
Use of Patient-Derived Organoids as a Treatment Selection Model for Colorectal Cancer: A Narrative Review

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Reviewing Methods
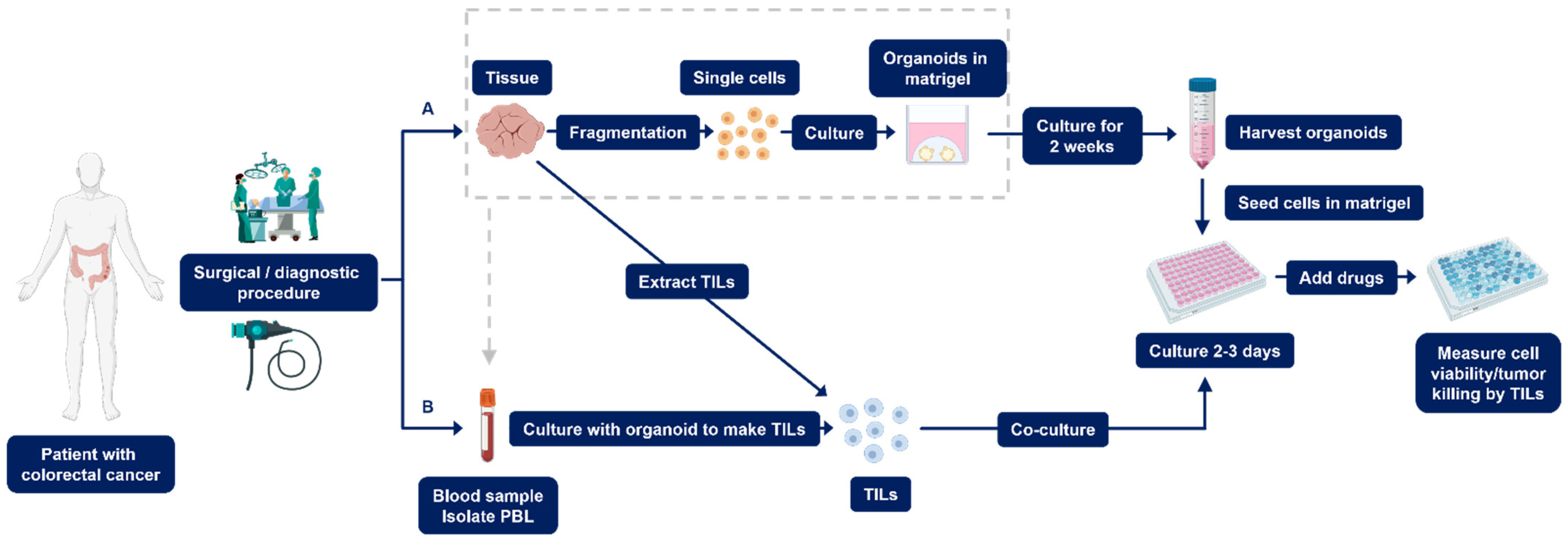
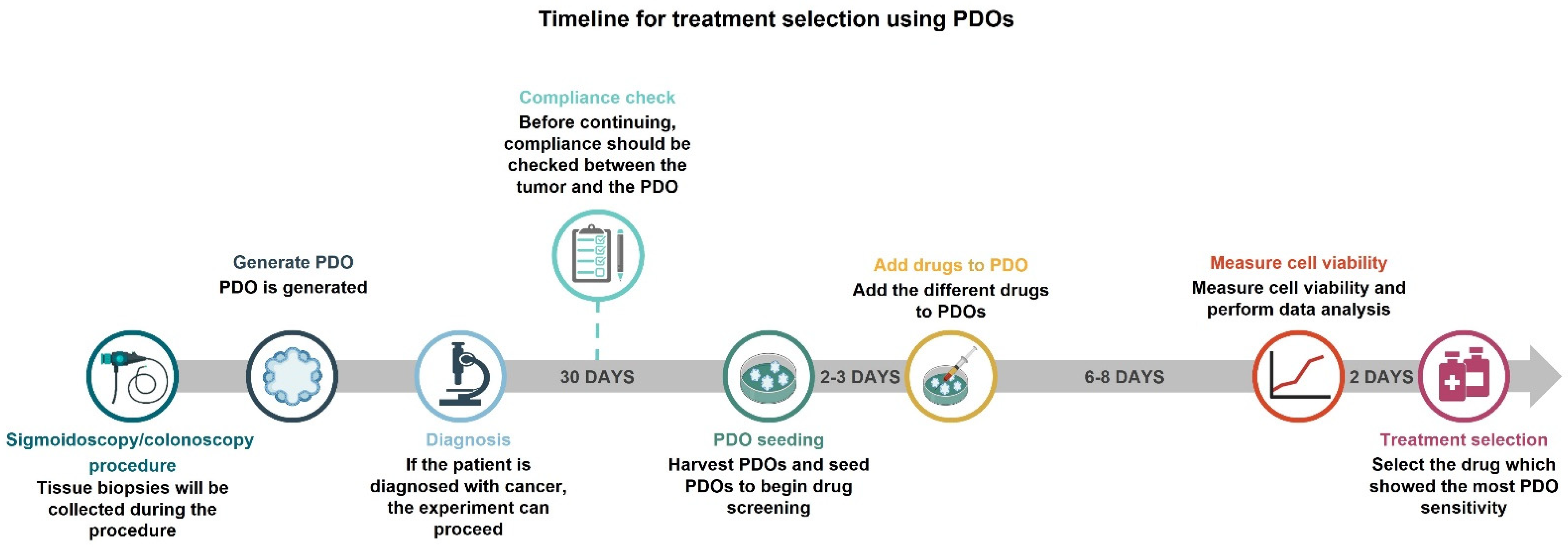
2.1. Establishment of PDOs
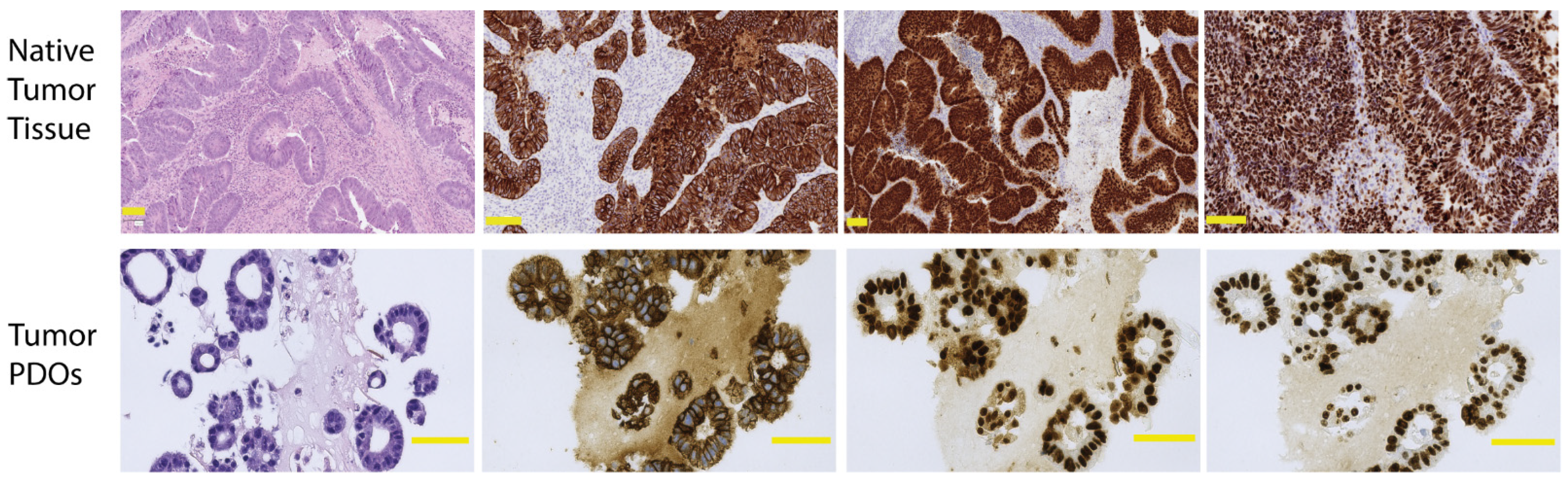
2.1.1. Quality Control to Ensure Compliance
2.1.2. Setting up PDO Drug Screening Assays
2.2. Establishment of Co-Culture PDOs
Setting up Co-Culture PDO Drug Screening Assays
3. Reviewing Organoid-Based Drug Screening Assays
3.1. PDOs
3.2. Co-Culture PDOs
4. Limitations
5. Potential
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Calon, A.; Lonardo, E.; Batlle, E. Determinants of metastatic competency in colorectal cancer. Mol. Oncol. 2017, 11, 97–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [Green Version]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.; Meyer, C.P.; Abdollah, F.; Sammon, J.D.; Sun, M.; Lipsitz, S.R.; Hollis, M.; Weissman, J.S.; Menon, M.; Trinh, Q.-D.; et al. Minimally invasive surgery and its impact on 30-day postoperative complications, unplanned readmissions and mortality. Br. J. Surg. 2017, 104, 1372–1381. [Google Scholar] [CrossRef]
- Schwenk, W.; Haase, O.; Neudecker, J.J.; Müller, J.M. Short term benefits for laparoscopic colorectal resection. Cochrane Database Syst. Rev. 2005, 2008, CD003145. [Google Scholar] [CrossRef]
- Renehan, A.G.; Malcomson, L.; Emsley, R.; Gollins, S.; Maw, A.; Myint, A.S.; Rooney, P.S.; Susnerwala, S.; Blower, A.; Saunders, M.P.; et al. Watch-and-wait approach versus surgical resection after chemoradiotherapy for patients with rectal cancer (the OnCoRe project): A propensity-score matched cohort analysis. Lancet Oncol. 2016, 17, 174–183. [Google Scholar] [CrossRef]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Buzzelli, J.N.; Ouaret, D.; Brown, G.; Allen, P.D.; Muschel, R.J. Colorectal cancer liver metastases organoids retain characteristics of original tumor and acquire chemotherapy resistance. Stem Cell Res. 2018, 27, 109–120. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Srinivasan, T.; Lin, C.; Tung, K.-L.; Gao, Z.; Hsu, D.S.; Lipkin, S.M.; Shen, X. Single-Cell Transcriptomics Reveals Heterogeneity and Drug Response of Human Colorectal Cancer Organoids Kai-Yuan. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2019, 2018, 2378–2381. [Google Scholar]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, N.; Clevers, H. Studying cellular heterogeneity and drug sensitivity in colorectal cancer using organoid technology. Curr. Opin. Genet. Dev. 2018, 52, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Arul, M.; Roslani, A.C.; Cheah, S.H. Heterogeneity in cancer cells: Variation in drug response in different primary and secondary colorectal cancer cell lines in vitro. In Vitro Cell. Dev. Biol. Anim. 2017, 53, 435–447. [Google Scholar] [CrossRef]
- Okazawa, Y.; Mizukoshi, K.; Koyama, Y.; Okubo, S.; Komiyama, H.; Kojima, Y.; Goto, M.; Habu, S.; Hino, O.; Sakamoto, K.; et al. High-sensitivity detection of micrometastases generated by GFP lentivirus-transduced organoids cultured from a patient-derived colon tumor. J. Vis. Exp. 2018, 2018, 57374. [Google Scholar] [CrossRef]
- Brown, K.; Xue, A.; Julovi, S.; Gill, A.; Pavlakis, N.; Samra, J.; Smith, R.; Hugh, T. Using patient-derived xenograft models of colorectal liver metastases to predict chemosensitivity. J. Surg. Res. 2018, 227, 158–167. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Hidalgo, M.; Kung, A.L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 2015, 15, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Inoue, M. Application of Cancer Organoid Model for Drug Screening and Personalized Therapy. Cells 2019, 8, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, M.; Ren, L.; He, G.; Lin, Q.; Tang, W.; Chen, Y.; Chen, J.; Liu, T.; Ji, M.; Wei, Y.; et al. A novel patient-derived organoids-based xenografts model for preclinical drug response testing in patients with colorectal liver metastases. J. Transl. Med. 2020, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Clohessy, J.G.; Paulo Pandolfi, P. Mouse hospital and co-clinical trial project—from bench to bedside. Nat. Rev. Clin. Oncol. 2015, 12, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, E.H.; Dale, J.; Griffin, G. Genetic engineering of animals: Ethical issues, including welfare concerns. Can. Vet. J. 2011, 52, 544. [Google Scholar]
- Yang, H.; Sun, L.; Liu, M.; Mao, Y. Patient-derived organoids: A promising model for personalized cancer treatment. Gastroenterol. Rep. 2018, 6, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.H.; Czerwinski, M.; Wu, A.; Dame, M.K.; Attili, D.; Hill, E.; Colacino, J.A.; Nowacki, L.M.; Shroyer, N.F.; Higgins, P.D.R.; et al. A Method for Cryogenic Preservation of Human Biopsy Specimens and Subsequent Organoid Culture. Cmgh 2018, 6, 218–222.e7. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt—Villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Li, J.; Xu, H.; Zhang, L.; Song, L.; Feng, D.; Peng, X.; Wu, M.; Zou, Y.; Wang, B.; Zhan, L.; et al. Malignant ascites-derived organoid (MADO) cultures for gastric cancer in vitro modelling and drug screening. J. Cancer Res. Clin. Oncol. 2019, 145, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clin. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M.L. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. npj Precis. Oncol. 2021, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, R.G. Rapid in vitro evaluation of immune responses to tumor-derived organoids as an adjunct to immunotherapy trials. J. Clin. Oncol. 2017, 35, 3573. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a Living Organoid Biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Weeber, F.; Van De Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-Van Hooijdonk, C.G.M.; Van Der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Schütte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef] [Green Version]
- Aberle, M.R.; Burkhart, R.A.; Tiriac, H.; Damink, S.W.M.O.; Dejong, C.H.C.; Tuveson, D.A.; van Dam, R.M. Patient-derived organoid models help define personalized management of gastro-intestinal cancer. Br. J. Surg. 2018, 105, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Francies, H.E.; Barthorpe, A.; McLaren-Douglas, A.; Barendt, W.J.; Garnett, M.J. Drug sensitivity assays of human cancer organoid cultures. Methods Mol. Biol. 2019, 1576, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleguezuelos-Manzano, C.; Puschhof, J.; van den Brink, S.; Geurts, V.; Beumer, J.; Clevers, H. Establishment and Culture of Human Intestinal Organoids Derived from Adult Stem Cells. Curr. Protoc. Immunol. 2020, 130, e106. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.; Kryeziu, K.; Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Langerud, J.; Røsok, B.; Totland, M.Z.; Brunsell, T.H.; Pellinen, T.; et al. Patient-Derived Organoids from Multiple Colorectal Cancer Liver Metastases Reveal Moderate Intra-patient Pharmacotranscriptomic Heterogeneity. Clin. Cancer Res. 2020, 26, 4107–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schäfer, R.; et al. Assay establishment and validation of a high-throughput screening platform for three-dimensional patient-derived colon cancer organoid cultures. J. Biomol. Screen. 2016, 21, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers Georgios. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Pathology Outlines—Stains & CD Markers. Available online: https://www.pathologyoutlines.com/stains.html (accessed on 22 December 2021).
- Young, L.; Sung, J.; Masters, J.R. Detection of mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef] [PubMed]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid–T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.M.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T.; et al. Medium-throughput Drug Screening of Patient-derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin. Cancer Res. 2020, 26, 3662–3670. [Google Scholar] [CrossRef]
- Kong, J.C.H.; Guerra, G.R.; Millen, R.M.; Roth, S.; Xu, H.; Neeson, P.J.; Darcy, P.K.; Kershaw, M.H.; Sampurno, S.; Malaterre, J.; et al. Tumor-Infiltrating Lymphocyte Function Predicts Response to Neoadjuvant Chemoradiotherapy in Locally Advanced Rectal Cancer. J. Clin. Oncol. 2020, 2, 1–15. [Google Scholar] [CrossRef]
- Grivicich, I.; Mans, D.R.A.; Peters, G.J.; Schwartsmann, C. Irinotecan and oxaliplatin: An overview of the novel chemotherapeutic options for the treatment of advanced colorectal cancer. Braz. J. Med. Biol. Res. 2001, 34, 1087–1103. [Google Scholar] [CrossRef] [Green Version]
- Kish, T.; Uppal, P. Trifluridine/tipiracil (Lonsurf) for the treatment of metastatic colorectal cancer. Phys. Ther. 2016, 41, 314–317. [Google Scholar]
- Zaman, M.H.; Trapani, L.M.; Sieminski, A.L.; Mackellar, D.; Gong, H.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Zaman, M.H.; Trapani, L.M.; et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 15–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.G. Nuclear Morphology and the Biology of Cancer Cells. Acta Cytol. 2020, 64, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Larsson, P.; Ljuslinder, I.; Öhlund, D.; Myte, R.; Löfgren-Burström, A.; Zingmark, C.; Ling, A.; Edin, S.; Palmqvist, R. Ex vivo organoid cultures reveal the importance of the tumor microenvironment for maintenance of colorectal cancer stem cells. Cancers 2020, 12, 923. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid technology and applications in cancer research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Reference | Cancer Type | Method | No of Organoids Investigated | No of pt Generated PDOs from | Investigating Intra-Pt Heterogeneity (Number of pt) | Treatment | Quality Control Check | Activation Before Assay | Time of Drug Testing | Endpoint | Endpoint Target | Endpoint Detection METHOD | No of PDO Correlating with Clinical pt Response | % Correlation Observed |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ganesh et al. [49] | pRC | PDO | 7 | 7 | 0 | 5-FU | Exon sequencing, IHC | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 7 | 100 |
| Ganesh et al. [49] | pRC | PDO | 7 | 7 | 0 | FOLFOX | Exon sequencing, IHC | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 7 | 100 |
| Ganesh et al. [49] | pRC | PDO | 19 | 7 | NA | Radiation | Exon sequencing, IHC | NA | 8 days | Cell viability | ATP | CellTiter-Glo 3D | 7 | 100 |
| Yao et al. [48] | pRC | PDO | 80 | 80 | 0 | 5-FU and radiation | IHC, CNV | NA | 24 days | Organoid size, cell viability | Size (uM), ATP | Image-Pro Plus 6.0, CellTiter-Glo 3D | 68 | 85 |
| Vlachogiannis et al. [47] | mCRC | PDO | 6 | 4 | 1 | TAS-102 | IHC, NGS | NA | 6–8 days | Cell viability | Metabolic capacity | CellTiter-Blue | 4 | 100 |
| Vlachogiannis et al. [47] | mCRC | PDO | 5 | 5 | 0 | Cetuximab | IHC, NGS | NA | 6–8 days | Cell viability | Metabolic capacity | CellTiter-Blue | 3 | 60 |
| Ooft et al. [57] | mCRC | PDO | 10 | 10 | 0 | Irinotecan | SNP | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 10 | 100 |
| Ooft et al. [57] | mCRC | PDO | 12 | 12 | 0 | 5-FU and irinotecan | SNP | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 12 | 100 |
| Ooft et al. [57] | mCRC | PDO | 16 | 10 | 0 | FOLFOX | SNP | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 0 | 0 |
| Ooft et al. [57] | mCRC | PDO | 16 | 10 | 0 | 5-FU | SNP | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 0 | 0 |
| Ooft et al. [57] | mCRC | PDO | 16 | 10 | 0 | Oxaliplatin | SNP | NA | 6 days | Cell viability | ATP | CellTiter-Glo 3D | 0 | 0 |
| Narasimhan et al. [58] | mCRC | PDO | 9 | 3 | FOLFOX, FOLFIRI | STR, IHC | NA | 6 days | Cell viability | ATP | CellTiter-Glo 2.0 | 0 | 0 | |
| Kong et al. [59] | mRC | Co-culture PDO | 17 | 17 | 0 | 5-FU and radiation | STR, IHC | NA | 3 days | Killing assay | Caspase 3/7, Propidium Iodide | Caspase activity, ScEM | 17 | 100 |
| Chalabi et al. [55] | pCRC | Co-culture PDO | 13 | 12 | 1 | Nivolumab and ipilimumab | SNP | Organoid with IFN-g | 14 days | T-cell activity | IFN-γ | Cytometric Bead Array | 9 | 75 |
| Ramsay [36] | pCRC | Co-culture PDO | 12 | 12 | NA | NA | NA | NA | NA | Killing assay, T-cell activity | Caspase, IFN-γ | Caspase activity, NA | NA | NA |
| Ramsay [36] | mCRC | Co-culture PDO | 20 | 20 | NA | NA | NA | NA | NA | Killing assay, T-cell activity | Caspase, IFN-γ | Caspase activity, NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furbo, S.; Urbano, P.C.M.; Raskov, H.H.; Troelsen, J.T.; Kanstrup Fiehn, A.-M.; Gögenur, I. Use of Patient-Derived Organoids as a Treatment Selection Model for Colorectal Cancer: A Narrative Review. Cancers 2022, 14, 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041069
Furbo S, Urbano PCM, Raskov HH, Troelsen JT, Kanstrup Fiehn A-M, Gögenur I. Use of Patient-Derived Organoids as a Treatment Selection Model for Colorectal Cancer: A Narrative Review. Cancers. 2022; 14(4):1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041069
Chicago/Turabian StyleFurbo, Sara, Paulo César Martins Urbano, Hans Henrik Raskov, Jesper Thorvald Troelsen, Anne-Marie Kanstrup Fiehn, and Ismail Gögenur. 2022. "Use of Patient-Derived Organoids as a Treatment Selection Model for Colorectal Cancer: A Narrative Review" Cancers 14, no. 4: 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041069