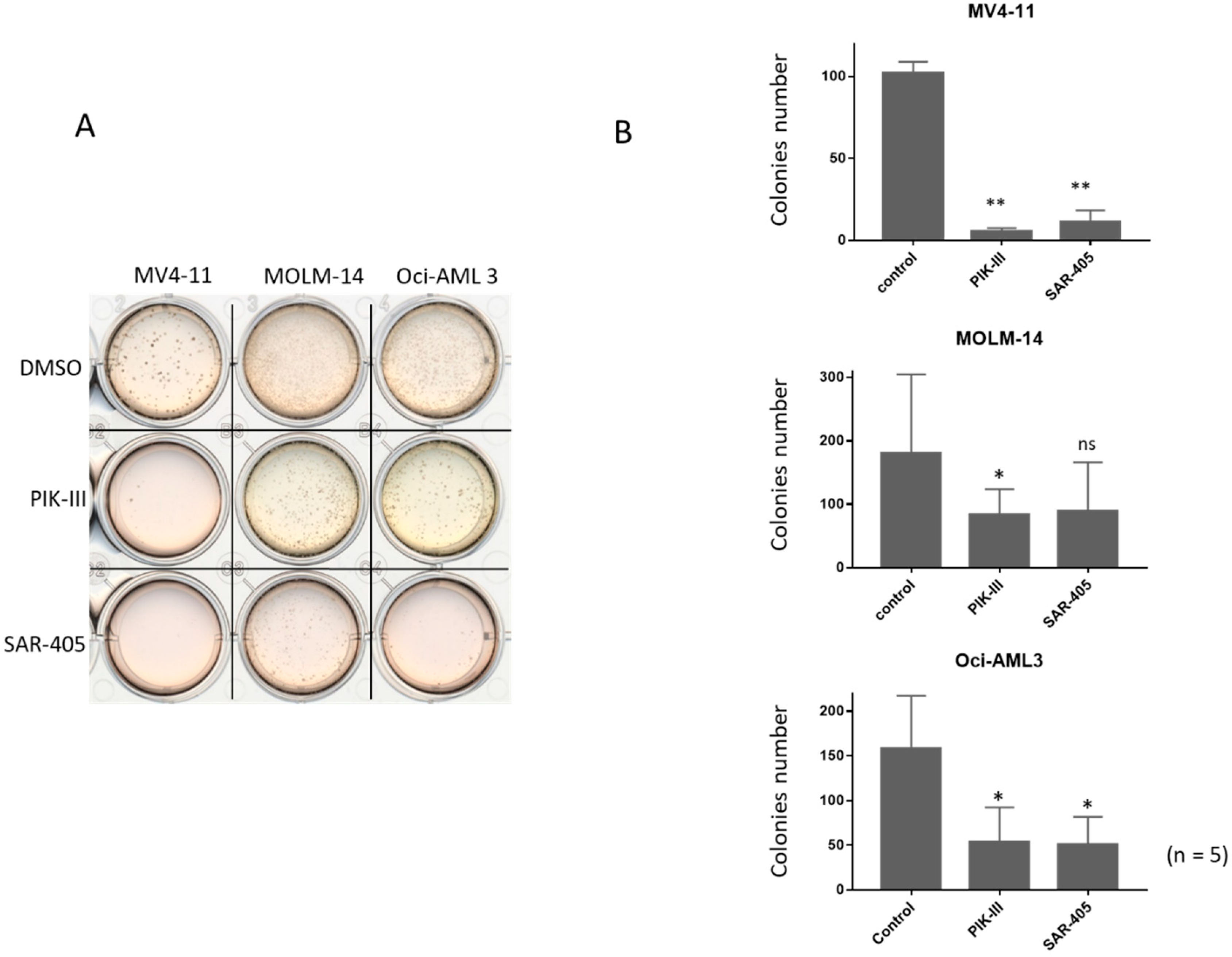
3.3. Inhibition of Autophagy Limits Repopulating Capacity of FLT3-ITD Cells in CFC Assay
3.4. Ex Vivo Autophagy Inhibition of Primary FLT3-ITD AML Cells Increases Apoptosis upon Transfer to Growth-Permissive Cultures
3.5. In Vivo, Combined Inhibition of Autophagy by PIK-III and SAR-405 and Mobilization Decrease Relapse Rate in Xenograft Model of FLT3-ITD AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilliland, D.G.; Griffin, J.D. Role of FLT3 in leukemia. Curr. Opin. Hematol. 2002, 9, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2017, 31, 505. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenk, R.F.; Döhner, K.; Krauter, J.; Fröhling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Späth, D.; Morgan, M.; Benner, A.; et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Ghiaur, G.; Levis, M. Mechanisms of Resistance to FLT3 Inhibitors and the Role of the Bone Marrow Microenvironment. Hematol. Oncol. Clin. N. Am. 2017, 31, 681–692. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Levis, M.J.; et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Sironi, S.; Wagner, M.; Kuett, A.; Drolle, H.; Polzer, H.; Spiekermann, K.; Rieger, C.; Fiegl, M. Microenvironmental Hypoxia regulates FLT3 expression and biology in AML. Sci. Rep. 2015, 5, 17550. [Google Scholar] [CrossRef]
- van Oosterwijk, J.G.; Buelow, D.R.; Drenberg, C.D.; Vasylieva, A.; Li, L.; Shi, L.; Wang, Y.D.; Janke, J.L.; Pounds, S.; Baker, S.D.; et al. Hypoxia-induced upregulation of BMX kinase mediates therapeutic resistance in acute myeloid leukemia. J. Clin. Investig. 2018, 128, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Dumas, P.-Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Bidet, A.; Kosmider, O.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5- and hypoxia- dependent up-regulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [Green Version]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Levis, M. FLT3 dancing on the stem cell. J. Exp. Med. 2017, 214, 1857–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergez, F.; Green, A.S.; Tamburini, J.; Sarry, J.E.; Gaillard, B.; Lacombe, C.; Bouscary, D.; Manenti, S.; Recher, C.; Demur, C.; et al. High levels of CD34+/CD38low/CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: A Groupe Ouest-Est des Leucémies Aigues et Maladies du Sang (GOELAMS) study. Haematologica 2011, 96, 1792–1798. [Google Scholar] [CrossRef]
- Dumas, P.-Y.; Bertoli, S.; Bérard, E.; Largeaud, L.; Bidet, A.; Delabesse, E.; Leguay, T.; Leroy, H.; Gadaud, N.; Rieu, J.B.; et al. Real-World Outcomes of Patients with Refractory or Relapsed FLT3-ITD Acute Myeloid Leukemia: A Toulouse-Bordeaux DATAML Registry Study. Cancers 2020, 12, 2044. [Google Scholar] [CrossRef] [PubMed]
- Cavazos, A.; Ma, H.; Benito, J.M.; Levis, M.J.; Daver, N.; Ashoorzadeh, A.; Anderson, R.; Patterson, A.; Smaill, J.; Konopleva, M. Pre-Clinical Activity of Novel Hypoxia-Activated FLT3 Inhibitors in FLT3-Mutated AML. Blood 2016, 128, 5210. [Google Scholar] [CrossRef]
- Sarry, J.-E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Cavelier, C.; Carroll, M.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Investig. 2010, 121, 384–395. [Google Scholar] [CrossRef]
- Qiu, S.; Yan, C.; Paterson, A.J.; Li, H.; Anderson, N.; Abraham, A.; He, J.; Shah, M.; Yang, J.; Xie, M.; et al. Role of Autophagy in Resistance of FLT3-ITD AML Stem Cells to FLT3 TKI Treatment. Blood 2019, 134, 2548. [Google Scholar] [CrossRef]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulliam, A.C.; Hobson, M.J.; Ciccone, S.L.; Li, Y.; Chen, S.; Srour, E.F.; Yang, F.-C.; Broxmeyer, H.E.; Clapp, D.W. AMD3100 synergizes with G-CSF to mobilize repopulating stem cells in Fanconi anemia knockout mice. Exp. Hematol. 2008, 36, 1084–1090. [Google Scholar] [CrossRef] [Green Version]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; McQueen, T.; Chen, Y.; Jacamo, R.; Konopleva, M.; Shinojima, N.; Shpall, E.; Huang, X.; Andreeff, M. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1α–mediated down-regulation of CXCL12. Blood 2011, 118, 4431–4439. [Google Scholar] [CrossRef] [Green Version]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Ianniciello, A.; Dumas, P.-Y.; Drullion, C.; Guitart, A.; Villacreces, A.; Peytour, Y.; Chevaleyre, J.; De La Grange, P.B.; Vigon, I.; Desplat, V.; et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 2017, 8, 96984–96992. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.K.; Nepstad, I.; Bruserud, O. Mesenchymal Stem Cells Support Survival and Proliferation of Primary Human Acute Myeloid Leukemia Cells through Heterogeneous Molecular Mechanisms. Front. Immunol. 2017, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Kantarjian, H.; Ravandi, F.; Daver, N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mas, V.M.-D. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2017, 37, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.-C.; Chang, C.-M.; Sun, H.S. Hypoxia Induces Autophagy through Translational Up-Regulation of Lysosomal Proteins in Human Colon Cancer Cells. PLoS ONE 2016, 11, e0153627. [Google Scholar] [CrossRef] [Green Version]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.; Glanville, J.; Knight, S.; Jacobsen, S.E.W.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.-F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Tormo, D.; Chęcińska, A.; Curbelo, D.A.; Pérez-Guijarro, E.; Cañón, E.; Riveiro-Falkenbach, E.; Calvo, T.G.; Larribere, L.; Megias, D.; Mulero, F.; et al. Targeted Activation of Innate Immunity for Therapeutic Induction of Autophagy and Apoptosis in Melanoma Cells. Cancer Cell 2009, 16, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Kayser, S.; Levis, M.J. FLT3tyrosine kinase inhibitors in acute myeloid leukemia: Clinical implications and limitations. Leuk. Lymphoma 2013, 55, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef]
- Ueno, Y.; Mori, M.; Kamiyama, Y.; Kaneko, N.; Isshiki, E.; Takeuchi, M. Gilteritinib (ASP2215), a Novel FLT3/AXL Inhibitor: Preclinical Evaluation in Combination with Azacitidine in Acute Myeloid Leukemia. Blood 2016, 128, 2830. [Google Scholar] [CrossRef]
- Levis, M.; Perl, A.E. Gilteritinib: Potent targeting of FLT3 mutations in AML. Blood Adv. 2020, 4, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Nechiporuk, T.; Bottomly, D.; Piehowski, P.D.; Reisz, J.A.; Pittsenbarger, J.; Kaempf, A.; Gosline, S.J.; Wang, Y.-T.; Hansen, J.R.; et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell 2021, 39, 999–1014.e8. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2021, 6, a026120. [Google Scholar]
- Pecoraro, A.; Carotenuto, P.; Franco, B.; De Cegli, R.; Russo, G.; Russo, A. Role of uL3 in the Crosstalk between Nucleolar Stress and Autophagy in Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2143. [Google Scholar] [CrossRef] [Green Version]
- Meunier, G.; Birsen, R.; Cazelles, C.; Belhadj, M.; Cantero-Aguilar, L.; Kosmider, O.; Fontenay, M.; Azar, N.; Mayeux, P.; Chapuis, N.; et al. Antileukemic activity of the VPS34-IN1 inhibitor in acute myeloid leukemia. Oncogenesis 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Flomenberg, N.; Devine, S.M.; Dipersio, J.F.; Liesveld, J.L.; McCarty, J.M.; Rowley, S.D.; Calandra, G.; Vesole, D.H.; Badel, K. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 2005, 106, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Karpova, D.; Ritchey, J.K.; Holt, M.S.; Abou-Ezzi, G.; Monlish, D.; Batoon, L.; Millard, S.; Spohn, G.; Wiercinska, E.; Chendamarai, E.; et al. Continuous blockade of CXCR4 results in dramatic mobilization and expansion of hematopoietic stem and progenitor cells. Blood 2017, 129, 2939–2949. [Google Scholar] [CrossRef] [Green Version]
- Dumas, P.-Y.; Villacreces, A.; Guitart, A.V.; El-Habhab, A.; Massara, L.; Mansier, O.; Bidet, A.; Martineau, D.; Fernandez, S.; Leguay, T.; et al. Dual Inhibition of FLT3 and AXL by Gilteritinib Overcomes Hematopoietic Niche-Driven Resistance Mechanisms in FLT3-ITD Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 6012–6025. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients N° | Sex | Age | Ratio | % Blasts | Mutation | Phenotype |
|---|---|---|---|---|---|---|
| 1 | w | 57 | 45 | 70 | NPM1 | AML4 |
| 2 | w | 74 | 42 | 83 | NPM1 | AML4 |
| 3 | w | 37 | 170 | 79 | NPM1 | AML1 |
| 4 | w | 71 | 72 | 98 | NPM1 | AML1 |
| 5 | m | 49 | 48 | 89 | NPM1 | AML5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupont, M.; Huart, M.; Lauvinerie, C.; Bidet, A.; Guitart, A.V.; Villacreces, A.; Vigon, I.; Desplat, V.; El Habhab, A.; Pigneux, A.; et al. Autophagy Targeting and Hematological Mobilization in FLT3-ITD Acute Myeloid Leukemia Decrease Repopulating Capacity and Relapse by Inducing Apoptosis of Committed Leukemic Cells. Cancers 2022, 14, 453. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020453
Dupont M, Huart M, Lauvinerie C, Bidet A, Guitart AV, Villacreces A, Vigon I, Desplat V, El Habhab A, Pigneux A, et al. Autophagy Targeting and Hematological Mobilization in FLT3-ITD Acute Myeloid Leukemia Decrease Repopulating Capacity and Relapse by Inducing Apoptosis of Committed Leukemic Cells. Cancers. 2022; 14(2):453. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020453
Chicago/Turabian StyleDupont, Marine, Mathilde Huart, Claire Lauvinerie, Audrey Bidet, Amélie Valérie Guitart, Arnaud Villacreces, Isabelle Vigon, Vanessa Desplat, Ali El Habhab, Arnaud Pigneux, and et al. 2022. "Autophagy Targeting and Hematological Mobilization in FLT3-ITD Acute Myeloid Leukemia Decrease Repopulating Capacity and Relapse by Inducing Apoptosis of Committed Leukemic Cells" Cancers 14, no. 2: 453. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020453