
Identification of New Vulnerabilities in Conjunctival Melanoma Using Image-Based High Content Drug Screening

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines
2.2. Next Generation Sequencing and Variant Calling
2.3. Variant Annotation and Filtering
2.4. Drug Screening
2.5. Apoptosis Assay
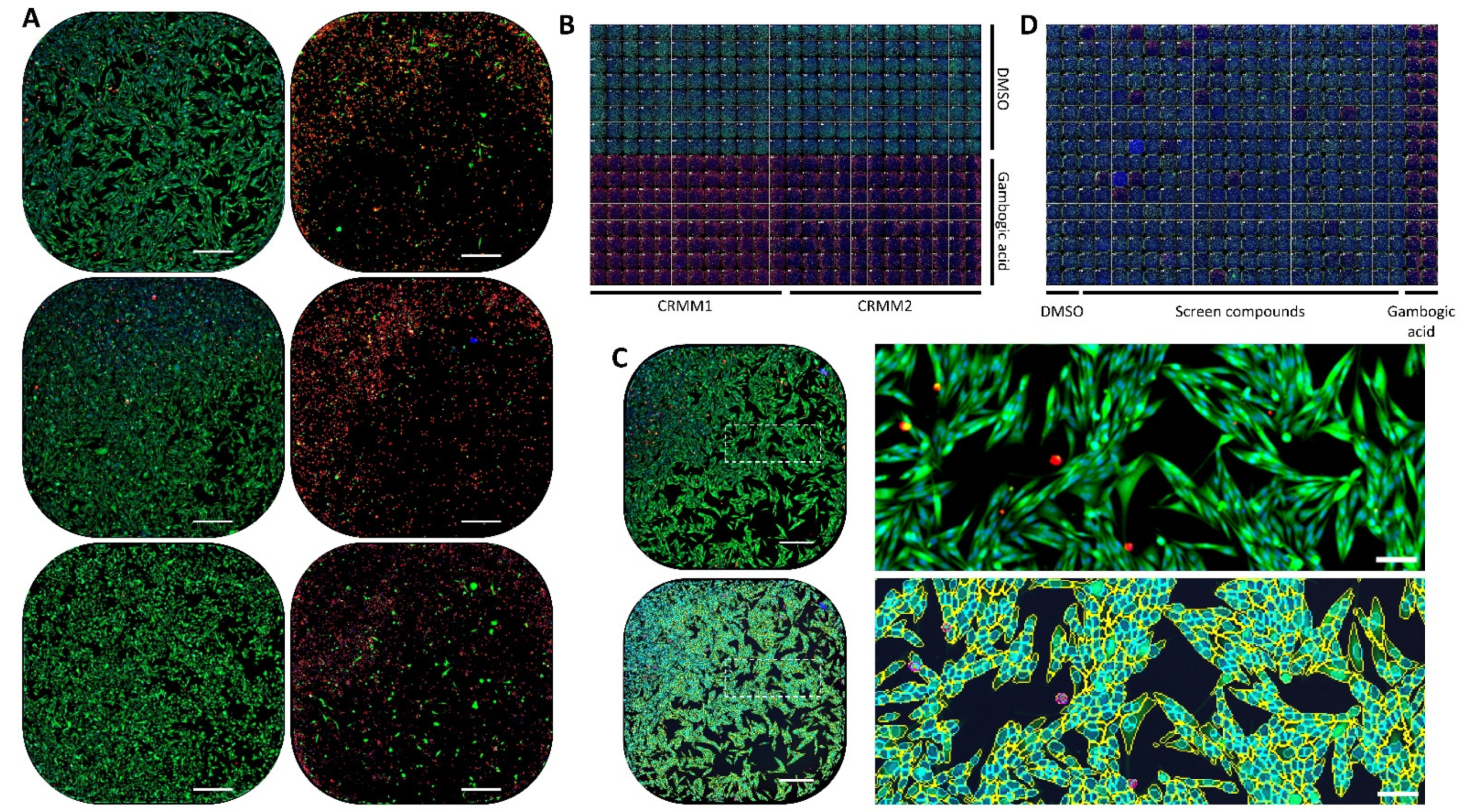
2.6. Automated Image Analysis Pipeline and Data-Analysis Workflow
3. Results
3.1. Genomic Background
3.2. Screening Assay Validation and Hits Assignment
3.3. Dose–Response Curves
3.4. Apoptosis Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shields, C.L.; Markowitz, J.S.; Belinsky, I.; Schwartzstein, H.; George, N.S.; Lally, S.E.; Mashayekhi, A.; Shields, J.A. Conjunctival melanoma: Outcomes based on tumor origin in 382 consecutive cases. Ophthalmology 2011, 118, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Missotten, G.S.; Keijser, S.; De Keizer, R.J.; De Wolff-Rouendaal, D. Conjunctival melanoma in the Netherlands: A nationwide study. Investig. Ophthalmol. Vis. Sci. 2005, 46, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Tuomaala, S.; Eskelin, S.; Tarkkanen, A.; Kivela, T. Population-based assessment of clinical characteristics predicting outcome of conjunctival melanoma in whites. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3399–3408. [Google Scholar]
- Yu, G.P.; Hu, D.N.; McCormick, S.; Finger, P.T. Conjunctival melanoma: Is it increasing in the United States? Am. J. Ophthalmol. 2003, 135, 800–806. [Google Scholar] [CrossRef]
- Triay, E.; Bergman, L.; Nilsson, B.; All-Ericsson, C.; Seregard, S. Time trends in the incidence of conjunctival melanoma in Sweden. Br. J. Ophthalmol. 2009, 93, 1524–1528. [Google Scholar] [CrossRef]
- Larsen, A.C.; Dahl, C.; Dahmcke, C.M.; Lade-Keller, J.; Siersma, V.D.; Toft, P.B.; Coupland, S.E.; Prause, J.U.; Guldberg, P.; Heegaard, S. BRAF mutations in conjunctival melanoma: Investigation of incidence, clinicopathological features, prognosis and paired premalignant lesions. Acta Ophthalmol. 2016, 94, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Rivolta, C.; Royer-Bertrand, B.; Rimoldi, D.; Schalenbourg, A.; Zografos, L.; Leyvraz, S.; Moulin, A. UV light signature in conjunctival melanoma; not only skin should be protected from solar radiation. J. Hum. Genet. 2016, 61, 361–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisarova, K.; Folcher, M.; El Zaoui, I.; Pescini-Gobert, R.; Peter, V.G.; Royer-Bertrand, B.; Zografos, L.; Schalenbourg, A.; Nicolas, M.; Rimoldi, D.; et al. Genomic and transcriptomic landscape of conjunctival melanoma. PLoS Genet. 2020, 16, e1009201. [Google Scholar] [CrossRef]
- Demirci, H.; Demirci, F.Y.; Ciftci, S.; Elner, V.M.; Wu, Y.M.; Ning, Y.; Chinnaiyan, A.; Robinson, D.R. Integrative Exome and Transcriptome Analysis of Conjunctival Melanoma and Its Potential Application for Personalized Therapy. JAMA Ophthalmol. 2019, 137, 1444–1448. [Google Scholar] [CrossRef]
- Larsen, A.C. Conjunctival malignant melanoma in Denmark: Epidemiology, treatment and prognosis with special emphasis on tumorigenesis and genetic profile. Acta Ophthalmol. 2016, 94, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Griewank, K.G.; Westekemper, H.; Murali, R.; Mach, M.; Schilling, B.; Wiesner, T.; Schimming, T.; Livingstone, E.; Sucker, A.; Grabellus, F.; et al. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3143–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, S.L.; Cosgarea, I.; Susskind, D.; Murali, R.; Moller, I.; Reis, H.; Leonardelli, S.; Schilling, B.; Schimming, T.; Hadaschik, E.; et al. NF1 mutations in conjunctival melanoma. Br. J. Cancer 2018, 118, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Gardrat, S.; Houy, A.; Brooks, K.; Cassoux, N.; Barnhill, R.; Dayot, S.; Bieche, I.; Raynal, V.; Baulande, S.; Marais, R.; et al. Definition of Biologically Distinct Groups of Conjunctival Melanomas According to Etiological Factors and Implications for Precision Medicine. Cancers 2021, 13, 3836. [Google Scholar] [CrossRef]
- Van Poppelen, N.M.; van Ipenburg, J.A.; van den Bosch, Q.; Vaarwater, J.; Brands, T.; Eussen, B.; Magielsen, F.; Dubbink, H.J.; Paridaens, D.; Brosens, E.; et al. Molecular Genetics of Conjunctival Melanoma and Prognostic Value of TERT Promoter Mutation Analysis. Int. J. Mol. Sci. 2021, 22, 5784. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Lally, S.E.; Milman, T.; Orloff, M.; Dalvin, L.A.; Eberhart, C.G.; Heaphy, C.M.; Rodriguez, F.J.; Lin, C.C.; Dockery, P.W.; Shields, J.A.; et al. Mutational Landscape and Outcomes of Conjunctival Melanoma in 101 Patients. Ophthalmology 2022. [Google Scholar] [CrossRef]
- Kenawy, N.; Kalirai, H.; Sacco, J.J.; Lake, S.L.; Heegaard, S.; Larsen, A.C.; Finger, P.T.; Milman, T.; Chin, K.; Mosci, C.; et al. Conjunctival melanoma copy number alterations and correlation with mutation status, tumor features, and clinical outcome. Pigment. Cell Melanoma Res. 2019, 32, 564–575. [Google Scholar] [CrossRef] [Green Version]
- El Zaoui, I.; Bucher, M.; Rimoldi, D.; Nicolas, M.; Kaya, G.; Pescini Gobert, R.; Bedoni, N.; Schalenbourg, A.; Sakina, E.; Zografos, L.; et al. Conjunctival Melanoma Targeted Therapy: MAPK and PI3K/mTOR Pathways Inhibition. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2764–2772. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Heijkants, R.C.; Jochemsen, A.G.; Dogrusoz, M.; de Lange, M.J.; van der Velden, P.A.; van der Burg, S.H.; Jager, M.J.; Verdijk, R.M. Targeting of the MAPK and AKT pathways in conjunctival melanoma shows potential synergy. Oncotarget 2017, 8, 58021–58036. [Google Scholar] [CrossRef]
- Brouwer, N.J.; Verdijk, R.M.; Heegaard, S.; Marinkovic, M.; Esmaeli, B.; Jager, M.J. Conjunctival melanoma: New insights in tumour genetics and immunology, leading to new therapeutic options. Prog. Retin. Eye. Res. 2022, 86, 100971. [Google Scholar] [CrossRef]
- Kim, J.M.; Weiss, S.; Sinard, J.H.; Pointdujour-Lim, R. Dabrafenib and Trametinib for BRAF-Mutated Conjunctival Melanoma. Ocul. Oncol. Pathol. 2020, 6, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855 e819. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.; et al. Modernizing reference genome assemblies. PLoS. Biol. 2011, 9, e1001091. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Quinodoz, M.; Peter, V.G.; Cisarova, K.; Royer-Bertrand, B.; Stenson, P.D.; Cooper, D.N.; Unger, S.; Superti-Furga, A.; Rivolta, C. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am. J. Hum. Genet. 2022, 109, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Simoneschi, D.; Rona, G.; Zhou, N.; Jeong, Y.T.; Jiang, S.; Milletti, G.; Arbini, A.A.; O’Sullivan, A.; Wang, A.A.; Nithikasem, S.; et al. CRL4(AMBRA1) is a master regulator of D-type cyclins. Nature 2021, 592, 789–793. [Google Scholar] [CrossRef]
- Cohen, R.B.; Aamdal, S.; Nyakas, M.; Cavallin, M.; Green, D.; Learoyd, M.; Smith, I.; Kurzrock, R. A phase I dose-finding, safety and tolerability study of AZD8330 in patients with advanced malignancies. Eur. J. Cancer 2013, 49, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, C.; Italiano, A.; Houede, N.; Awada, A.; Aftimos, P.; Lesimple, T.; Dinulescu, M.; Schellens, J.H.M.; Leijen, S.; Rottey, S.; et al. Selective Oral MEK1/2 Inhibitor Pimasertib in Metastatic Melanoma: Antitumor Activity in a Phase I, Dose-Escalation Trial. Target Oncol. 2021, 16, 47–57. [Google Scholar] [CrossRef]
- Micel, L.N.; Tentler, J.J.; Tan, A.C.; Selby, H.M.; Brunkow, K.L.; Robertson, K.M.; Davis, S.L.; Klauck, P.J.; Pitts, T.M.; Gangolli, E.; et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol. Cancer Ther. 2015, 14, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S.; et al. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, A.; Soria, J.C.; Hollebecque, A.; LoRusso, P.; Bendell, J.; Huang, S.A.; Wagle, M.C.; Okrah, K.; Liu, L.; Murray, E.; et al. A First-in-Human Phase I Study to Evaluate the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1229–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.; van der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.; Stayner, L.A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Gokmen-Polar, Y.; Liu, Y.; Toroni, R.A.; Sanders, K.L.; Mehta, R.; Badve, S.; Rommel, C.; Sledge, G.W., Jr. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res. Treat. 2012, 136, 673–682. [Google Scholar] [CrossRef]
- Hernandez-Prat, A.; Rodriguez-Vida, A.; Juanpere-Rodero, N.; Arpi, O.; Menendez, S.; Soria-Jimenez, L.; Martinez, A.; Iarchouk, N.; Rojo, F.; Albanell, J.; et al. Novel Oral mTORC1/2 Inhibitor TAK-228 Has Synergistic Antitumor Effects When Combined with Paclitaxel or PI3Kalpha Inhibitor TAK-117 in Preclinical Bladder Cancer Models. Mol. Cancer Res. 2019, 17, 1931–1944. [Google Scholar] [CrossRef]
- Ingels, A.; Zhao, H.; Thong, A.E.; Saar, M.; Valta, M.P.; Nolley, R.; Santos, J.; Peehl, D.M. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int. J. Cancer J. Int. Du Cancer 2014, 134, 2322–2329. [Google Scholar] [CrossRef]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.M.; Villanueva, J.; Nguyen, T.T.; Lioni, M.; Xiao, M.; Kong, J.; Krepler, C.; Vultur, A.; Flaherty, K.T.; Nathanson, K.L.; et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS ONE 2013, 8, e59588. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Cismas, S.; Crudden, C.; Trocme, E.; Worrall, C.; Suleymanova, N.; Lin, T.; Zheng, H.; Seregard, S.; Girnita, A.; et al. IGF-1R is a molecular determinant for response to p53 reactivation therapy in conjunctival melanoma. Oncogene 2022, 41, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Danilov, A.V.; Hu, S.; Orr, B.; Godek, K.; Mustachio, L.M.; Sekula, D.; Liu, X.; Kawakami, M.; Johnson, F.M.; Compton, D.A.; et al. Dinaciclib Induces Anaphase Catastrophe in Lung Cancer Cells via Inhibition of Cyclin-Dependent Kinases 1 and 2. Mol. Cancer Ther. 2016, 15, 2758–2766. [Google Scholar] [CrossRef] [Green Version]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef]
- Stephenson, J.J.; Nemunaitis, J.; Joy, A.A.; Martin, J.C.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Yao, S.L.; Zhu, Y.; Zhou, H.; et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer 2014, 83, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Scarfo, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs. ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Eshima, S.; Kato, S.; Fisher, D.E.; Sakurai, H.; Hayakawa, Y.; Yokoyama, S. Rational Combination Therapy for Melanoma with Dinaciclib by Targeting BAK-Dependent Cell Death. Mol. Cancer Ther. 2020, 19, 627–636. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Jalili, A.; Moser, A.; Pashenkov, M.; Wagner, C.; Pathria, G.; Borgdorff, V.; Gschaider, M.; Stingl, G.; Ramaswamy, S.; Wagner, S.N. Polo-like kinase 1 is a potential therapeutic target in human melanoma. J. Investig. Dermatol. 2011, 131, 1886–1895. [Google Scholar] [CrossRef] [Green Version]
- Cholewa, B.D.; Ndiaye, M.A.; Huang, W.; Liu, X.; Ahmad, N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett. 2017, 385, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Schoffski, P.; Awada, A.; Dumez, H.; Gil, T.; Bartholomeus, S.; Wolter, P.; Taton, M.; Fritsch, H.; Glomb, P.; Munzert, G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 2012, 48, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M. Aurora kinases. Int. J. Biochem. Cell Biol. 2005, 37, 1572–1577. [Google Scholar] [CrossRef]
- Puig-Butille, J.A.; Vinyals, A.; Ferreres, J.R.; Aguilera, P.; Cabre, E.; Tell-Marti, G.; Marcoval, J.; Mateo, F.; Palomero, L.; Badenas, C.; et al. AURKA Overexpression Is Driven by FOXM1 and MAPK/ERK Activation in Melanoma Cells Harboring BRAF or NRAS Mutations: Impact on Melanoma Prognosis and Therapy. J. Investig. Dermatol. 2017, 137, 1297–1310. [Google Scholar] [CrossRef] [Green Version]
- Kaestner, P.; Stolz, A.; Bastians, H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol. Cancer 2009, 8, 2046–2056. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Moschos, S.J.; Becker, D. Functional analysis and molecular targeting of aurora kinases a and B in advanced melanoma. Genes. Cancer 2010, 1, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathria, G.; Garg, B.; Borgdorff, V.; Garg, K.; Wagner, C.; Superti-Furga, G.; Wagner, S.N. Overcoming MITF-conferred drug resistance through dual AURKA/MAPK targeting in human melanoma cells. Cell Death Dis. 2016, 7, e2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.C.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M.; et al. Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. J. Natl. Cancer Inst. 2016, 108, djv406. [Google Scholar] [CrossRef]
- Punt, S.; Malu, S.; McKenzie, J.A.; Manrique, S.Z.; Doorduijn, E.M.; Mbofung, R.M.; Williams, L.; Silverman, D.A.; Ashkin, E.L.; Dominguez, A.L.; et al. Aurora kinase inhibition sensitizes melanoma cells to T-cell-mediated cytotoxicity. Cancer Immunol. Immunother. 2021, 70, 1101–1113. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Ozcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamura, T.; Lowell, A.M.; Engelman, J.A.; Shapiro, G.I. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005, 65, 6401–6408. [Google Scholar] [CrossRef] [Green Version]
- Normant, E.; Paez, G.; West, K.A.; Lim, A.R.; Slocum, K.L.; Tunkey, C.; McDougall, J.; Wylie, A.A.; Robison, K.; Caliri, K.; et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011, 30, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Revi. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Marmarelis, M.E.; Hodi, F.S. Activity of the heat shock protein 90 inhibitor ganetespib in melanoma. PLoS ONE 2013, 8, e56134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquaviva, J.; Smith, D.L.; Jimenez, J.P.; Zhang, C.; Sequeira, M.; He, S.; Sang, J.; Bates, R.C.; Proia, D.A. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol. Cancer Ther. 2014, 13, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Mbofung, R.M.; McKenzie, J.A.; Malu, S.; Zhang, M.; Peng, W.; Liu, C.; Kuiatse, I.; Tieu, T.; Williams, L.; Devi, S.; et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 2017, 8, 451. [Google Scholar] [CrossRef]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable BRAF (V600E)-Mutant Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5516–5524. [Google Scholar] [CrossRef] [Green Version]
- Niu, L.; Yang, J.; Yan, W.; Yu, Y.; Zheng, Y.; Ye, H.; Chen, Q.; Chen, L. Reversible binding of the anticancer drug KXO1 (tirbanibulin) to the colchicine-binding site of beta-tubulin explains KXO1’s low clinical toxicity. J. Biol. Chem. 2019, 294, 18099–18108. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Min, A.; Lee, K.H.; Yang, Y.; Kim, T.Y.; Lim, J.M.; Park, S.J.; Nam, H.J.; Kim, J.E.; Song, S.H.; et al. Antitumor Effect of KX-01 through Inhibiting Src Family Kinases and Mitosis. Cancer Res. Treat. 2017, 49, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbalagan, M.; Carrier, L.; Glodowski, S.; Hangauer, D.; Shan, B.; Rowan, B.G. KX-01, a novel Src kinase inhibitor directed toward the peptide substrate site, synergizes with tamoxifen in estrogen receptor alpha positive breast cancer. Breast Cancer Res. Treat. 2012, 132, 391–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hu, W.; Dalton, H.J.; Choi, H.J.; Huang, J.; Kang, Y.; Pradeep, S.; Miyake, T.; Song, J.H.; Wen, Y.; et al. Targeting SRC and tubulin in mucinous ovarian carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6532–6543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauvelt, A.; Kempers, S.; Lain, E.; Schlesinger, T.; Tyring, S.; Forman, S.; Ablon, G.; Martin, G.; Wang, H.; Cutler, D.L.; et al. Phase 3 Trials of Tirbanibulin Ointment for Actinic Keratosis. N. Engl. J. Med. 2021, 384, 512–520. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Other Name | Drug Class | CM2005.1 IC50 nM | CM2005.1 Apoptosis | CRMM1 IC50 nM | CRMM1 Apoptosis | CRMM2 IC50 nM | CRMM2 Apoptosis |
|---|---|---|---|---|---|---|---|---|
| Tivantinib 1 | MET I | 452.25 | N | 756.47 | N | 390.85 | N | |
| Tivantinib 2 | MET I | 352.81 | N | 843.04 | N | 378.37 | Y | |
| PLX4720 | BRAF I | ns | 25.6 | Y | 5.72 | N | ||
| Dabrafenib | BRAF I | 44.16 | Y | 988.87 | Y | ns | ||
| RAF-265 | CHIR-265 | BRAF I | 1012.16 | nt | 1001.5 | nt | 1048.7 | nt |
| Trametinib 3 | MEK I | 475.61 | Y | 4.71 | N | ns | ||
| Trametinib 4 | MEK I | 22.09 | Y | ns | 107.1 | Y | ||
| PD0325901 | Mirdametinib | MEK I | 24.52 | Y | 0.73 | N | 431.59 | Y |
| Cobimetinib | MEK I | 44.42 | Y | 349.57 | Y | 488.53 | Y | |
| AS703026 | Pimasertib | MEK I | 52.4 | Y | 292.37 | Y | 439.79 | Y |
| AZD8330 | MEK I | 12.5 | Y | 103.94 | Y | 62.94 | Y | |
| TAK-733 | MEK I | 31.85 | Y | 640.35 | Y | 134.27 | Y | |
| AZD6244 | Selumetinib | MEK I | 229.98 | Y | 1.85 | N | ns | |
| PD318088 | MEK I | 261.88 | Y | 247.98 | N | ns | ||
| PD184352 | MEK I | 748.43 | Y | 33.64 | N | ns | ||
| GDC-0994 | Ravoxertinib | ERK I | 816.06 | Y | 32.25 | N | ns | |
| VRT752271 | Ulixertinib | ERK I | ns | 294 | N | ns | ||
| A-674563 | AKT I | 1001.89 | Y | 630.25 | N | ns | ||
| GSK2126458 | Omipalisib | PI3k/mTOR | 883.6 | Y | 657.42 | Y | 164.6 | Y |
| PIK-75 | PI3K I | 389.79 | Y | 701.89 | Y | 439.64 | Y | |
| PI-103 | PI3K I | ns | ns | ns | ||||
| PF-04691502 | PI3K I | 948.41 | Y | ns | 648.29 | Y | ||
| GDC-0980 | Apitolisib | PI3K I | 938.46 | Y | ns | 871.75 | Y | |
| BKM120 | Buparlisib | PI3K I | 3000 | nt | 1220.8 | nt | 867.2 | nt |
| Rapamycin | Sirolimus | mTOR I | ns | 115.15 | Y | ns | ||
| INK-128 | Sapanisertib | mTOR I | 536.15 | Y | 427.96 | Y | 219.17 | Y |
| AZD8055 | mTOR I | 603.26 | Y | 769.45 | Y | 322.95 | Y | |
| WYE-125132 | mTOR I | 604.89 | Y | 778.8 | Y | 286.97 | Y | |
| AZD2014 | Vistusertib | mTOR I | ns | ns | 740.23 | Y | ||
| Torin 2 | mTOR I | ns | 558.31 | Y | 419.91 | Y | ||
| Dinaciclib | CDK I | 67.66 | Y | 113.84 | Y | 38.82 | Y | |
| SNS-032 | CDK I | ns | ns | 609.51 | Y | |||
| Flavopiridol | Alvocidib | CDK I | 594.29 | Y | 427.26 | Y | 540.87 | Y |
| RGB-286638 | CDK I | 361.93 | Y | 366.39 | Y | 259.59 | Y | |
| Volasertib | PLK I | 108.5 | N | 37.14 | N | 29.32 | Y | |
| GSK461364 | PLK I | 799.39 | N | 58.1 | Y | 12.77 | Y | |
| ON-01910 | Rigosertib | PLK I | 363.93 | Y | 528.75 | Y | 55.24 | Y |
| NMS-1286937 | Onvansertib | PLK I | 105.48 | N | 297.29 | Y | 36.6 | N |
| HMN-214 | PLK I | 340.78 | N | 952.54 | Y | 775.01 | Y | |
| Alisertib | Aurora K I | ns | 467.01 | N | 32.77 | Y | ||
| MLN8054 | Aurora K I | 213.36 | N | 975.24 | N | 335.6 | N | |
| Ganetespib | Hsp 90 I | 94.41 | Y | 282.74 | Y | 38.65 | Y | |
| AT9283 | JAK I | ns | ns | 542.92 | Y | |||
| AZ 960 | JAK I | 373 | N | 390.16 | N | 371.03 | Y | |
| SB1317 | Zotiraciclib | JAK I/CDK I | 355.6 | Y | 461.27 | Y | 422.11 | Y |
| AZD7772 | chK I | ns | 777 | Y | 309.93 | Y | ||
| LY2603618 | Rabusertib | chK I | ns | ns | 934.34 | Y | ||
| CHIR-124 | chK I | 960.17 | Y | 652.17 | Y | 301.9 | Y | |
| KX2-391 | Tirbanibulin | SRC I | 466.1 | Y | 185.14 | Y | 32.6 | Y |
| PD-166285 | SRC/FGFR I | 3000 | ns | 1220.8 | nt | 867.2 | nt | |
| Birinipant | IAP I | 322.95 | N | 294 | Y | ns | ||
| KG-5 | PDGFR I | 2005.1 | ns | 1237.2 | nt | 941.7 | nt | |
| Hypericin | multiK I | 114.6 | N | 115 | N | 115.6 | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardou, K.; Nicolas, M.; Kuttler, F.; Cisarova, K.; Celik, E.; Quinodoz, M.; Riggi, N.; Michielin, O.; Rivolta, C.; Turcatti, G.; et al. Identification of New Vulnerabilities in Conjunctival Melanoma Using Image-Based High Content Drug Screening. Cancers 2022, 14, 1575. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061575
Nardou K, Nicolas M, Kuttler F, Cisarova K, Celik E, Quinodoz M, Riggi N, Michielin O, Rivolta C, Turcatti G, et al. Identification of New Vulnerabilities in Conjunctival Melanoma Using Image-Based High Content Drug Screening. Cancers. 2022; 14(6):1575. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061575
Chicago/Turabian StyleNardou, Katya, Michael Nicolas, Fabien Kuttler, Katarina Cisarova, Elifnaz Celik, Mathieu Quinodoz, Nicolo Riggi, Olivier Michielin, Carlo Rivolta, Gerardo Turcatti, and et al. 2022. "Identification of New Vulnerabilities in Conjunctival Melanoma Using Image-Based High Content Drug Screening" Cancers 14, no. 6: 1575. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061575