
Addressing the Elephant in the Immunotherapy Room: Effector T-Cell Priming versus Depletion of Regulatory T-Cells by Anti-CTLA-4 Therapy


Abstract
:Simple Summary
Abstract
1. Introduction
2. Tregs and CTLA-4 in Cancer
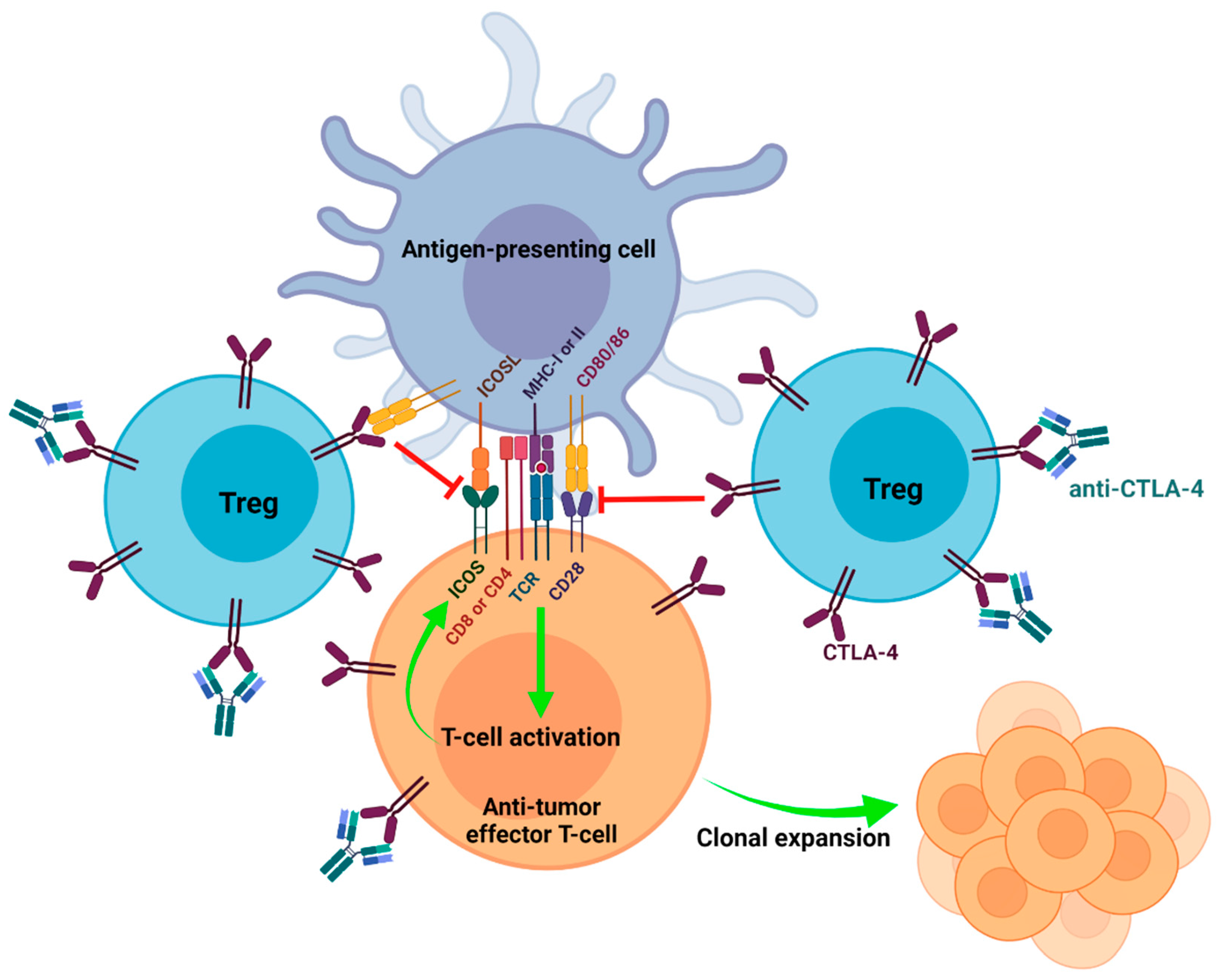
3. Blockading CD80/86-CTLA-4 Interactions
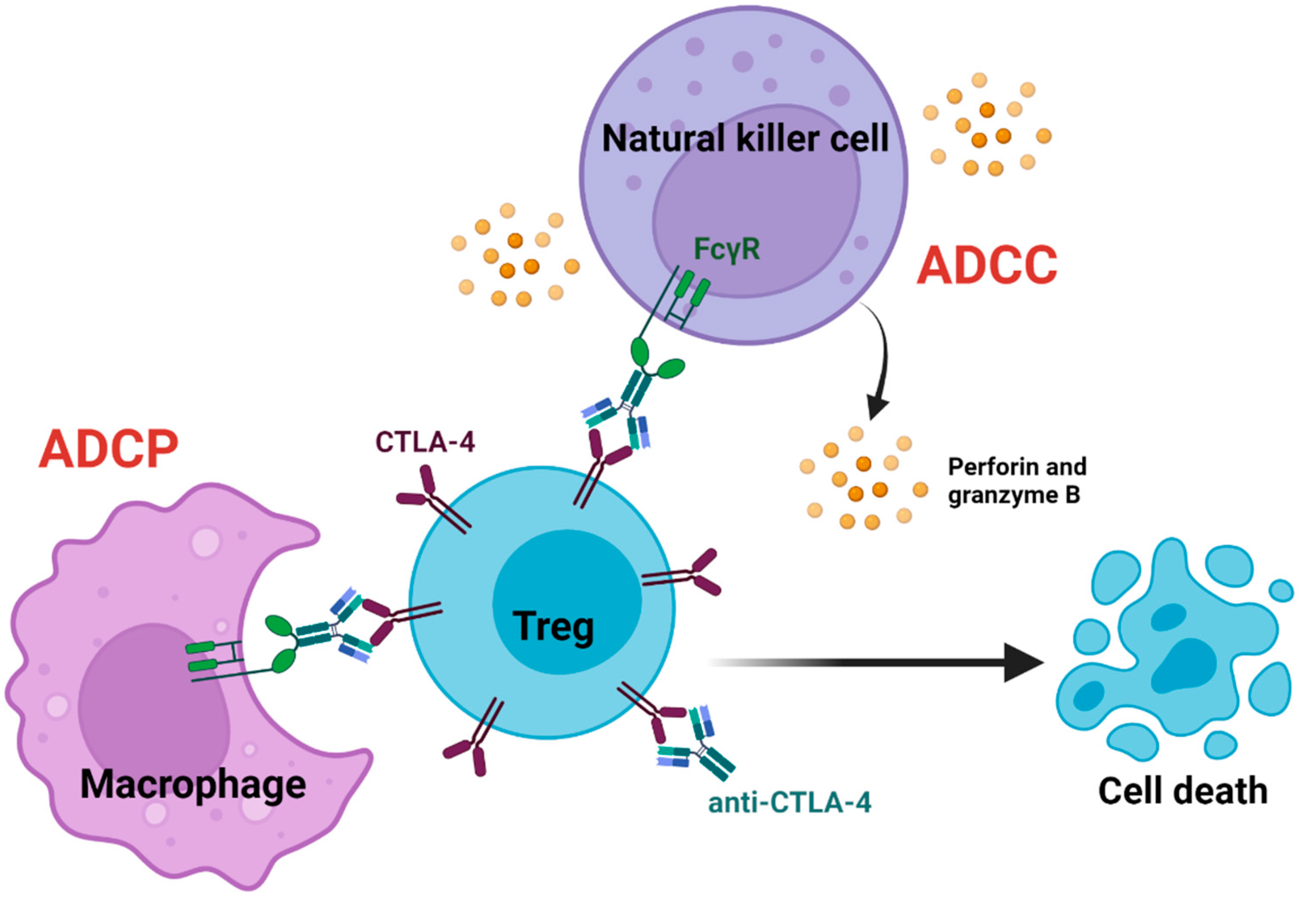
4. Anti-CTLA-4-Mediated Antibody-Dependent Cellular Cytotoxicity and Phagocytosis
5. Alteration of Treg Metabolism and Plasticity
6. Clinical Applications
7. Next-Generation CTLA-4 Targeting and Strategies to Improve Therapeutic Efficacy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on Both Effector and Regulatory T Cell Compartments Contributes to the Antitumor Activity of Anti–CTLA-4 Antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 666–677.e6. [Google Scholar] [CrossRef] [Green Version]
- Laouar, Y.; Town, T.; Jeng, D.; Tran, E.; Wan, Y.; Kuchroo, V.K.; Flavell, R.A. TGF-β Signaling in Dendritic Cells Is a Prerequisite for the Control of Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 10865–10870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S. Regulatory T Cells: History and Perspective. In Regulatory T Cells: Methods and Protocols; Kassiotis, G., Liston, A., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 3–17. ISBN 978-1-61737-979-6. [Google Scholar]
- Baatar, D.; Olkhanud, P.; Sumitomo, K.; Taub, D.; Gress, R.; Biragyn, A. Human Peripheral Blood T Regulatory Cells (Tregs), Functionally Primed CCR4+ Tregs and Unprimed CCR4− Tregs, Regulate Effector T Cells Using FasL. J. Immunol. 2007, 178, 4891–4900. [Google Scholar] [CrossRef] [Green Version]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective Depletion of Foxp3+ Regulatory T Cells Induces a Scurfy-like Disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Perea, A.L.; Arcia, E.D.; Rueda, C.M.; Velilla, P.A. Phenotypical Characterization of Regulatory T Cells in Humans and Rodents. Clin. Exp. Immunol. 2016, 185, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Taams, L.S.; Palmer, D.B.; Akbar, A.N.; Robinson, D.S.; Brown, Z.; Hawrylowicz, C.M. Regulatory T Cells in Human Disease and Their Potential for Therapeutic Manipulation. Immunology 2006, 118, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kuehnemuth, B.; Piseddu, I.; Wiedemann, G.M.; Lauseker, M.; Kuhn, C.; Hofmann, S.; Schmoeckel, E.; Endres, S.; Mayr, D.; Jeschke, U.; et al. CCL1 Is a Major Regulatory T Cell Attracting Factor in Human Breast Cancer. BMC Cancer 2018, 18, 1278. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc Decreases CCL8-Mediated Migration of CCR5+ Regulatory T Cells and Reduces Metastatic Tumor Growth in the Lungs. OncoImmunology 2016, 5, e1150398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righi, E.; Kashiwagi, S.; Yuan, J.; Santosuosso, M.; Leblanc, P.; Ingraham, R.; Forbes, B.; Edelblute, B.; Collette, B.; Xing, D.; et al. CXCL12/CXCR4 Blockade Induces Multimodal Antitumor Effects That Prolong Survival in an Immunocompetent Mouse Model of Ovarian Cancer. Cancer Res. 2011, 71, 5522–5534. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Kono, K.; Kawagushi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 Chemokines within Tumor Microenvironment Are Related to Accumulation of Foxp3+ Regulatory T Cells in Gastric Cancer. Int. J. Cancer 2008, 122, 2286–2293. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Liu, F.-F.; Zhang, X.-M.; Guo, X.-J.; Ren, M.-J.; Fu, L. Tumor Secretion of CCL22 Activates Intra-tumoral Treg Infiltration and Is Independent Prognostic Predictor of Breast Cancer. PLoS ONE 2013, 8, e76379. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour Hypoxia Promotes Tolerance and Angiogenesis via CCL28 and T(Reg) Cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Kotsakis, A.; Koinis, F.; Katsarou, A.; Gioulbasani, M.; Aggouraki, D.; Kentepozidis, N.; Georgoulias, V.; Vetsika, E.-K. Prognostic Value of Circulating Regulatory T Cell Subsets in Untreated Non-Small Cell Lung Cancer Patients. Sci. Rep. 2016, 6, 39247. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An Increased Abundance of Tumor-Infiltrating Regulatory T Cells Is Correlated with the Progression and Prognosis of Pancreatic Ductal Adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, G.; Jones, E.; Junaid, S.; El-Shanawany, T.; Scurr, M.; Mizen, P.; Kumar, M.; Jones, S.; Rees, B.; Williams, G.; et al. Suppression of Tumour-Specific CD4+ T Cells by Regulatory T Cells Is Associated with Progression of Human Colorectal Cancer. Gut 2012, 61, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza-Gonzalez, A.; Verhoef, C.; Ijzermans, J.N.M.; Peppelenbosch, M.P.; Kwekkeboom, J.; Verheij, J.; Janssen, H.L.A.; Sprengers, D. Activated Tumor-Infiltrating CD4+ Regulatory T Cells Restrain Antitumor Immunity in Patients with Primary or Metastatic Liver Cancer. Hepatology 2013, 57, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.; Tanaka, A.; Kibayashi, T.; Tanemura, A.; Sugiyama, D.; Wing, J.B.; Lim, E.L.; Teng, K.W.W.; Adeegbe, D.; Newell, E.W.; et al. Differential Control of Human Treg and Effector T Cells in Tumor Immunity by Fc-Engineered Anti–CTLA-4 Antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Jie, H.-B.; Gildener-Leapman, N.; Li, J.; Srivastava, R.M.; Gibson, S.P.; Whiteside, T.L.; Ferris, R.L. Intra-tumoral Regulatory T Cells Upregulate Immunosuppressive Molecules in Head and Neck Cancer Patients. Br. J. Cancer 2013, 109, 2629–2635. [Google Scholar] [CrossRef]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [Green Version]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells within B16 Melanoma Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of Tumor Immunity by Removing CD25+CD4+ T Cells: A Common Basis Between Tumor Immunity and Autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Maleki Vareki, S. High and Low Mutational Burden Tumors versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Rowell, E.A.; Thomas, R.M.; Hancock, W.W.; Wells, A.D. Transcriptional Regulation by Foxp3 Is Associated with Direct Promoter Occupancy and Modulation of Histone Acetylation. J. Biol. Chem. 2006, 281, 36828–36834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Josefowicz, S.Z.; Kas, A.; Chu, T.-T.; Gavin, M.A.; Rudensky, A.Y. Genome-Wide Analysis of Foxp3 Target Genes in Developing and Mature Regulatory T Cells. Nature 2007, 445, 936–940. [Google Scholar] [CrossRef]
- Zheng, Y.; Manzotti, C.N.; Burke, F.; Dussably, L.; Qureshi, O.; Walker, L.S.K.; Sansom, D.M. Acquisition of Suppressive Function by Activated Human CD4+CD25− T Cells Is Associated with the Expression of CTLA-4 Not FoxP3. J. Immunol. 2008, 181, 1683–1691. [Google Scholar] [CrossRef] [Green Version]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. Pillars Article: CTLA-4 Can Function as a Negative Regulator of T Cell Activation. J. Immunol. 2011, 187, 3466–3474. [Google Scholar]
- Krummel, M.F.; Allison, J.P. CTLA-4 Engagement Inhibits IL-2 Accumulation and Cell Cycle Progression upon Activation of Resting T Cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Walanus, T.L.; Bakker, C.Y.; Bluestone, J.A. CTLA-4 Ligation Blocks CD28-Dependent T Cell Activation. J. Exp. Med. 1996, 183, 2541–2550. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Beckermann, K.E.; Hongo, R.; Ye, X.; Young, K.; Carbonell, K.; Healey, D.C.C.; Siska, P.J.; Barone, S.; Roe, C.E.; Smith, C.C.; et al. CD28 Costimulation Drives Tumor-Infiltrating T Cell Glycolysis to Promote Inflammation. JCI Insight 2020, 5, e138729. [Google Scholar] [CrossRef] [PubMed]
- Shelby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intra-tumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Lu, J.; An, H. The Relative Change in Regulatory T Cells / T Helper Lymphocytes Ratio as Parameter for Prediction of Therapy Efficacy in Metastatic Colorectal Cancer Patients. Oncotarget 2017, 8, 109079–109093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.L.; Blair, P.J.; Musser, J.T.; Abe, R.; Tezuka, K.; Tsuji, T.; June, C.H. ICOS Costimulation Requires IL-2 and Can Be Prevented by CTLA-4 Engagement. J. Immunol. 2001, 166, 4943–4948. [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS Is an Inducible T-Cell Co-Stimulator Structurally and Functionally Related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Chen, Q.; Mo, L.; Cai, X.; Wei, L.; Xie, Z.; Li, H.; Li, J.; Hu, Z. ICOS Signal Facilitates Foxp3 Transcription to Favor Suppressive Function of Regulatory T Cells. Int. J. Med. Sci. 2018, 15, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL Pathway Is Required for Optimal Antitumor Responses Mediated by Anti–CTLA-4 Therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS Pathway Markedly Enhances Efficacy of CTLA-4 Blockade in Cancer Immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 Blockade Increases IFNgamma-Producing CD4+ICOShi Cells to Shift the Ratio of Effector to Regulatory T Cells in Cancer Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [Green Version]
- Carthon, B.C.; Wolchok, J.D.; Yuan, J.; Kamat, A.; Tang, D.S.N.; Sun, J.; Ku, G.; Troncoso, P.; Logothetis, C.J.; Allison, J.P.; et al. Preoperative CTLA-4 Blockade: Tolerability and Immune Monitoring in the Setting of a Presurgical Clinical Trial. Clin. Cancer Res. 2010, 16, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Vandenborre, K.; Van Gool, S.W.; Kasran, A.; Ceuppens, J.L.; Boogaerts, M.A.; Vandenberghe, P. Interaction of CTLA-4 (CD152) with CD80 or CD86 Inhibits Human T-cell Activation. Immunology 1999, 98, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.L.; Carter, K.P.; Wagner, E.K.; Asensio, M.A.; Benzie, E.; Chiang, Y.Y.; Coles, G.L.; Edgar, C.; Gautam, B.K.; Gras, A.; et al. Lack of Blocking Activity in Anti-CTLA-4 Antibodies Reduces Toxicity, but Not Anti-Tumor Efficacy. BioRxiv 2021. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M. A Reappraisal of CTLA-4 Checkpoint Blockade in Cancer Immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Girard, T.; Gaucher, D.; El-Far, M.; Breton, G.; Sékaly, R.-P. CD80 and CD86 IgC Domains Are Important for Quaternary Structure, Receptor Binding and Co-Signaling Function. Immunol. Lett. 2014, 161, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Simone, R.; Brizzolara, R.; Chiappori, A.; Milintenda-Floriani, F.; Natale, C.; Greco, L.; Schiavo, M.; Bagnasco, M.; Pesce, G.; Saverino, D. A Functional Soluble Form of CTLA-4 Is Present in the Serum of Celiac Patients and Correlates with Mucosal Injury. Int. Immunol. 2009, 21, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. From the Cover: PNAS Plus: Structural Basis for Cancer Immunotherapy by the First-in-Class Checkpoint Inhibitor Ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, E4223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Chai, Y.; Qi, J.; Zhang, C.W.H.; Tong, Z.; Shi, Y.; Yan, J.; Tan, S.; Gao, G.F. Remarkably Similar CTLA-4 Binding Properties of Therapeutic Ipilimumab and Tremelimumab Antibodies. Oncotarget 2017, 8, 67129–67139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulliard, Y.; Jolicoeur, R.; Zhang, J.; Dranoff, G.; Wilson, N.S.; Brogdon, J.L. OX40 Engagement Depletes Intra-tumoral Tregs via Activating FcγRs, Leading to Antitumor Efficacy. Immunol. Cell Biol. 2014, 92, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Zahavi, D.; AlDeghaither, D.; O’Connell, A.; Weiner, L.M. Enhancing Antibody-Dependent Cell-Mediated Cytotoxicity: A Strategy for Improving Antibody-Based Immunotherapy. Antib. Ther. 2018, 1, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Rosskopf, S.; Leitner, J.; Zlabinger, G.J.; Steinberger, P. CTLA-4 Antibody Ipilimumab Negatively Affects CD4+ T-Cell Responses in Vitro. Cancer Immunol. Immunother. 2019, 68, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-Dependent Cell-Mediated Cytotoxicity of Regulatory T Cells Ex Vivo by Nonclassical Monocytes in Melanoma Patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-Dependent Depletion of Tumor-Infiltrating Regulatory T Cells Co-Defines the Efficacy of Anti–CTLA-4 Therapy against Melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of Mouse and Human IgG Receptors and Their Contribution to Disease Models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti–CTLA-4 Activates Intra-tumoral NK Cells and Combined with IL15/IL15Rα Complexes Enhances Tumor Control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis-Marcisak, E.F.; Fitzgerald, A.A.; Kessler, M.D.; Danilova, L.; Jaffee, E.M.; Zaidi, N.; Weiner, L.M.; Fertig, E.J. A Novel Mechanism of Natural Killer Cell Response to Anti-CTLA-4 Therapy Identified by Integrative Analysis of Mouse and Human Tumors. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, A.; Ren, Z.; Tseng, K.-F.; Liu, X.; Li, H.; Lu, C.; Cai, Y.; Minna, J.D.; Fu, Y.-X. Dual Targeting of CTLA-4 and CD47 on Treg Cells Promotes Immunity against Solid Tumors. Sci. Transl. Med. 2021, 13, eabg8693:1–eabg8693:15. [Google Scholar] [CrossRef]
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Gall, C.L.; Crowley, S.; et al. Anti–CTLA-4 Therapy Requires an Fc Domain for Efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Comin-Anduix, B.; Economou, J.S.; Donahue, T.R.; de la Rocha, P.; Morris, L.F.; Jalil, J.; Dissette, V.B.; Shintaku, I.P.; Glaspy, J.A.; et al. Intra-tumoral Immune Cell Infiltrates, FoxP3, and Indoleamine 2,3-Dioxygenase in Patients with Melanoma Undergoing CTLA4 Blockade. Clin. Cancer Res. 2009, 15, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, B.; O’Brien, S.; Lee, D.; Hou, Y.; Weinberg, V.; Rini, B.; Allison, J.P.; Small, E.J.; Fong, L. CTLA4 Blockade Expands FoxP3+ Regulatory and Activated Effector CD4+ T Cells in a Dose-Dependent Fashion. Blood 2008, 112, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Maker, A.V.; Attia, P.; Rosenberg, S.A. Analysis of the Cellular Mechanism of Antitumor Responses and Autoimmunity in Patients Treated with CTLA-4 Blockade. J. Immunol. 2005, 175, 7746–7754. [Google Scholar] [CrossRef] [Green Version]
- Tarhini, A.A.; Edington, H.; Butterfield, L.H.; Lin, Y.; Shuai, Y.; Tawbi, H.; Sander, C.; Yin, Y.; Holzman, M.; Johnson, J.; et al. Immune Monitoring of the Circulation and the Tumor Microenvironment in Patients with Regionally Advanced Melanoma Receiving Neoadjuvant Ipilimumab. PLoS ONE 2014, 9, e87705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoni, F.; Zhakyp, A.; Corsini, M.; Geels, S.N.; Carrizosa, E.; Thelen, M.; Mani, V.; Prüßmann, J.N.; Warner, R.D.; Ozga, A.J.; et al. Expansion of Tumor-Associated Treg Cells upon Disruption of a CTLA-4-Dependent Feedback Loop. Cell 2021, 184, 3998–4015.e19. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Wang, C.J.; Ryan, G.A.; Clough, L.E.; Qureshi, O.S.; Goodall, M.; Abbas, A.K.; Sharpe, A.H.; Sansom, D.M.; Walker, L.S.K. CTLA-4 Controls Regulatory T Cell Peripheral Homeostasis and Is Required for Suppression of Pancreatic Islet Autoimmunity. J. Immunol. 2009, 182, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, A.; Nguyen, L.T.; Stone, S.C.; Yang, S.Y.C.; Katz, S.R.; Shaw, P.A.; Clarke, B.A.; Ghazarian, D.; Al-Habeeb, A.; Easson, A.; et al. Regulatory T Cells in Ovarian Cancer Are Characterized by a Highly Activated Phenotype Distinct from That in Melanoma. Clin. Cancer Res. 2018, 24, 5685–5696. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; Cronin, J.G.; Dolton, G.; Panetti, S.; Schauenburg, A.J.; Galloway, S.A.E.; Sewell, A.K.; Cole, D.K.; Thornton, C.A.; Francis, N.J. Metabolic Adaptation of Human CD4+ and CD8+ T-Cells to T-Cell Receptor-Mediated Stimulation. Front. Immunol. 2017, 8, 1516:1–1516:12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Chi, H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front. Immunol. 2019, 10, 2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 Blockade Drives Loss of T Reg Stability in Glycolysis-Low Tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Howie, D.; Cobbold, S.P.; Adams, E.; Bokum, A.T.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 Drives Oxidative Phosphorylation and Protection from Lipotoxicity. JCI Insight 2017, 2, e89160:1–e89160:18. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like Receptor Signaling Balance T Reg Cell Anabolic Metabolism for Suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential Role of Mitochondrial Energy Metabolism in Foxp3+ T-Regulatory Cell Function and Allograft Survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Valmori, D.; Raffin, C.; Raimbaud, I.; Ayyoub, M. Human RORγt+ TH17 Cells Preferentially Differentiate from Naive FOXP3+Treg in the Presence of Lineage-Specific Polarizing Factors. Proc. Natl. Acad. Sci. USA 2010, 107, 19402–19407. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic Modeling of Single Th17 Cells Reveals Regulators of Autoimmunity. Cell 2021, 184, 4168–4185.e21. [Google Scholar] [CrossRef]
- Koenen, H.J.P.M.; Smeets, R.L.; Vink, P.M.; van Rijssen, E.; Boots, A.M.H.; Joosten, I. Human CD25highFoxp3pos Regulatory T Cells Differentiate into IL-17–Producing Cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Wu, K.; Zhao, E.; Wei, S.; Vatan, L.; Szeliga, W.; Huang, E.; Greenson, J.; Chang, A.; Rolinski, J.; et al. IL-17+ Regulatory T Cells in the Microenvironments of Chronic Inflammation and Cancer. J. Immunol. 2011, 186, 4388–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Yang, L.; Qiao, G.; Li, Z.; Zhang, L.; Yin, F.; Xie, D.; Zhang, J. Cutting Edge: CTLA-4–B7 Interaction Suppresses Th17 Cell Differentiation. J. Immunol. 2010, 185, 1375–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Ye, J.; Hsueh, E.C.; Zhang, Y.; Hoft, D.F.; Peng, G. Tumor Microenvironments Direct the Recruitment and Expansion of Human Th17 Cells. J. Immunol. 2010, 184, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, Distribution, Generation, and Functional and Clinical Relevance of Th17 Cells in the Human Tumor Environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Qi, Y.; Hu, J.; Tang, L.; Zhao, S.; Shan, B. Expression of Th17 Cells in Breast Cancer Tissue and Its Association with Clinical Parameters. Cell Biochem. Biophys. 2012, 62, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, W.; Lian, J.; Wu, Q.; Liu, S.; Yang, L.; Li, F.; Huang, L.; Chen, X.; Zhang, Z.; et al. Th17 Cells Inhibit CD8+ T Cell Migration by Systematically Downregulating CXCR3 Expression via IL-17A/STAT3 in Advanced-Stage Colorectal Cancer Patients. J. Hematol. Oncol.J Hematol Oncol 2020, 13, 68. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef]
- Ye, Z.-J.; Zhou, Q.; Gu, Y.-Y.; Qin, S.-M.; Ma, W.-L.; Xin, J.-B.; Tao, X.-N.; Shi, H.-Z. Generation and Differentiation of IL-17–Producing CD4+ T Cells in Malignant Pleural Effusion. J. Immunol. 2010, 185, 6348–6354. [Google Scholar] [CrossRef] [Green Version]
- Lida, T.; Iwahashi, M.; Katsuda, M.; Ishida, K.; Nakamori, M.; Nakamura, M.; Naka, T.; Ojima, T.; Ueda, K.; Hayata, K.; et al. Tumor-Infiltrating CD4+ Th17 Cells Produce IL-17 in Tumor Microenvironment and Promote Tumor Progression in Human Gastric Cancer. Oncol. Rep. 2011, 25, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 Enhances BFGF-, HGF- and VEGF-Induced Growth of Vascular Endothelial Cells. Immunol. Lett. 2005, 98, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijik, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4–Ig Regulates Tryptophan Catabolism in Vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.-L.; Puccetti, P. Modulation of Tryptophan Catabolism by Regulatory T Cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO Activates Regulatory T Cells and Blocks Their Conversion into Th17-Like T Cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- von Euw, E.; Chodon, T.; Attar, N.; Jalil, J.; Koya, R.C.; Comin-Anduix, B.; Ribas, A. CTLA4 Blockade Increases Th17 Cells in Patients with Metastatic Melanoma. J. Transl. Med. 2009, 7, 35:1–35:13. [Google Scholar] [CrossRef] [Green Version]
- Tarhini, A.A.; Zahoor, H.; Lin, Y.; Malhotra, U.; Sander, C.; Butterfield, L.H.; Kirkwood, J.M. Baseline Circulating IL-17 Predicts Toxicity While TGF-Β1 and IL-10 Are Prognostic of Relapse in Ipilimumab Neoadjuvant Therapy of Melanoma. J. Immunother. Cancer 2015, 3, 39:1–39:6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-Specific Th17-Polarized Cells Eradicate Large Established Melanoma. Blood 2008, 112, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Olsen, D.J.; Eroglu, Z.; Brockstein, B.; Poklepovic, A.S.; Bajaj, M.; Babu, S.; Hallmeyer, S.; Velasco, M.; Lutzky, J.; Higgs, E.; et al. Pembrolizumab Plus Ipilimumab Following Anti-PD-1/L1 Failure in Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 2647–2655. [Google Scholar] [CrossRef]
- Chen, J.; Li, S.; Yao, Q.; Du, N.; Fu, X.; Lou, Y.; Wang, M.; Mao, F.; Mao, D.; Khadaroo, P.A.; et al. The Efficacy and Safety of Combined Immune Checkpoint Inhibitors (Nivolumab plus Ipilimumab): A Systematic Review and Meta-Analysis. World J. Surg. Oncol. 2020, 18, 150:1–150:11. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Caro, R.B.; Zurawski, B.; Kim, S.-W.; Costa, E.C.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-Line Nivolumab plus Ipilimumab Combined with Two Cycles of Chemotherapy in Patients with Non-Small-Cell Lung Cancer (CheckMate 9LA): An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Lond. Engl. 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination Anti-CTLA-4 plus Anti-PD-1 Checkpoint Blockade Utilizes Cellular Mechanisms Partially Distinct from Monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V.; et al. Combination Therapy with Anti-CTLA4 and Anti-PD1 Leads to Distinct Immunologic Changes in-Vivo. J. Immunol. 2015, 194, 950–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus Adjuvant Ipilimumab plus Nivolumab in Macroscopic Stage III Melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; William, W.N.; Weissferdt, A.; Leung, C.H.; Lin, H.Y.; Pataer, A.; Godoy, M.C.B.; Carter, B.W.; Federico, L.; Reuben, A.; et al. Neoadjuvant Nivolumab or Nivolumab plus Ipilimumab in Operable Non-Small Cell Lung Cancer: The Phase 2 Randomized NEOSTAR Trial. Nat. Med. 2021, 27, 504–514. [Google Scholar] [CrossRef]
- Gao, J.; Navai, N.; Alhalabi, O.; Siefker-Radtke, A.; Campbell, M.T.; Tidwell, R.S.; Guo, C.C.; Kamat, A.M.; Matin, S.F.; Araujo, J.C.; et al. Neoadjuvant PD-L1 plus CTLA-4 Blockade in Patients with Cisplatin-Ineligible Operable High-Risk Urothelial Carcinoma. Nat. Med. 2020, 26, 1845–1851. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Synder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Scholling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A Prospective Phase II Trial Exploring the Association between Tumor Microenvironment Biomarkers and Clinical Activity of Ipilimumab in Advanced Melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Semmrich, M.; Marchand, J.-B.; Fend, L.; Rehn, M.; Remy, C.; Holmkvist, P.; Silvestre, N.; Svensson, C.; Kleinpeter, P.; Deforges, J.; et al. Vectorized Treg-Depleting ACTLA-4 Elicits Antigen Cross-Presentation and CD8+ T Cell Immunity to Reject ‘Cold’ Tumors. J. Immunother. Cancer 2022, 10, e003488. [Google Scholar] [CrossRef]
- Lussier, D.M.; Alspach, E.; Ward, J.P.; Miceli, A.P.; Runci, D.; White, J.M.; Mpoy, C.; Arthur, C.D.; Kohlmiller, H.N.; Jacks, T.; et al. From the Cover: Radiation-Induced Neoantigens Broaden the Immunotherapeutic Window of Cancers with Low Mutational Loads. Proc. Natl. Acad. Sci. USA 2021, 118, e2102611118. [Google Scholar] [CrossRef]
- Krombach, J.; Hennel, R.; Brix, N.; Orth, M.; Schoetz, U.; Ernst, A.; Schuster, J.; Zuchtriegel, G.; Reichel, C.A.; Bierschenk, S.; et al. Priming Anti-Tumor Immunity by Radiotherapy: Dying Tumor Cell-Derived DAMPs Trigger Endothelial Cell Activation and Recruitment of Myeloid Cells. Oncoimmunology 2019, 8, e1523097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudqvist, N.-P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.P.; van der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti–CTLA-4 Therapy Broadens the Melanoma-Reactive CD8+ T Cell Response. Sci. Transl. Med. 2014, 6, 254ra128:1–254ra128:15. [Google Scholar] [CrossRef] [PubMed]
- Pilones, K.A.; Golden, E.B.; Kraynak, J.; Formenti, S.C. Intra-tumoral Anti-CTLA-4 Blockade Can Potentiate Radiation-Induced Anti-Tumor Immunity In Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e537. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-Mediated Inhibition of Metastases after Treatment with Local Radiation and CTLA-4 Blockade in a Mouse Model of Breast Cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III Randomized Clinical Trial Comparing Tremelimumab With Standard-of-Care Chemotherapy in Patients With Advanced Melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Bondarenko, I.; Garbe, C.; Testori, A.; Richards, J.; Miller, W.H.; Harmankaya, K.; Chen, T.-T.; Wolchok, J.D. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, Z.; Kim, D.W.; Wang, X.; Camacho, L.H.; Chmielowski, B.; Seja, E.; Villanueva, A.; Ruchalski, K.; Glaspy, J.A.; Kim, K.B.; et al. Long Term Survival with CTLA-4 Blockade Using Tremelimumab. Eur. J. Cancer Oxf. Engl. 1990 2015, 51, 2689–2697. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Camacho, L.H.; Lopez-Berestein, G.; Pavlov, D.; Bulanhagui, C.A.; Millham, R.; Comin-Anduix, B.; Reuben, J.M.; Seja, E.; Parker, C.A.; et al. Antitumor Activity in Melanoma and Anti-Self Responses in a Phase I Trial With the Anti-Cytotoxic T Lymphocyte–Associated Antigen 4 Monoclonal Antibody CP-675,206. J. Clin. Oncol. 2005, 23, 8968–8977. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, Y.-P.; Du, X.-J.; Liu, J.-Q.; Huang, C.-L.; Chen, L.; Zhou, G.-Q.; Li, W.-F.; Mao, Y.-P.; Hsu, C.; et al. Comparative Safety of Immune Checkpoint Inhibitors in Cancer: Systematic Review and Network Meta-Analysis. BMJ 2018, 363, k4226. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, N.; van Elsas, A.; Verbeek, J.S.; Zaiss, D.M.W. Isotype Selection for Antibody-Based Cancer Therapy. Clin. Exp. Immunol. 2021, 203, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, D.W.-M.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Bar, J.; Rasco, D.W.; Ahn, M.; Yoh, K.; Kim, D.; Nagrial, A.; Satouchi, M.; Lee, D.H.; Spigel, D.R.; et al. Safety and Efficacy of Quavonlimab, a Novel Anti-CTLA-4 Antibody (MK-1308), in Combination with Pembrolizumab in First-Line Advanced Non-Small-Cell Lung Cancer. Ann. Oncol. 2020, 32, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Liu, M.; Su, J.; Zhang, P.; Tang, F.; Ye, P.; Devenport, M.; Wang, X.; Zhang, Y.; Liu, Y.; et al. Uncoupling Therapeutic from Immunotherapy-Related Adverse Effects for Safer and Effective Anti-CTLA-4 Antibodies in CTLA4 Humanized Mice. Cell Res. 2018, 28, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valzasina, B.; Guiducci, C.; Dislich, H.; Killeen, N.; Weinberg, A.D.; Colombo, M.P. Triggering of OX40 (CD134) on CD4+CD25+ T Cells Blocks Their Inhibitory Activity: A Novel Regulatory Role for OX40 and Its Comparison with GITR. Blood 2005, 105, 2845–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, M.D.; Xiao, X.; Gao, W.; Degauque, N.; Chen, M.; Kroemer, A.; Killeen, N.; Ishii, N.; Chang Li, X. OX40 Costimulation Turns off Foxp3+ Tregs. Blood 2007, 110, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Piconese, S.; Valzasina, B.; Colombo, M.P. OX40 Triggering Blocks Suppression by Regulatory T Cells and Facilitates Tumor Rejection. J. Exp. Med. 2008, 205, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschhorn-Cymerman, D.; Rizzuto, G.A.; Merghoub, T.; Cohen, A.D.; Avogadri, F.; Lesokhin, A.M.; Weinberg, A.D.; Wolchok, J.D.; Houghton, A.N. OX40 Engagement and Chemotherapy Combination Provides Potent Antitumor Immunity with Concomitant Regulatory T Cell Apoptosis. J. Exp. Med. 2009, 206, 1103–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumi, K.; Miyakawa, R.; Shibasaki, C.; Henmi, M.; Mizoguchi, Y.; Ueda, R.; Hashimoto, H.; Hiraoka, N.; Yoshida, T.; Aoki, K. Local Administration of GITR Agonistic Antibody Induces a Stronger Antitumor Immunity than Systemic Delivery. Sci. Rep. 2019, 9, 5562. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Schaer, D.A.; Liu, C.; Li, Y.; Hirschhorn-Cymmerman, D.; Kim, S.C.; Diab, A.; Rizzuto, G.; Duan, F.; Perales, M.A.; et al. Agonist Anti-GITR Monoclonal Antibody Induces Melanoma Tumor Immunity in Mice by Altering Regulatory T Cell Stability and Intra-Tumor Accumulation. PLoS ONE 2010, 5, e10436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, J.; Nishikawa, H.; Muraoka, D.; Wang, L.; Noguchi, T.; Sato, E.; Kondo, S.; Allison, J.P.; Sakaguchi, S.; Old, L.J.; et al. Two Distinct Mechanisms of Augmented Antitumor Activity by Modulation of Immunostimulatory/Inhibitory Signals. Clin. Cancer Res. 2010, 16, 2781–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvarnhammar, A.M.; Veitonmäki, N.; Hägerbrand, K.; Dahlman, A.; Smith, K.E.; Fritzell, S.; von Schantz, L.; Thagesson, M.; Werchau, D.; Smedenfors, K.; et al. The CTLA-4 x OX40 Bispecific Antibody ATOR-1015 Induces Anti-Tumor Effects through Tumor-Directed Immune Activation. J. Immunother. Cancer 2019, 7, 103:1–103:14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmond, W.L.; Linch, S.N.; Kasiewicz, M.J. Combined Targeting of Co-Stimulatory (OX40) and Co-Inhibitory (CTLA-4) Pathways Elicits Potent Effector T Cells Capable of Driving Robust Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutmuller, R.P.; van Duivenvoorde, L.M.; van Elsas, A.; Schumacher, T.N.; Wildenberg, M.E.; Allison, J.P.; Toes, R.E.; Offringa, R.; Melief, C.J. Synergism of Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade and Depletion of CD25(+) Regulatory T Cells in Antitumor Therapy Reveals Alternative Pathways for Suppression of Autoreactive Cytotoxic T Lymphocyte Responses. J. Exp. Med. 2001, 194, 823–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Du, X.; Liu, M.; Tang, F.; Zhang, P.; Ai, C.; Fields, J.K.; Sundberg, E.J.; Latinovic, O.S.; Devenport, M.; et al. Hijacking Antibody-Induced CTLA-4 Lysosomal Degradation for Safer and More Effective Cancer Immunotherapy. Cell Res. 2019, 29, 609–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | Isotype | |
|---|---|---|
| Human | Ipilimumab | Human IgG1 |
| Tremelimumab | Human IgG2 | |
| Mouse | 9H10 | Syrian Hamster IgG |
| 9D9 | Mouse IgG2b | |
| 4F10 | Armenian Hamster IgG |
| Trial | Cancer | Treatments | Overall Response Rate (%) 1 | Overall Survival Rate (%) 2 | |
|---|---|---|---|---|---|
| NCT00094653 [2] | Unresectable or metastatic melanoma | gp100 (n = 136) | 1.5 | 13.7 (2-yr) | |
| Ipilimumab (3 mg/kg) (n = 137) | 10.9 | 23.5 | |||
| Ipilimumab (3 mg/kg) + gp100 (n = 403) | 5.7 | 21.6 | |||
| NCT01844505 [114] | Unresectable or metastatic melanoma (BRAF V600—wildtype or mutant) | BRAF-wildtype | BRAF-mutant | ||
| Nivolumab (3 mg/kg) (n-316) | 45 | 22 (6.5-yr) | 25 (6.5-yr) | ||
| Ipilimumab (3 mg/kg) (n = 315) | 19 | 42 | 43 | ||
| Ipilimumab (3 mg/kg) + nivolumab (3 mg/kg) (n = 314) | 58 | 46 | 57 | ||
| NCT00636168 [3] | Stage III melanoma—adjuvant therapy | Placebo (n = 476) | N/A | 54.4 (5-yr) | |
| Ipilimumab (10 mg/kg) (n = 475) | 65.4 | ||||
| NCT02231749 [115] | Advanced renal cell carcinoma | Sunitinib (n = 422) | 26.5 | 60 (1.5-yr) | |
| Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 425) | 41.6 | 75 | |||
| NCT02231749 [116] | MSI-H/dMMR metastatic colorectal cancer | Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 119) | 55 | 85 (1-yr) | |
| NCT01658878 [117] | Hepatocellular carcinoma | Nivolumab (1 mg/kg) + ipilimumab (3 mg/kg) (n = 50) | 32 | 48 (2-yr) | |
| Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 49) | 31 | 30 | |||
| Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 49) | 31 | 42 | |||
| NCT02477826 [118] | Metastatic NSCLC (≥1% PD-L1) | Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 396) | 35.9 | 40 (2-yr) | |
| Platinum-doublet chemotherapy (n = 397) | 30 | 32.8 | |||
| NCT03215706 [119] | Metastatic or recurrent NSCLC | Nivolumab (360 mg) + ipilimumab (1 mg/kg) + platinum-doublet chemotherapy (n = 361) | 38 | 63 (1-yr) | |
| Platinum-doublet chemotherapy (n = 358) | 25 | 47 | |||
| NCT02899299 [120] | Unresectable malignant pleural mesothelioma | Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) (n = 303) | 40 | 41 (2-yr) | |
| Platinum + pemetrexed chemotherapy (n = 302) | 43 | 27 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, M.M.Y.; Maleki Vareki, S. Addressing the Elephant in the Immunotherapy Room: Effector T-Cell Priming versus Depletion of Regulatory T-Cells by Anti-CTLA-4 Therapy. Cancers 2022, 14, 1580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061580
Hong MMY, Maleki Vareki S. Addressing the Elephant in the Immunotherapy Room: Effector T-Cell Priming versus Depletion of Regulatory T-Cells by Anti-CTLA-4 Therapy. Cancers. 2022; 14(6):1580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061580
Chicago/Turabian StyleHong, Megan M Y, and Saman Maleki Vareki. 2022. "Addressing the Elephant in the Immunotherapy Room: Effector T-Cell Priming versus Depletion of Regulatory T-Cells by Anti-CTLA-4 Therapy" Cancers 14, no. 6: 1580. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061580