
Comprehensive Approach to Distinguish Patients with Solid Tumors from Healthy Controls by Combining Androgen Receptor Mutation p.H875Y with Cell-Free DNA Methylation and Circulating miRNAs

 ,
, 
Abstract
:Simple Summary
Abstract
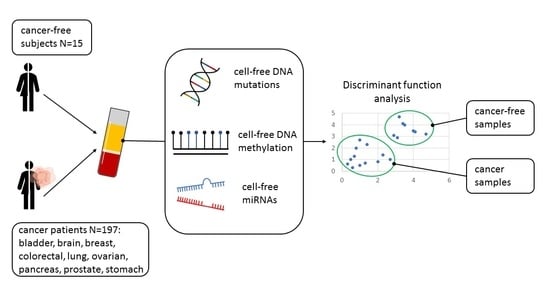
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and Liquid Biopsy
2.3. Cell-Free DNA Extraction, Processing, and Analysis
2.4. Mutation Analysis
2.5. Methylation Analysis
2.6. RNA Extraction, Processing, and microRNA Analysis
2.7. Statistical Analysis
2.8. Identification of Candidate Biomarkers
3. Results
3.1. Patient Characteristics
3.2. Plasma cfDNA Levels
3.3. Plasma cfDNA Mutation Detection
3.4. Androgen Receptor p.H875Y Mutation
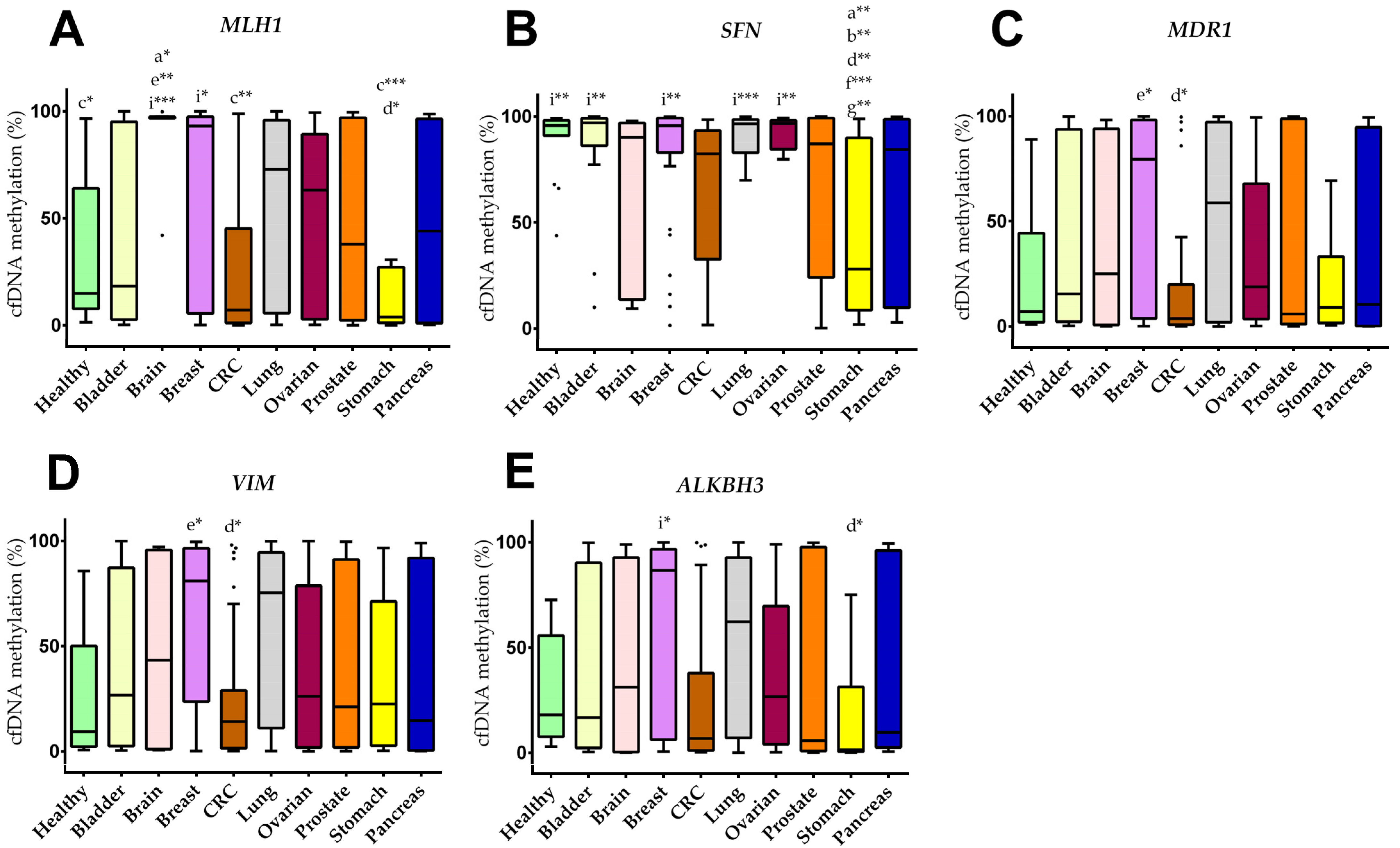
3.5. Plasma cfDNA Methylation
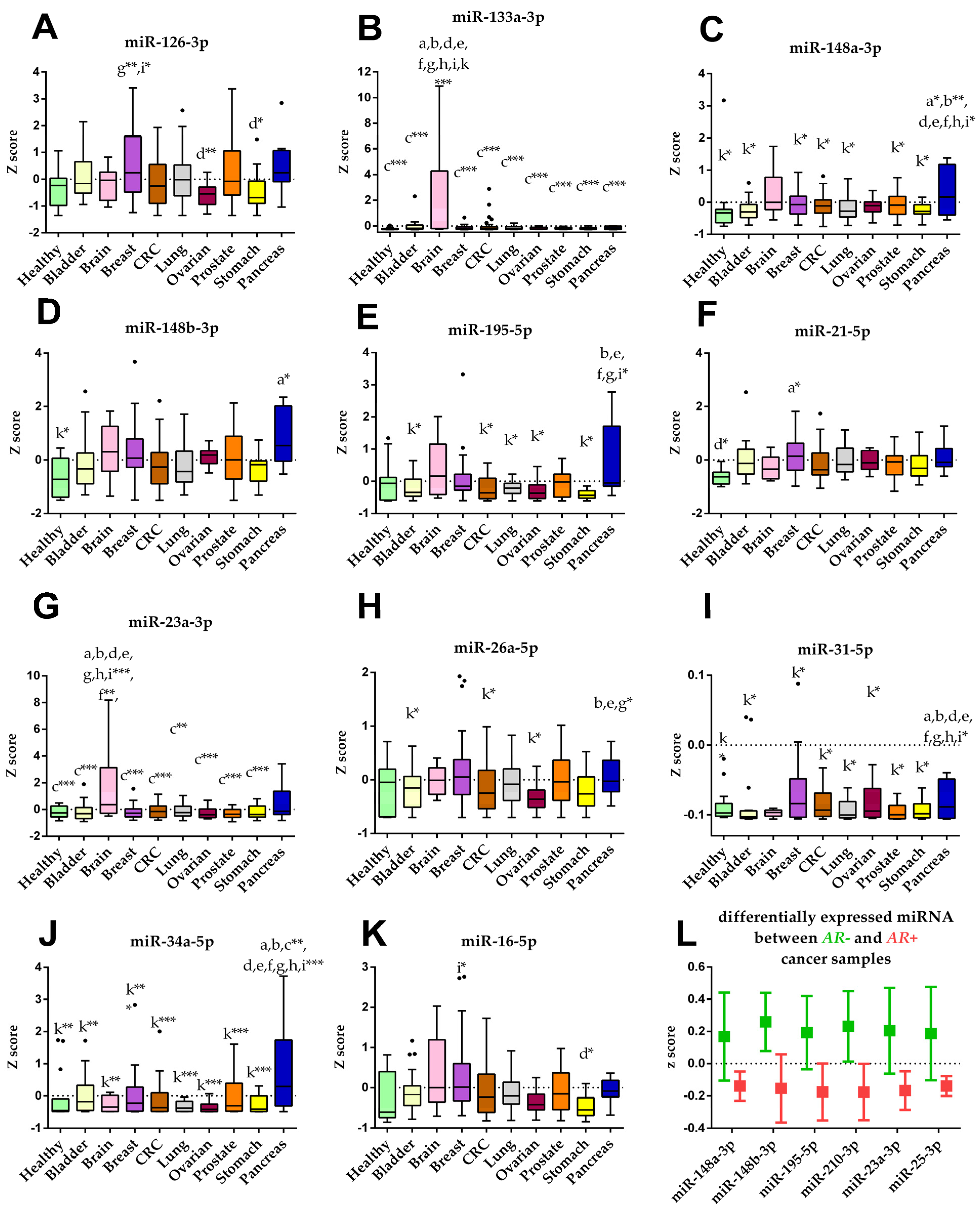
3.6. Identification of Differently Expressed Circulating miRNAs
3.7. Identification of Cancer Type Specific Biomarkers
3.8. Classification of Tumor Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter Methylation of ADAMTS1 and BNC1 as Potential Biomarkers for Early Detection of Pancreatic Cancer in Blood. Clin. Epigenet. 2019, 11, 59. [Google Scholar] [CrossRef]
- Verna, E.C.; Hwang, C.; Stevens, P.D.; Rotterdam, H.; Stavropoulos, S.N.; Sy, C.D.; Prince, M.A.; Chung, W.K.; Fine, R.L.; Chabot, J.A.; et al. Pancreatic Cancer Screening in a Prospective Cohort of High-Risk Patients: A Comprehensive Strategy of Imaging and Genetics. Clin. Cancer Res. 2010, 16, 5028–5037. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Carpenter, E.; Issadore, D. Detection and Isolation of Circulating Exosomes and Microvesicles for Cancer Monitoring and Diagnostics Using Micro-/Nano-Based Devices. Analyst 2016, 141, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Hood, S.; Kallam, E.; Overman, D.; Barker, K.; Rutledge, D.; Riojas, J.; Best, C.; Eisenberg, M.; Morgan, L.K. Epi ProColon®: Use of a Non-Invasive SEPT9 Gene Methylation Blood Test for Colorectal Cancer Screening: A National Laboratory Experience. J. Clin. Epigenet. 2018, 4, 7. [Google Scholar] [CrossRef]
- Liles, E.G.; Coronado, G.D.; Perrin, N.; Harte, A.H.; Nungesser, R.; Quigley, N.; Potter, N.T.; Weiss, G.; Koenig, T.; deVos, T. Uptake of a Colorectal Cancer Screening Blood Test Is Higher than of a Fecal Test Offered in Clinic: A Randomized Trial. Cancer Treat. Res. Commun. 2017, 10, 27–31. [Google Scholar] [CrossRef]
- Danilova, L.; Wrangle, J.; Herman, J.G.; Cope, L. DNA-Methylation for the Detection and Distinction of 19 Human Malignancies. Epigenetics 2021, 1–11, Online ahead of print. [Google Scholar] [CrossRef]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-Invasive Cancer Diagnosis and Tissue-of-Origin Prediction Using Methylation Profiles of Cell-Free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef] [Green Version]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of Tissue-Specific Cell Death Using Methylation Patterns of Circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Gai, W.; Sun, K. Epigenetic Biomarkers in Cell-Free DNA and Applications in Liquid Biopsy. Genes 2019, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Shigeyasu, K.; Toden, S.; Zumwalt, T.J.; Okugawa, Y.; Goel, A. Emerging Role of MicroRNAs as Liquid Biopsy Biomarkers in Gastrointestinal Cancers. Clin. Cancer Res. 2017, 23, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Fendler, W.; Stawiski, K.; Fiascone, S.J.; Vitonis, A.F.; Berkowitz, R.S.; Frendl, G.; Konstantinopoulos, P.; Crum, C.P.; Kedzierska, M.; et al. Diagnostic Potential for a Serum MiRNA Neural Network for Detection of Ovarian Cancer. eLife 2017, 6, e28932. [Google Scholar] [CrossRef]
- Eastham, J.A.; Riedel, E.; Scardino, P.T.; Shike, M.; Fleisher, M.; Schatzkin, A.; Lanza, E.; Latkany, L.; Begg, C.B.; for the Polyp Prevention Trial Study Group. Variation of Serum Prostate-Specific Antigen Levels: An Evaluation of Year-to-Year Fluctuations. JAMA 2003, 289, 2695. [Google Scholar] [CrossRef] [Green Version]
- Esposito, A.; Criscitiello, C.; Locatelli, M.; Milano, M.; Curigliano, G. Liquid Biopsies for Solid Tumors: Understanding Tumor Heterogeneity and Real Time Monitoring of Early Resistance to Targeted Therapies. Pharmacol. Ther. 2016, 157, 120–124. [Google Scholar] [CrossRef]
- Fu, Q.; Schoenhoff, F.S.; Savage, W.J.; Zhang, P.; Van Eyk, J.E. Multiplex Assays for Biomarker Research and Clinical Application: Translational Science Coming of Age. Prot. Clin. Appl. 2010, 4, 271–284. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined Circulating Tumor DNA and Protein Biomarker-Based Liquid Biopsy for the Earlier Detection of Pancreatic Cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [Green Version]
- Liou, Y.-L.; Zhang, T.-L.; Yan, T.; Yeh, C.-T.; Kang, Y.-N.; Cao, L.; Wu, N.; Chang, C.-F.; Wang, H.-J.; Yen, C.; et al. Combined Clinical and Genetic Testing Algorithm for Cervical Cancer Diagnosis. Clin. Epigenet. 2016, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; LaRiviere, M.J.; Ko, J.; Till, J.E.; Christensen, T.E.; Yee, S.S.; Black, T.A.; Tien, K.; Lin, A.; Shen, H.; et al. A Multi-Analyte Panel Consisting of Extracellular Vesicle MiRNAs and MRNAs, CfDNA, and CA19-9 Shows Utility for Diagnosis and Staging of Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 35, 3248–3258. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Baldassano, S.N.; Loh, P.-L.; Kording, K.; Litt, B.; Issadore, D. Machine Learning to Detect Signatures of Disease in Liquid Biopsies—A User’s Guide. Lab Chip 2018, 18, 395–405. [Google Scholar] [CrossRef]
- Sisson, B.A.; Uvalic, J.; Kelly, K.; Selvam, P.; Hesse, A.N.; Ananda, G.; Chandok, H.; Bergeron, D.; Holinka, L.; Reddi, H.V. Technical and Regulatory Considerations for Taking Liquid Biopsy to the Clinic: Validation of the JAX PlasmaMonitorTM Assay. Biomark. Insights 2019, 14, 1177271919826545. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Busch, J.; Jung, M.; Rabenhorst, S.; Ralla, B.; Kilic, E.; Mergemeier, S.; Budach, N.; Fendler, A.; Jung, K. Diagnostic and Prognostic Potential of Circulating Cell-Free Genomic and Mitochondrial DNA Fragments in Clear Cell Renal Cell Carcinoma Patients. Clin. Chim. Acta 2016, 452, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Harris, L.; Mamon, H.; Kulke, M.H.; Liu, W.-H.; Zhu, P.; Mike Makrigiorgos, G. Whole Genome Amplification of Plasma-Circulating DNA Enables Expanded Screening for Allelic Imbalance in Plasma. J. Mol. Diagn. 2006, 8, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Mestdagh, P.; Van Vlierberghe, P.; De Weer, A.; Muth, D.; Westermann, F.; Speleman, F.; Vandesompele, J. A Novel and Universal Method for MicroRNA RT-QPCR Data Normalization. Genome Biol. 2009, 10, R64. [Google Scholar] [CrossRef] [Green Version]
- Jernberg, E.; Bergh, A.; Wikström, P. Clinical Relevance of Androgen Receptor Alterations in Prostate Cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [CrossRef] [Green Version]
- Obinata, D.; Lawrence, M.G.; Takayama, K.; Choo, N.; Risbridger, G.P.; Takahashi, S.; Inoue, S. Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 581515. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Spratt, D.E.; Pierce, L.J.; Speers, C.W. ARe we there yet? Understanding androgen receptor signaling in breast cancer. NPJ Breast Cancer 2020, 6, 47. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Stefaniuk, C.; Jakubowski, M.A. Real-time PCR and targeted next-generation sequencing in the detection of low level EGFR mutations: Instructive case analyses. Respir. Med. Case Rep. 2019, 28, 100901. [Google Scholar] [CrossRef]
- Hughes, G. On the Mean Accuracy of Statistical Pattern Recognizers. IEEE Trans. Inform. Theory 1968, 14, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Kanal, L.; Chandrasekaran, B. On Dimensionality and Sample Size in Statistical Pattern Classification. Pattern Recognit. 1971, 3, 225–234. [Google Scholar] [CrossRef]
- Keup, C.; Suryaprakash, V.; Hauch, S.; Storbeck, M.; Hahn, P.; Sprenger-Haussels, M.; Kolberg, H.-C.; Tewes, M.; Hoffmann, O.; Kimmig, R.; et al. Integrative Statistical Analyses of Multiple Liquid Biopsy Analytes in Metastatic Breast Cancer. Genome Med. 2021, 13, 85. [Google Scholar] [CrossRef]
- Yan, Y.; Guo, Q.; Wang, F.; Adhikari, R.; Zhu, Z.; Zhang, H.; Zhou, W.; Yu, H.; Li, J.; Zhang, J. Cell-Free DNA: Hope and Potential Application in Cancer. Front. Cell Dev. Biol. 2021, 9, 639233. [Google Scholar] [CrossRef]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; et al. Cell-Free DNA Concentration and Fragment Size as a Biomarker for Prostate Cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Chik, K.-W.; Chiu, R.W.; Ho, C.-Y.; Lam, C.W.; Lo, Y.D. Predominant Hematopoietic Origin of Cell-Free DNA in Plasma and Serum after Sex-Mismatched Bone Marrow Transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Guo, Y.A.; Ho, D.; Poon, P.; Poh, Z.W.; Wong, P.M.; Gan, A.; Chang, M.M.; Kleftogiannis, D.; Lau, Y.T.; et al. Tissue-Specific Cell-Free DNA Degradation Quantifies Circulating Tumor DNA Burden. Nat. Commun. 2021, 12, 2229. [Google Scholar] [CrossRef]
- Dou, Y.; Gold, H.D.; Luquette, L.J.; Park, P.J. Detecting Somatic Mutations in Normal Cells. Trends Genet. 2018, 34, 545–557. [Google Scholar] [CrossRef]
- De Oliveira, I.B.D.; Hirata, R.D.C. Circulating Cell-Free DNA as a Biomarker in the Diagnosis and Prognosis of Colorectal Cancer. Braz. J. Pharm. Sci. 2018, 54, 17368. [Google Scholar] [CrossRef]
- Adler, A.; Geiger, S.; Keil, A.; Bias, H.; Schatz, P.; deVos, T.; Dhein, J.; Zimmermann, M.; Tauber, R.; Wiedenmann, B. Improving Compliance to Colorectal Cancer Screening Using Blood and Stool Based Tests in Patients Refusing Screening Colonoscopy in Germany. BMC Gastroenterol. 2014, 14, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.; Kaye, S.; Banerjee, S. Delivering Widespread BRCA Testing and PARP Inhibition to Patients with Ovarian Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.P.; Li, S.; Cheng, H. Circulating DNA in EGFR-Mutated Lung Cancer. Ann. Transl. Med. 2017, 5, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The Performance of the SEPT9 Gene Methylation Assay and a Comparison with Other CRC Screening Tests: A Meta-Analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | n | Age Mean (95% CI) | Sex n (%) | Cancer stage n (%) | Family History of Cancer n (%) | BMI Mean | Current Infection n (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Female | Male | I | II | III | ||||||
| Healthy | 15 | 54.60 (46.23–62.97) b,c,e,f | 8 (53) | 7 (47) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 25.47 | 0(0) |
| Bladder | 20 | 70.75 (66.05–75.45) a | 3 (15) | 17 (85) | 9 (45) | 6 (30) | 5 (25) | 0 (0) | 26.26 | 0 (0) |
| Brain | 9 | 63.22 (54.33–72.12) | 3 (33) | 6 (67) | 2 (22) | 4 (44) | 3 (33) | 0 (0) | 25.22 | 0 (0) |
| Breast | 30 | 63.67 (59.39–67.94) | 29 (97) | 1 (3) | 8 (27) | 16 (53) | 6 (20) | 0 (0) | 25.5 | 0 (0) |
| Colorectal | 28 | 66.57 (62.81–70.34) a | 14 (50) | 14 (50) | 3 (11) | 19 (68) | 6 (21) | 0 (0) | 24.65 d | 0 (0) |
| Lung | 29 | 62.62 (59.05–66.19) | 4 (14) | 25 (86) | 1 (3) | 12 (41) | 16 (55) | 5 (17) | 25.77 | 1 (3) |
| Ovarian | 19 | 60.95 (56.44–65.46) | 19 (100) | 0 (0) | 0 (0) | 4 (21) | 15 (79) | 0 (0) | 28.19 c | 0 (0) |
| Prostate | 27 | 66.70 (63.15–70.26) a | 0 (0) | 27 (100) | 16 (59) | 7 (26) | 4 (15) | 1 (4) | 26.08 | 2 (7) |
| Stomach | 23 | 69.35 (65.04–73.65) a | 15 (65) | 8 (35) | 0 (0) | 8 (35) | 15 (65) | 0 (0) | 22.98 | 0 (0) |
| Pancreas | 12 | 67.08 (61.77–72.40) | 6 (50) | 6 (50) | 0 (0) | 5 (42) | 7 (58) | 0 (0) | 24.41 | 0 (0) |
| Total | 212 | 64.55 (63.08–66.01) | 101 (48) | 111 (52) | 39 (18) | 81 (38) | 77 (36) | 6 (3) | 25.45 | 3 (1) |
| Cancer Type | cfDNA Mutations | cfDNA Methylation | miRNAs |
|---|---|---|---|
| Bladder | AR (COSM238555), TP53 (COSM10758) | - | miR-17-5p |
| Brain | - | MLH1 m%, GATA5 m% | miR-133a-3p |
| Breast | AR (COSM238555), TP53 (COSM10758) | MDR1 m% | miR-17-5p |
| CRC | AR (COSM238555), TP53 (COSM10758) | - | miR-17-5p, |
| Lung | TP53 (COSM10758) | - | miR-17-5p, miR-92a-3p, miR-155-5p |
| Ovarian | - | - | miR-29c-3p, miR-92a-3p, miR-101-3p, miR-148b-3p |
| Pancreas | - | SFN m% | miR-27a-3p, miR-29c-3p, miR-148b-3p |
| Prostate | AR (COSM238555) | - | miR-17-5p, miR-26a-5p |
| Stomach | APC (COSM18561) | - | miR-20a-5p, miR-21-5p |
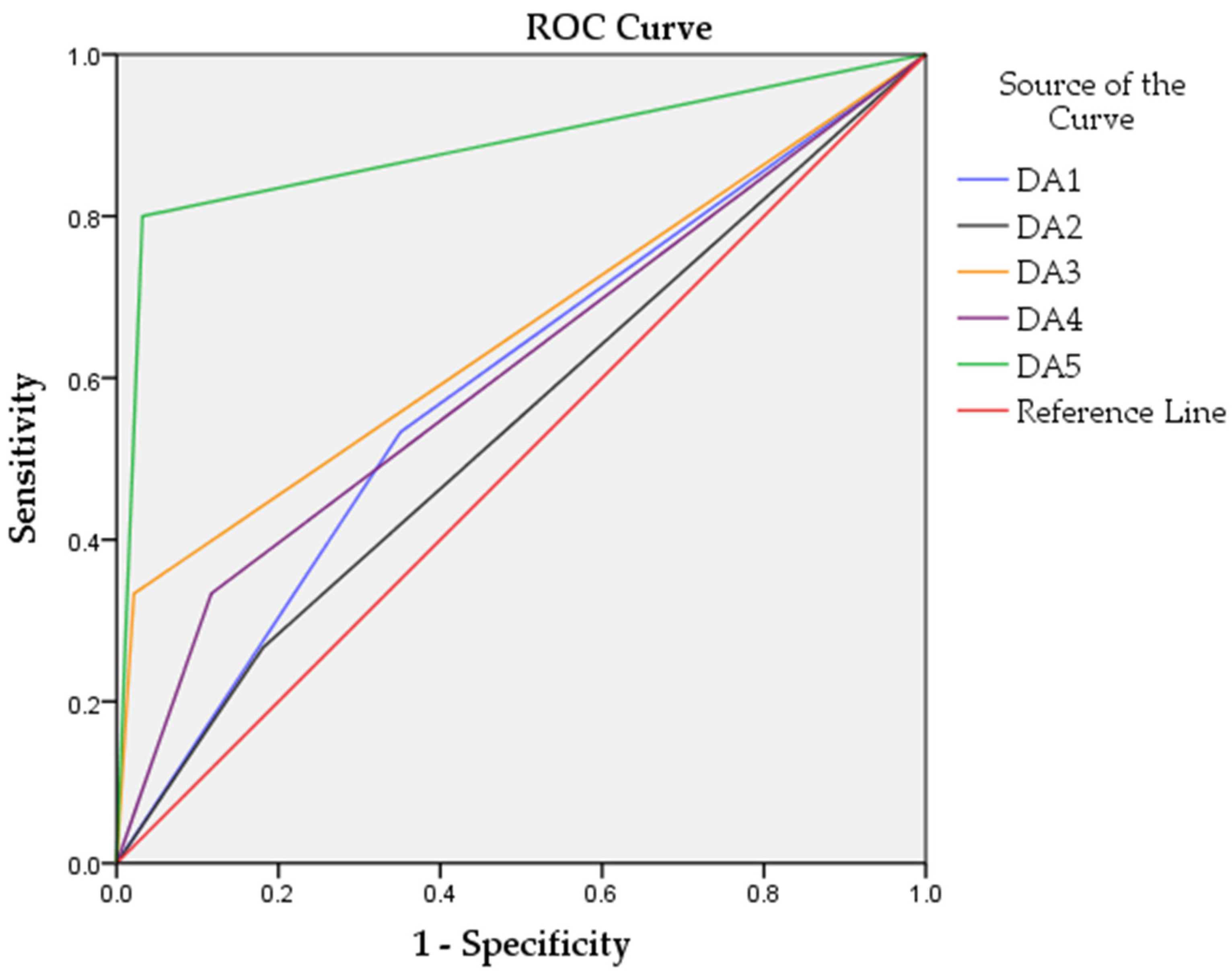
| Discriminant Analysis Model | Accuracy % | Sensitivity % | Specificity % | ROC AUC | n of Biomarkers |
|---|---|---|---|---|---|
| DA1 | 55.9 | 46.7 | 57.3 | 0.591 | 119 |
| DA2 | 73.9 | 81.3 | 26.7 | 0.543 | 64 |
| DA3 | 89.2 | 97.9 | 33.3 | 0.656 | 12 |
| DA4 | 80.2 | 87.5 | 33.3 | 0.608 | 43 |
| DA5 | 95.4 | 97.9 | 80.0 | 0.884 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomeva, E.; Switzeny, O.J.; Heitzinger, C.; Hippe, B.; Haslberger, A.G. Comprehensive Approach to Distinguish Patients with Solid Tumors from Healthy Controls by Combining Androgen Receptor Mutation p.H875Y with Cell-Free DNA Methylation and Circulating miRNAs. Cancers 2022, 14, 462. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020462
Tomeva E, Switzeny OJ, Heitzinger C, Hippe B, Haslberger AG. Comprehensive Approach to Distinguish Patients with Solid Tumors from Healthy Controls by Combining Androgen Receptor Mutation p.H875Y with Cell-Free DNA Methylation and Circulating miRNAs. Cancers. 2022; 14(2):462. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020462
Chicago/Turabian StyleTomeva, Elena, Olivier J. Switzeny, Clemens Heitzinger, Berit Hippe, and Alexander G. Haslberger. 2022. "Comprehensive Approach to Distinguish Patients with Solid Tumors from Healthy Controls by Combining Androgen Receptor Mutation p.H875Y with Cell-Free DNA Methylation and Circulating miRNAs" Cancers 14, no. 2: 462. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020462