Testing Metal–Organic Framework Catalysts in a Microreactor for Ethyl Paraoxon Hydrolysis

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
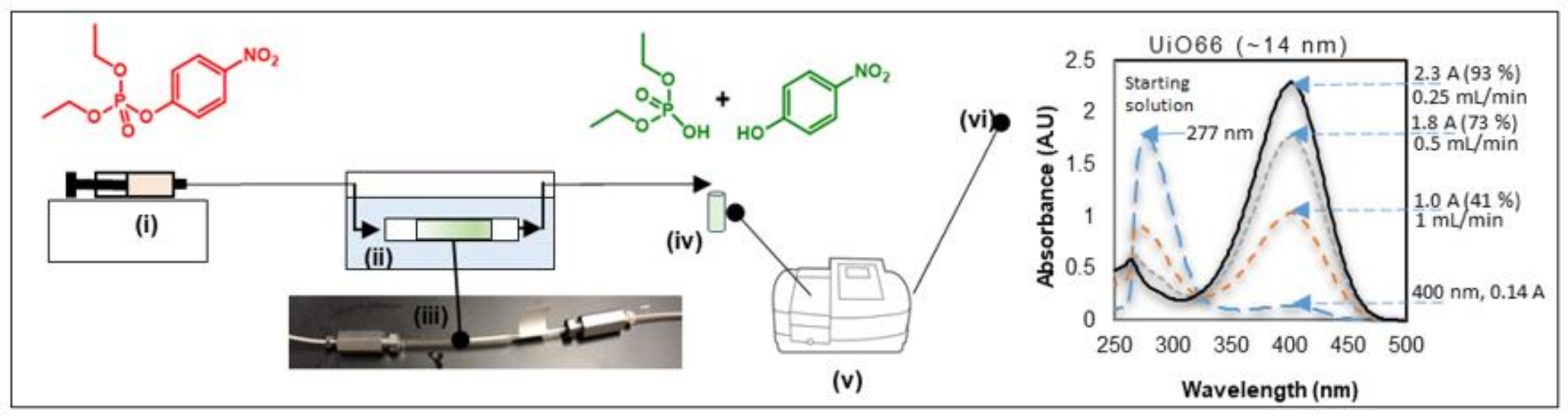
2.1. MOF Synthesis, Loading Sieved Catalysts in the Capillary Flow Reactor and Analysis of the Catalyst Bed
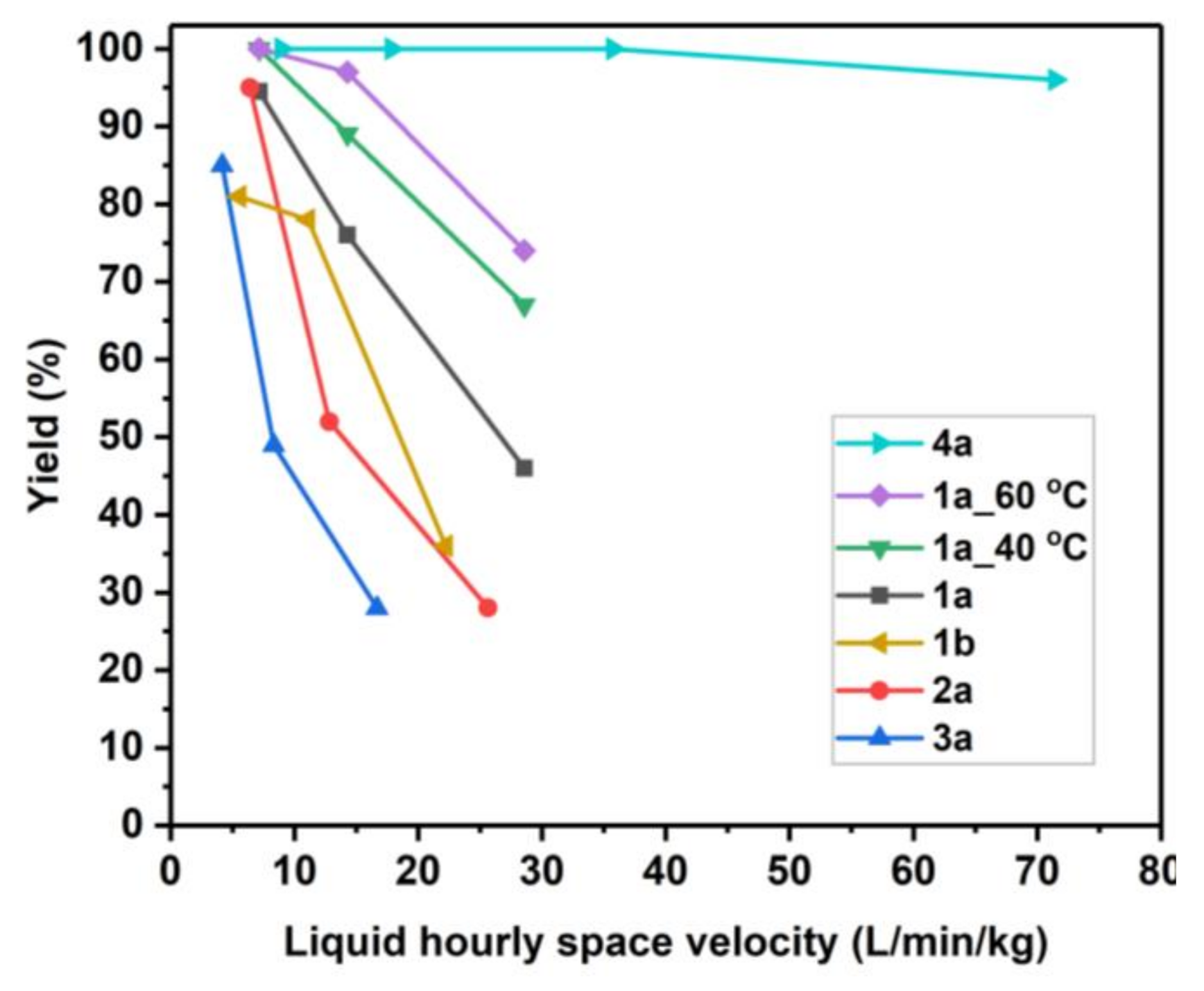
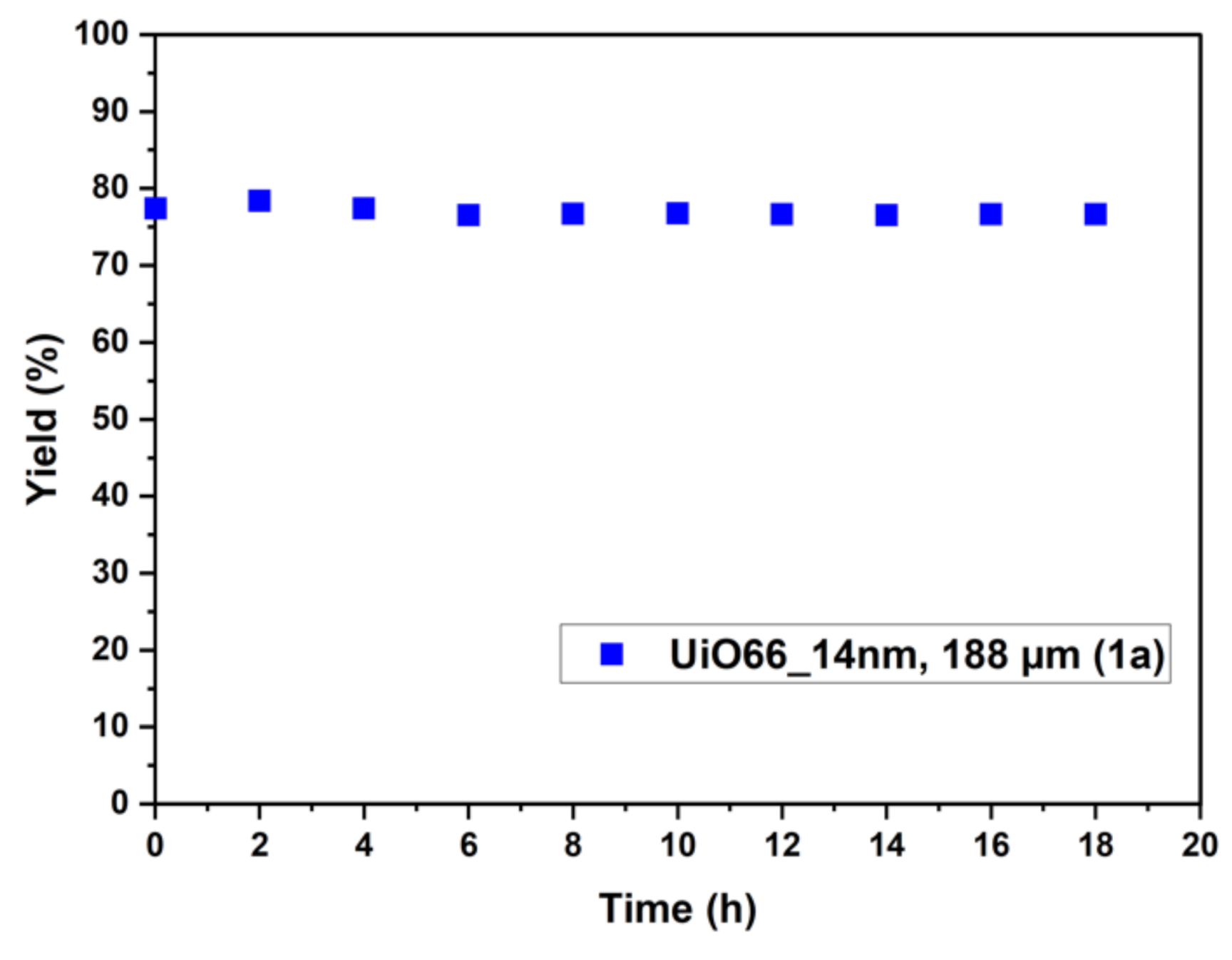
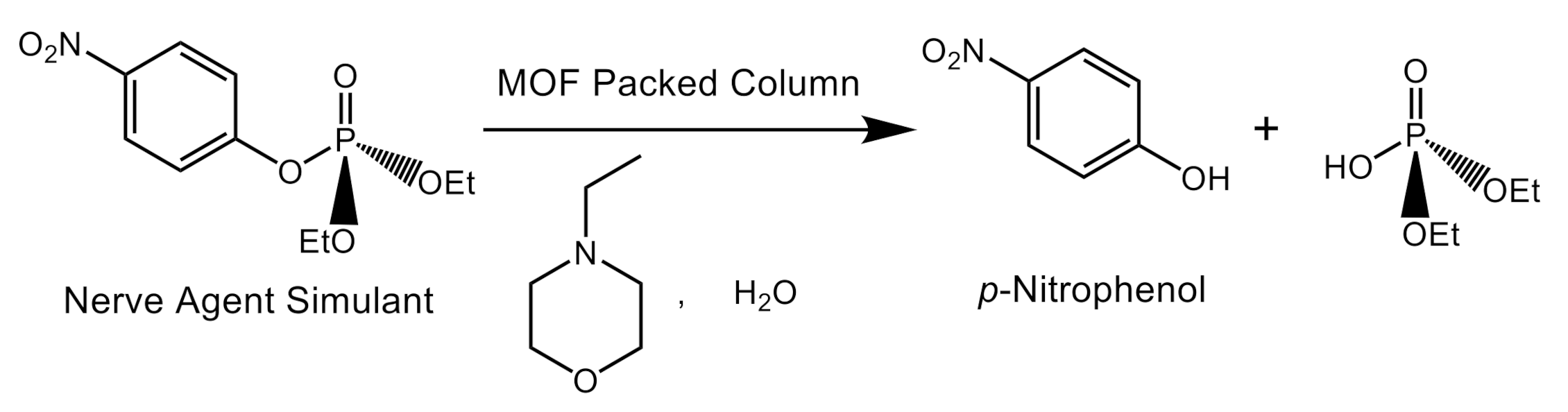
2.2. Catalyst Testing for Ethyl Paraoxon Hydrolysis and Analysis
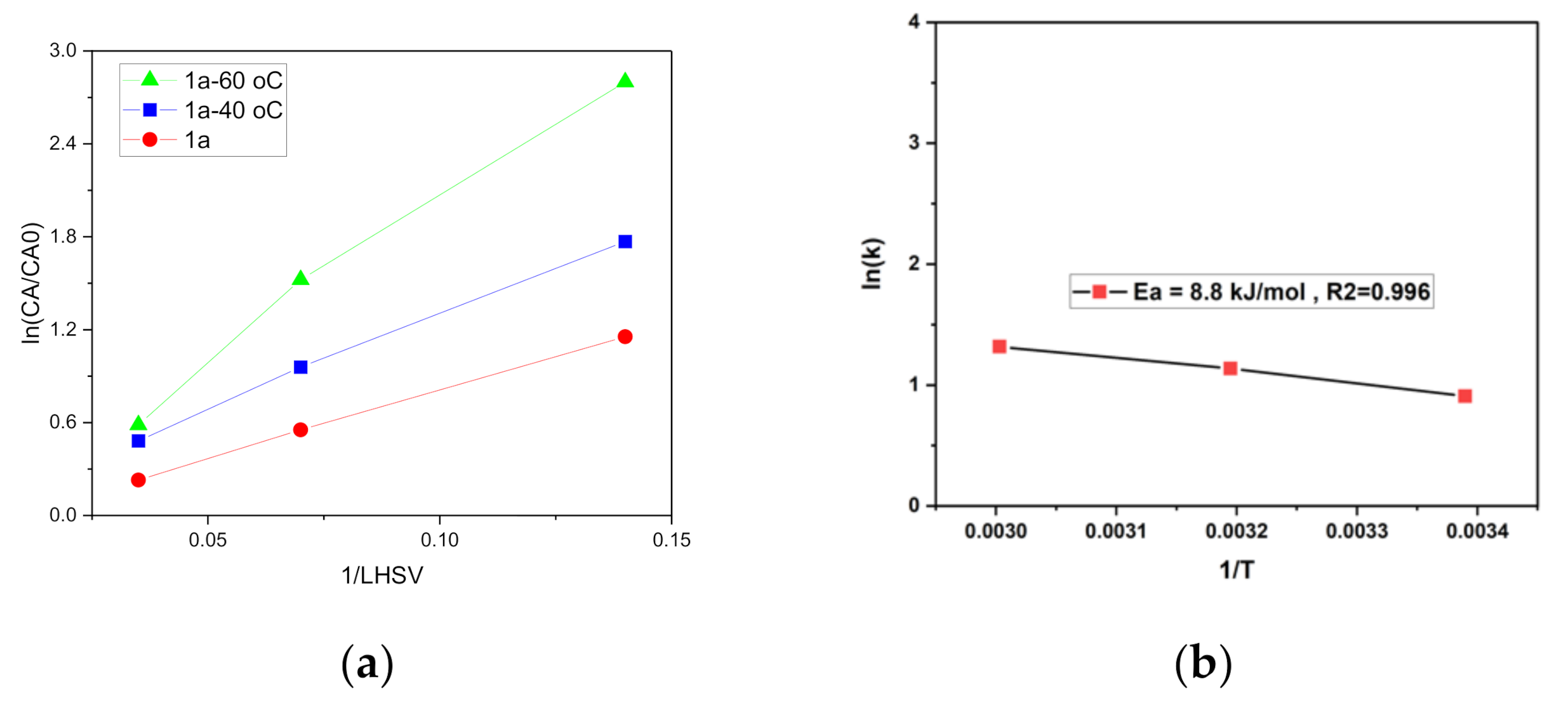
2.3. Catalyst Kinetic Evaluation Based on Initial Rates
3. Materials and Methods
3.1. Materials
3.2. Synthesis
3.2.1. UiO66 MOF Nano-Catalyst Synthesis
Synthesis of UiO-66_14 nm (1) and UiO-66-NH2_14 nm (4)
Synthesis of UiO-66_200 nm (2) and UiO-66_540 nm (3)
3.2.2. Synthesis of Paraoxon-Ethyl
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Inoue, T.; Schmidt, M.A.; Jensen, K.F. Microfabricated multiphase reactors for the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Ind. Eng. Chem. Res. 2007, 46, 1153–1160. [Google Scholar] [CrossRef]
- Plouffe, P.; Macchi, A.; Roberge, D.M. From batch to continuous chemical synthesis-a toolbox approach. Org. Process Res. Dev. 2014, 18, 1286–1294. [Google Scholar] [CrossRef]
- Wada, Y.; Schmidt, M.A.; Jensen, K.F. Flow distribution and ozonolysis in gas-liquid multichannel microreactors. Ind. Eng. Chem. Res. 2006, 45, 8036–8042. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology-A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Chambers, R.D.; Fox, M.A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Elemental fluorine Part 16. Versatile thin-film gas-liquid multi-channel microreactors for effective scale-out. Lab Chip 2005, 5, 191–198. [Google Scholar] [CrossRef]
- Leclerc, A.; Alame, M.; Schweich, D.; Pouteau, P.; Delattre, C.; de Bellefon, C. Gas-liquid selective oxidations with oxygen under explosive conditions in a micro-structured reactor. Lab Chip 2008, 8, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Schwolow, S.; Hollmann, J.; Schenkel, B.; Röder, T. Application-oriented analysis of mixing performance in microreactors. Org. Process Res. Dev. 2012, 16, 1513–1522. [Google Scholar] [CrossRef]
- Gobert, S.R.L.; Kuhn, S.; Braeken, L.; Thomassen, L.C.J. Characterization of Milli- and Microflow Reactors: Mixing Efficiency and Residence Time Distribution. Org. Process Res. Dev. 2017, 21, 531–542. [Google Scholar] [CrossRef]
- Reckamp, J.M.; Bindels, A.; Duffield, S.; Liu, Y.C.; Bradford, E.; Ricci, E.; Susanne, F.; Rutter, A. Mixing Performance Evaluation for Commercially Available Micromixers Using Villermaux-Dushman Reaction Scheme with the Interaction by Exchange with the Mean Model. Org. Process Res. Dev. 2017, 21, 816–820. [Google Scholar] [CrossRef]
- Hartman, R.L. Managing solids in microreactors for the upstream continuous processing of fine chemicals. Org. Process Res. Dev. 2012, 16, 870–887. [Google Scholar] [CrossRef]
- Nakamura, H. Nanoparticle Synthesis in Microreactors. In Encyclopedia of Microfluidics and Nanofluidics; Springer US: Berlin/Heidelberg, Germany, 2008; pp. 1437–1446. [Google Scholar]
- Wagner, J.; Köhler, J.M. Continuous synthesis of gold nanoparticles in a microreactor. Nano Lett. 2005, 5, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Reizman, B.J.; Jensen, K.F. Feedback in Flow for Accelerated Reaction Development. Acc. Chem. Res. 2016, 49, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.P.; Jensen, K.F. Rapid determination of reaction kinetics with an automated microfluidic system. Org. Process Res. Dev. 2011, 15, 398–407. [Google Scholar] [CrossRef]
- May, S.A. Flow Chemistry, Continuous Processing, and Continuous Manufacturing: A Pharmaceutical Perspective. J. Flow Chem. 2017, 7, 137–145. [Google Scholar] [CrossRef]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef]
- Kalaj, M.; Denny, M.S.; Bentz, K.C.; Palomba, J.M.; Cohen, S.M. Nylon–MOF Composites through Postsynthetic Polymerization. Angew. Chem. Int. Ed. 2019, 58, 2336–2340. [Google Scholar] [CrossRef]
- Chen, Z.; Hanna, S.L.; Redfern, L.R.; Alezi, D.; Islamoglu, T.; Farha, O.K. Reticular chemistry in the rational synthesis of functional zirconium cluster-based MOFs. Coord. Chem. Rev. 2019, 386, 32–49. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, K.; Mahle, J.J.; Wang, H.; Syed, Z.H.; Atilgan, A.; Chen, Y.; Xin, J.H.; Islamoglu, T.; Peterson, G.W.; et al. Integration of Metal-Organic Frameworks on Protective Layers for Destruction of Nerve Agents under Relevant Conditions. J. Am. Chem. Soc. 2019, 141, 20016–20021. [Google Scholar] [CrossRef]
- Ji, P.; Feng, X.; Oliveres, P.; Li, Z.; Murakami, A.; Wang, C.; Lin, W. Strongly Lewis Acidic Metal-Organic Frameworks for Continuous Flow Catalysis. J. Am. Chem. Soc. 2019, 141, 14878–14888. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Navalon, S.; Asiri, A.M.; Garcia, H. Metal organic frameworks as solid catalysts for liquid-phase continuous flow reactions. Chem. Commun. 2019, 56, 26–45. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, X.; Zhang, S.; Li, S.; Cao, S.; Pei, X.; Zhou, J.; Feng, X.; Wang, B. Shaping of Metal–Organic Frameworks: From Fluid to Shaped Bodies and Robust Foams. J. Am. Chem. Soc. 2016, 138, 10810–10813. [Google Scholar] [CrossRef]
- Furukawa, S.; Reboul, J.; Phane Diring, S.; Sumida, K.; Kitagawa, S. Structuring of metal-organic frameworks at the mesoscopic/macroscopic scale. Chem. Soc. Rev. 2014, 5700, 5700. [Google Scholar] [CrossRef] [PubMed]
- Gascon, J.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F.X. Metal Organic Framework Catalysis: Quo vadis? ACS Catal. 2014, 4, 361–378. [Google Scholar] [CrossRef]
- Tan, J.C.; Cheetham, A.K. Mechanical properties of hybrid inorganic-organic framework materials: Establishing fundamental structure-property relationships. Chem. Soc. Rev. 2011, 40, 1059–1080. [Google Scholar] [CrossRef] [PubMed]
- Serra-Crespo, P.; Stavitski, E.; Kapteijn, F.; Gascon, J. High compressibility of a flexible metal-organic framework. Rsc Adv. 2012, 2, 5051–5053. [Google Scholar] [CrossRef]
- Montoro, C.; Linares, F.; Quartapelle Procopio, E.; Senkovska, I.; Kaskel, S.; Galli, S.; Masciocchi, N.; Barea, E.; Navarro, J.A.R. Capture of nerve agents and mustard gas analogues by hydrophobic robust MOF-5 type metal-organic frameworks. J. Am. Chem. Soc. 2011, 133, 11888–11891. [Google Scholar] [CrossRef]
- Yot, P.G.; Yang, K.; Ragon, F.; Dmitriev, V.; Devic, T.; Horcajada, P.; Serre, C.; Maurin, G. Exploration of the mechanical behavior of metal organic frameworks UiO-66(Zr) and MIL-125(Ti) and their NH2 functionalized versions. Dalt. Trans. 2016, 45, 4283–4288. [Google Scholar] [CrossRef]
- Wu, H.; Yildirim, T.; Zhou, W. Exceptional mechanical stability of highly porous zirconium metal-organic framework UiO-66 and its important implications. J. Phys. Chem. Lett. 2013, 4, 925–930. [Google Scholar] [CrossRef]
- Van De Voorde, B.; Stassen, I.; Bueken, B.; Vermoortele, F.; De Vos, D.; Ameloot, R.; Tan, J.C.; Bennett, T.D. Improving the mechanical stability of zirconium-based metal-organic frameworks by incorporation of acidic modulators. J. Mater. Chem. A 2015, 3, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Morris, W.; Briley, W.E.; Auyeung, E.; Cabezas, M.D.; Mirkin, C.A. Nucleic acid-metal organic framework (MOF) nanoparticle conjugates. J. Am. Chem. Soc. 2014, 136, 7261–7264. [Google Scholar] [CrossRef]
- Tamilselvi, A.; Mugesh, G. Hydrolysis of Organophosphate Esters: Phosphotriesterase Activity of Metallo-β-lactamase and Its Functional Mimics. Chem. A Eur. J. 2010, 16, 8878–8886. [Google Scholar] [CrossRef] [PubMed]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 6, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Azarifar, D.; Daliran, S.; Ghorbani-Vaghei, R.; Oveisi, R.A. A Multifunctional Zirconium-Based Metal–Organic Framework for the One-Pot Tandem Photooxidative Passerini Three-Component Reaction of Alcohols. ChemCatChem 2017, 9, 1992–2000. [Google Scholar] [CrossRef]
- Xuechuan, G.; Ruixue, C.; Jia, G.; Liu, Z. Size and surface controllable metal–organic frameworks (MOFs) for fluorescence imaging and cancer therapy. Nanoscale 2018, 10, 6205–6211. [Google Scholar]
- Užarević, K.; Wang, T.C.; Moon, S.Y.; Fidelli, A.M.; Hupp, J.T.; Farha, O.K.; Friščić, T. Mechanochemical and solvent-free assembly of zirconium-based metal-organic frameworks. Chem. Commun. 2016, 52, 2133–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, M.J.; Moon, S.Y.; Mondloch, J.E.; Beyzavi, M.H.; Stephenson, C.J.; Hupp, J.T.; Farha, O.K. Exploiting parameter space in MOFs: A 20-fold enhancement of phosphate-ester hydrolysis with UiO-66-NH2. Chem. Sci. 2015, 6, 2286–2291. [Google Scholar] [CrossRef] [Green Version]
- Marin, G.B.; Yablonsky, G.S. Kinetics of Chemical Reactions: Decoding Complexity; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 428. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactor | Loaded UiO66 | Amount (mg) | Length (mm) | Sieved Fraction (µm) |
|---|---|---|---|---|
| 1a | UiO66_14nm | 35 | 30 | 125–250 |
| 1b | UiO66_14nm | 45 | 28 | 45–125 |
| 2a | UiO66_200nm | 39 | 29 | 125–250 |
| 3a | UiO66_540nm | 60 | 37 | 125–250 |
| 4a | UiO66-NH2_14nm | 28 | 36 | 125–250 |
| 4b | UiO66-NH2_14nm | 35 | 30 | 45–125 |
| MOF | Langmuir Surface Area (m2 g−1) |
|---|---|
| 1 | 1280 |
| 1a | 1102 |
| 1a AR | 1218 |
| 2a | 628 |
| 3a | 230 |
| 4 | 1317 |
| 4a | 1246 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elumalai, P.; Elrefaei, N.; Chen, W.; Al-Rawashdeh, M.; Madrahimov, S.T. Testing Metal–Organic Framework Catalysts in a Microreactor for Ethyl Paraoxon Hydrolysis. Catalysts 2020, 10, 1159. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10101159
Elumalai P, Elrefaei N, Chen W, Al-Rawashdeh M, Madrahimov ST. Testing Metal–Organic Framework Catalysts in a Microreactor for Ethyl Paraoxon Hydrolysis. Catalysts. 2020; 10(10):1159. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10101159
Chicago/Turabian StyleElumalai, Palani, Nagat Elrefaei, Wenmiao Chen, Ma’moun Al-Rawashdeh, and Sherzod T. Madrahimov. 2020. "Testing Metal–Organic Framework Catalysts in a Microreactor for Ethyl Paraoxon Hydrolysis" Catalysts 10, no. 10: 1159. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10101159