
APOBEC SBS13 Mutational Signature—A Novel Predictor of Radioactive Iodine Refractory Papillary Thyroid Carcinoma

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Cohort
2.2. Imaging Analysis
2.3. Definition of Radioactive Iodine Refractoriness
- 131I uptake absent on diagnostic 131I scan of locoregional recurrence or distant metastasis.
- 131I uptake absent on a 131I scan performed several days after 131I treatment.
- 131I uptake present in some, but not all tumor foci.
- Disease progression despite a cumulative 131I activity of ≥600 mCi.
- Metastatic disease progression despite 131I uptake.
- Rising serum thyroglobulin levels ≥6 months after 131I treatment.
- Structural disease progression after 131I treatment.
2.4. Sample Processing
2.5. Whole-Exome Sequencing
2.6. Mutation Calling
2.7. Gene Copy Number Profiling
2.8. Telomerase Reverse Transcriptase Promoter Mutations
2.9. Driver Events
2.10. Mutational Signature Analysis
2.11. Statistical Analysis
3. Results
3.1. Clinicopathological Associations
3.2. Genomic Landscape
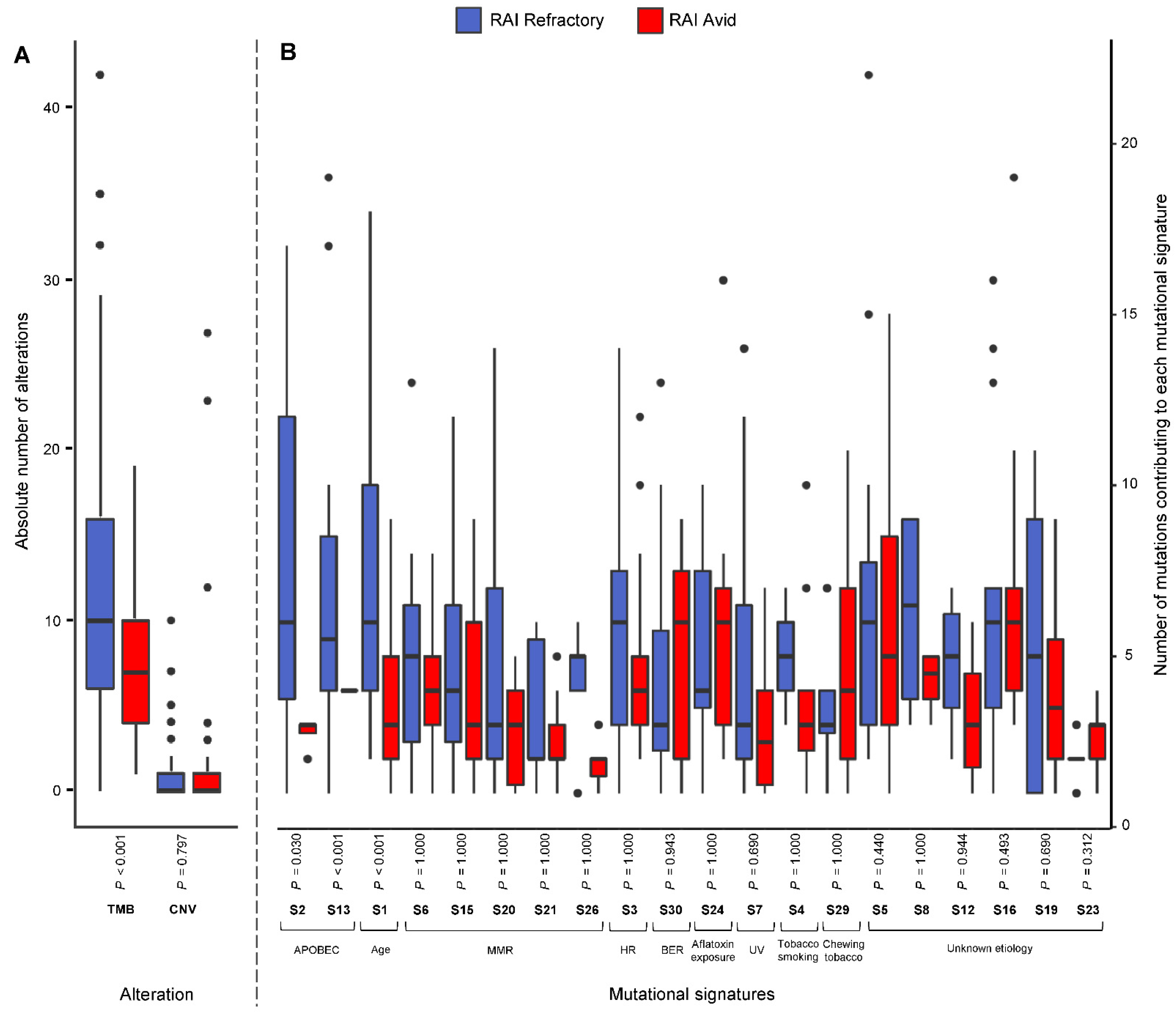
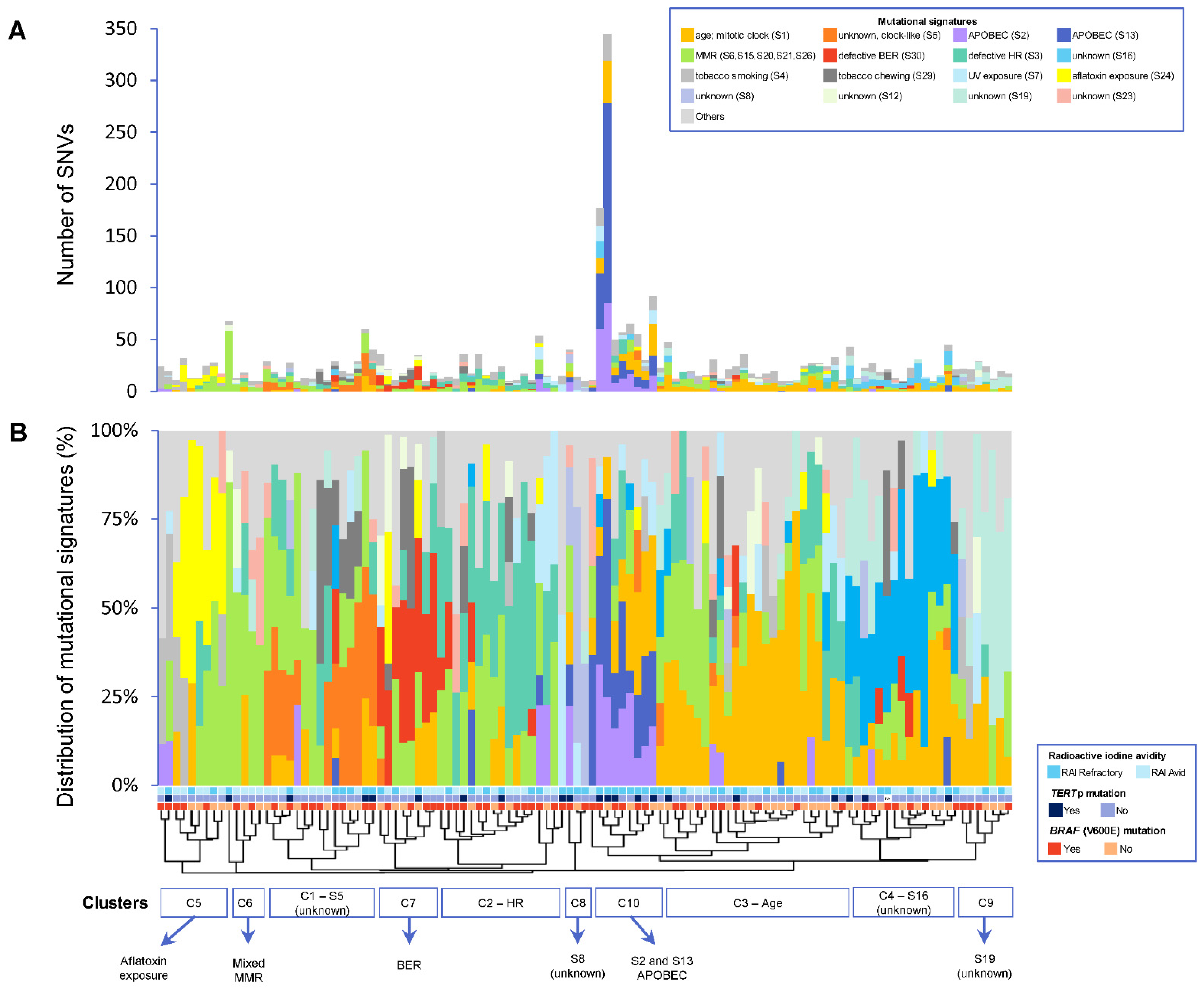
3.3. Frequency of Mutational Signatures
3.4. Patterns of Mutational Signatures
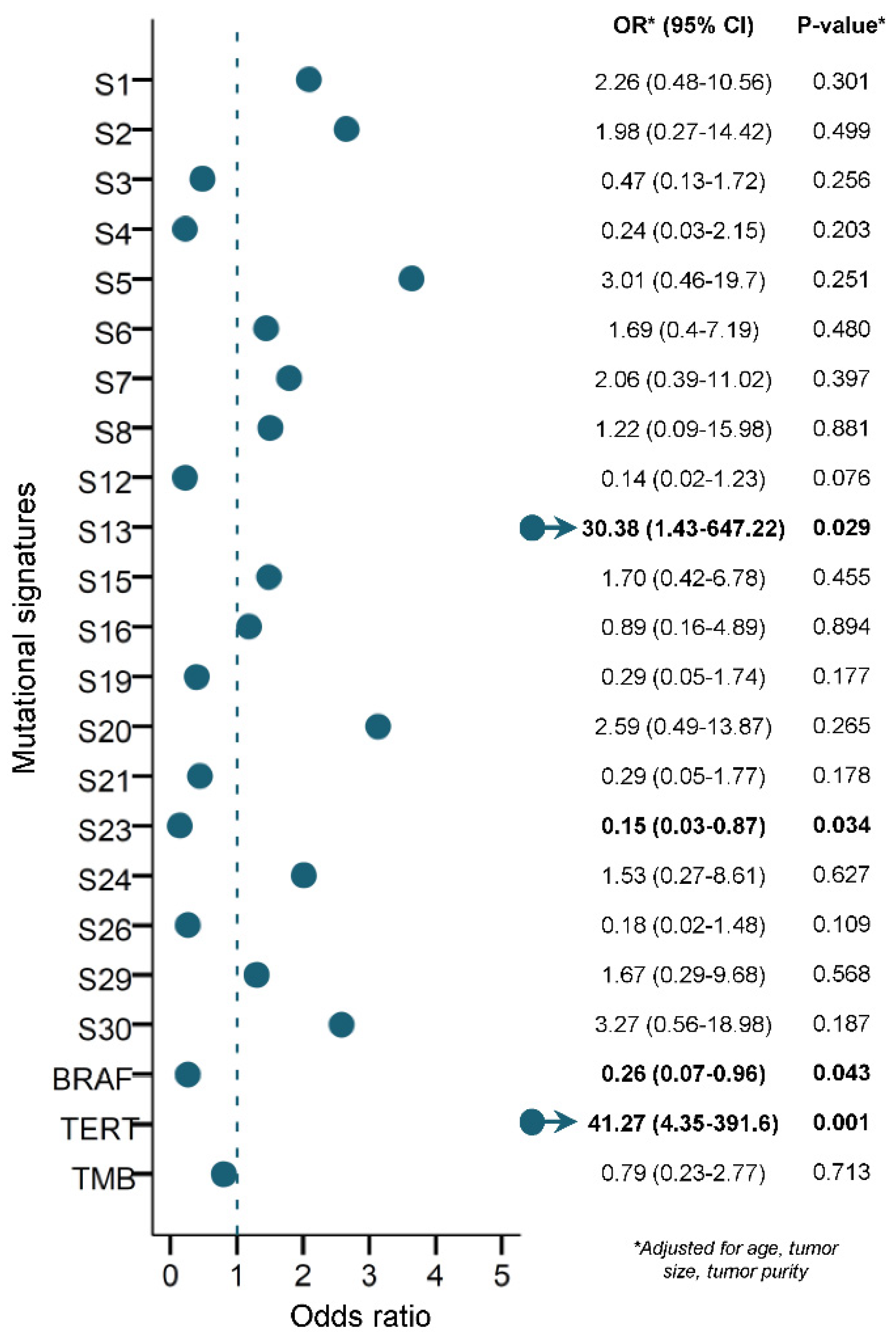
3.5. Prediction of RAI Refractoriness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiltshire, J.J.; Drake, T.M.; Uttley, L.; Balasubramanian, S.P. Systematic review of trends in the incidence rates of thyroid cancer. Thyroid 2016, 26, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Saudi Cancer Registry. Cancer Incidence Report. Saudi Arabia. 2015. Available online: https://nhic.gov.sa/eServices/Documents/ESCRfinal6NOV.pdf (accessed on 4 September 2020).
- Al-Zaher, N.; Al-Salam, S.; El Teraifi, H. Thyroid carcinoma in the United Arab Emirates: Perspectives and experience of a tertiary care hospital. Hematol. Oncol. Stem Cell Ther. 2008, 1, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.S. Recurrence of papillary thyroid cancer after optimized surgery. Gland. Surg. 2015, 4, 52. [Google Scholar]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Worden, F. Treatment strategies for radioactive iodine-refractory differentiated thyroid cancer. Ther. Adv. Med. Oncol. 2014, 6, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Haugen, B.R.; Schlumberger, M. Progress in molecular-based management of differentiated thyroid cancer. Lancet 2013, 381, 1058–1069. [Google Scholar] [CrossRef] [Green Version]
- Sciuto, R.; Romano, L.; Rea, S.; Marandino, F.; Sperduti, I.; Maini, C.L. Natural history and clinical outcome of differentiated thyroid carcinoma: A retrospective analysis of 1503 patients treated at a single institution. Ann. Oncol. 2009, 20, 1728–1735. [Google Scholar] [CrossRef]
- Nixon, I.J.; Whitcher, M.M.; Palmer, F.L.; Tuttle, R.M.; Shaha, A.R.; Shah, J.P.; Patel, S.G.; Ganly, I. The impact of distant metastases at presentation on prognosis in patients with differentiated carcinoma of the thyroid gland. Thyroid 2012, 22, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: Benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef]
- Faugeras, L.; Pirson, A.S.; Donckier, J.; Michel, L.; Lemaire, J.; Vandervorst, S.; D’Hondt, L. Refractory thyroid carcinoma: Which systemic treatment to use? Ther. Adv. Med. Oncol. 2018, 10, 1758834017752853. [Google Scholar] [CrossRef]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019, 11, 1382. [Google Scholar] [CrossRef] [Green Version]
- Temko, D.; Tomlinson, I.P.M.; Severini, S.; Schuster-Bockler, B.; Graham, T.A. The effects of mutational processes and selection on driver mutations across cancer types. Nat. Commun. 2018, 9, 1857. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Illingworth, C.J.; Campbell, P.J.; Mustonen, V. EMu: Probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol. 2013, 14, R39. [Google Scholar] [CrossRef] [Green Version]
- Gehring, J.S.; Fischer, B.; Lawrence, M.; Huber, W. SomaticSignatures: Inferring mutational signatures from single-nucleotide variants. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Tremmel, G.; Miyano, S.; Stephens, M. A Simple Model-Based Approach to Inferring and Visualizing Cancer Mutation Signatures. PLoS Genet. 2015, 11, e1005657. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Masoodi, T.; Siraj, A.K.; Siraj, S.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Al-Sobhi, S.S.; AlDawish, M.; Alkuraya, F.S.; Al-Kuraya, K.S. Evolution and Impact of Subclonal Mutations in Papillary Thyroid Cancer. Am. J. Hum. Genet. 2019, 105, 959–973. [Google Scholar] [CrossRef]
- Masoodi, T.; Siraj, A.K.; Siraj, S.; Azam, S.; Qadri, Z.; Albalawy, W.N.; Parvathareddy, S.K.; Al-Sobhi, S.S.; Al-Dayel, F.; Alkuraya, F.S.; et al. Whole-Exome Sequencing of Matched Primary and Metastatic Papillary Thyroid Cancer. Thyroid 2020, 30, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.W.; Gout, A.M.; Zhang, J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2022, 38, 194–208. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef] [PubMed]
- Hurst, Z.; Liyanarachchi, S.; He, H.; Brock, P.; Sipos, J.; Nabhan, F.; Kebebew, E.; Green, P.; Cote, G.J.; Sherman, S.; et al. Risk Haplotypes Uniquely Associated with Radioiodine-Refractory Thyroid Cancer Patients of High African Ancestry. Thyroid 2019, 29, 530–539. [Google Scholar] [CrossRef]
- Tuttle, R.M.; Ahuja, S.; Avram, A.M.; Bernet, V.J.; Bourguet, P.; Daniels, G.H.; Dillehay, G.; Draganescu, C.; Flux, G.; Fuhrer, D.; et al. Controversies, Consensus, and Collaboration in the Use of (131)I Therapy in Differentiated Thyroid Cancer: A Joint Statement from the American Thyroid Association, the European Association of Nuclear Medicine, the Society of Nuclear Medicine and Molecular Imaging, and the European Thyroid Association. Thyroid 2019, 29, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Gulec, S.A.; Ahuja, S.; Avram, A.M.; Bernet, V.J.; Bourguet, P.; Draganescu, C.; Elisei, R.; Giovanella, L.; Grant, F.; Greenspan, B.; et al. A Joint Statement from the American Thyroid Association, the European Association of Nuclear Medicine, the European Thyroid Association, the Society of Nuclear Medicine and Molecular Imaging on Current Diagnostic and Theranostic Approaches in the Management of Thyroid Cancer. Thyroid 2021, 31, 1009–1019. [Google Scholar] [CrossRef]
- Siraj, A.K.; Masoodi, T.; Bu, R.; Beg, S.; Al-Sobhi, S.S.; Al-Dayel, F.; Al-Dawish, M.; Alkuraya, F.S.; Al-Kuraya, K.S. Genomic Profiling of Thyroid Cancer Reveals a Role for Thyroglobulin in Metastasis. Am. J. Hum. Genet. 2016, 98, 1170–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Bu, R.; Siraj, A.K.; Divya, S.P.; Kong, Y.; Parvathareddy, S.K.; Al-Rasheed, M.; Al-Obaisi, K.A.S.; Victoria, I.G.; Al-Sobhi, S.S.; Al-Dawish, M.; et al. Telomerase reverse transcriptase mutations are independent predictor of disease-free survival in Middle Eastern papillary thyroid cancer. Int. J. Cancer 2018, 142, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Z.; Chen, T.; Zeng, W.; Guo, Y.; Huang, T. TERT promoter Mutation and Its Association with Clinicopathological Features and Prognosis of Papillary Thyroid Cancer: A Meta-analysis. Sci. Rep. 2016, 6, 36990. [Google Scholar] [CrossRef]
- McKelvey, B.A.; Umbricht, C.B.; Zeiger, M.A. Telomerase Reverse Transcriptase (TERT) Regulation in Thyroid Cancer: A Review. Front. Endocrinol. 2020, 11, 485. [Google Scholar] [CrossRef]
- Donati, B.; Ciarrocchi, A. Telomerase and Telomeres Biology in Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 2887. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, R.; Shen, X.; Zhu, G.; Li, B.; Xing, M. The Genetic Duet of BRAF V600E and TERT Promoter Mutations Robustly Predicts Loss of Radioiodine Avidity in Recurrent Papillary Thyroid Cancer. J. Nucl. Med. 2020, 61, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shobab, L.; Gomes-Lima, C.; Zeymo, A.; Feldman, R.; Jonklaas, J.; Wartofsky, L.; Burman, K.D. Clinical, Pathological, and Molecular Profiling of Radioactive Iodine Refractory Differentiated Thyroid Cancer. Thyroid 2019, 29, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.J.; Cho, S.W.; Won, J.K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Matsuse, M.; Saenko, V.; Yamashita, S.; Ren, P.; Zheng, X.; Jia, Q.; Tan, J.; Li, N.; Zheng, W.; et al. TERT promoter mutation in primary papillary thyroid carcinoma lesions predicts absent or lower (131) i uptake in metastases. IUBMB Life 2019, 71, 1030–1040. [Google Scholar] [CrossRef]
- Yang, X.; Li, J.; Li, X.; Liang, Z.; Gao, W.; Liang, J.; Cheng, S.; Lin, Y. TERT Promoter Mutation Predicts Radioiodine-Refractory Character in Distant Metastatic Differentiated Thyroid Cancer. J. Nucl. Med. 2017, 58, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Rodríguez, I.; De la Vieja, A.; Costamagna, E.; Carrasco, N.; Nistal, M.; Santisteban, P. The BRAFV600E oncogene induces transforming growth factor β secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009, 69, 8317–8325. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Shen, W.-z.; Yan, Y.-j.; Yi, J.-l.; Zhang, L. Effects of BRAFV600E mutation on Na+/I-symporter expression in papillary thyroid carcinoma. J. Huazhong Univ. Sci. Technol. Med. Sci. 2016, 36, 77–81. [Google Scholar] [CrossRef]
- Anekpuritanang, T.; Uataya, M.; Claimon, A.; Laokulrath, N.; Pongsapich, W.; Pithuksurachai, P. The Association Between Radioiodine Refractory in Papillary Thyroid Carcinoma, Sodium/Iodide Symporter Expression, and BRAFV600E Mutation. OncoTargets Ther. 2021, 14, 3959. [Google Scholar] [CrossRef]
- Zoghlami, A.; Roussel, F.; Sabourin, J.-C.; Kuhn, J.-M.; Marie, J.-P.; Dehesdin, D.; Choussy, O. BRAF mutation in papillary thyroid carcinoma: Predictive value for long-term prognosis and radioiodine sensitivity. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2014, 131, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef] [Green Version]
- von Morgen, P.; Maciejowski, J. The ins and outs of telomere crisis in cancer. Genome Med. 2018, 10, 89. [Google Scholar] [CrossRef]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Variables | RAI Refractory n (%) | RAI Avid n (%) | Odds Ratio (95% CI 1) | Multivariable p-Value |
|---|---|---|---|---|
| Age | 0.327 | |||
| Median | 44 | 39 | 1.0 (0.9–1.0) | |
| Range | 20–83 | 20–75 | ||
| Sex | 0.483 | |||
| Male | 18 (27.3) | 21 (22.8) | 0.7 (0.2–2.1) | |
| Female | 48 (72.7) | 71 (77.2) | ||
| Histopathological subtype | ||||
| Classical variant | 33 (55.9) | 45 (57.7) | 1 | |
| Follicular variant | 12 (20.3) | 24 (30.8) | 0.9 (0.2–4.6) | 0.918 |
| Tall cell variant | 14 (23.7) | 9 (11.5) | 0.9 (0.2–3.9) | 0.932 |
| Extrathyroidal extension | 0.155 | |||
| Absent | 26 (39.4) | 63 (68.5) | 1 | |
| Present | 40 (60.6) | 29 (31.5) | 4.5 (0.6–35.7) | |
| Lymphovascular invasion | 0.239 | |||
| Absent | 25 (50.0) | 47 (55.3) | 1 | |
| Present | 25 (50.0) | 38 (44.7) | 0.5 (0.2–1.6) | |
| Surgical margin involvement | 0.016 | |||
| Absent | 20 (34.5) | 66 (73.3) | 1 | |
| Present | 38 (65.5) | 24 (26.7) | 4.8 (1.3–17.0) | |
| Tumour focality | 0.577 | |||
| Unifocal | 39 (59.1) | 41 (44.6) | ||
| Multifocal | 27 (40.9) | 51 (55.4) | 1.4 (0.5–4.1) | |
| Stage | ||||
| I | 34 (51.5) | 65 (70.7) | 1 | |
| II | 7 (10.6) | 9 (9.8) | 0.3 (0.01–8.3) | 0.484 |
| III | 10 (15.2) | 13 (14.1) | 2.2 (0.3–15.0) | 0.413 |
| IV | 15 (22.7) | 5 (5.4) | 3.3 (0.1–72.2) | 0.453 |
| pT | ||||
| T1 | 8 (12.1) | 30 (32.6) | 1 | |
| T2 | 12 (18.2) | 23 (25.0) | 1.2 (0.2–6.3) | 0.864 |
| T3 | 35 (53.0) | 36 (39.1) | 0.7 (0.1–6.2) | 0.771 |
| T4 | 11 (16.7) | 3 (3.3) | 0.5 (0.01–24.3) | 0.745 |
| pN | 0.012 | |||
| N0 | 24 (37.5) | 58 (69.9) | 1 | |
| N1 | 40 (62.5) | 25 (30.1) | 4.4 (1.4–14.3) | |
| pM | ||||
| M0 | 54 (81.8) | 91 (98.9) | ||
| M1 | 12 (18.2) | 1 (1.1) | ||
| Types of surgery | 0.059 | |||
| Hemi-thyroidectomy | 6 (9.1) | 7 (7.6) | 1 | |
| Total thyroidectomy | 60 (90.9) | 85 (92.4) | 0.1 (0.01–1.1) |
| Genotype | Total Cases n | RAI Refractory n (%) | RAI Avid n (%) | p-Value (Alpha = 0.05) | Odds Ratio (95% Confidence Intervals) |
|---|---|---|---|---|---|
| SBS13 (−) and TERTp (−) | 83 | 27 (32.5) | 56 (67.5) | ||
| SBS13 (+) and TERTp (−) | 8 | 7 (87.5) | 1 (12.5) | 0.015 | 14.5 (1.7–124.0) |
| SBS13 (−) and TERTp (+) | 14 | 12 (85.7) | 2 (14.3) | 0.002 | 12.4 (2.6–59.6) |
| SBS13 (+) and TERTp (+) | 7 | 7 (100.0) | 0 (0.0) | 0.021 | 30.8 (1.7–559.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siraj, S.; Masoodi, T.; Siraj, A.K.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Bu, R.; Siddiqui, K.S.; Al-Sobhi, S.S.; AlDawish, M.; et al. APOBEC SBS13 Mutational Signature—A Novel Predictor of Radioactive Iodine Refractory Papillary Thyroid Carcinoma. Cancers 2022, 14, 1584. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061584
Siraj S, Masoodi T, Siraj AK, Azam S, Qadri Z, Parvathareddy SK, Bu R, Siddiqui KS, Al-Sobhi SS, AlDawish M, et al. APOBEC SBS13 Mutational Signature—A Novel Predictor of Radioactive Iodine Refractory Papillary Thyroid Carcinoma. Cancers. 2022; 14(6):1584. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061584
Chicago/Turabian StyleSiraj, Sarah, Tariq Masoodi, Abdul K. Siraj, Saud Azam, Zeeshan Qadri, Sandeep K. Parvathareddy, Rong Bu, Khawar S. Siddiqui, Saif S. Al-Sobhi, Mohammed AlDawish, and et al. 2022. "APOBEC SBS13 Mutational Signature—A Novel Predictor of Radioactive Iodine Refractory Papillary Thyroid Carcinoma" Cancers 14, no. 6: 1584. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061584