Dehydrogenation of Formic Acid to CO2 and H2 by Manganese(I)–Complex: Theoretical Insights for Green and Sustainable Route



Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Ponte P. Bucci cubo 14 C, Arcavacata di Rende, CAP 87036 Cosenza, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(1), 141; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010141
Submission received: 4 January 2021
/
Revised: 14 January 2021
/
Accepted: 16 January 2021
/
Published: 19 January 2021
(This article belongs to the Special Issue Transition Metal Catalysis for Biomass Transformation and Green Energy Production)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:In this work, a detailed computational study on a recently synthetized Mn(I)-dependent complex [(tBuPNNOP)Mn(CO)2]+ is reported. This species promotes the dehydrogenation of formic acid to carbon dioxide and hydrogen. The here proposed catalytic cycle proceeds through the formation of stabilized adduct between [(tBuPNNOPtBu)Mn(CO)2]+ and formate and the progressive release of CO2 and H2, mediated by the presence of trimethylamine. In order to evaluate the influence of the environment on the catalytic activity, different solvents have been taken into account. The computed barriers and the geometrical parameters account well for the available experimental data, confirming the robustness of the complex and reproducing its good catalytic performance. Outcomes from the present investigation can stimulate further experimental works in the design of new more efficient catalysts devoted to H2 production.
1. Introduction
The increasing need for new and sustainable energetic resources represents one of the most important challenges characterizing the current century [1,2]. Indeed, fossil fuels, gas, coal and nuclear energy are still widely used, but environmentally dangerous, energy sources [2,3,4,5]. The intensive use of fossil fuels, for example, has been directly linked to the increasing level of CO2 and greenhouse gas emissions, dramatically influencing the climate changes [3].
For these reasons, in the last fifty years, the interest devoted to possible “green” alternatives, like the use of sunlight-, wind- and water-based energies [6], have been increased, but, despite their promising efficiency, different technical issues are related to them and in particular to the storage of energy vectors on large scale [7].
One of the possible solutions is represented by the so-called sustainable hydrogen economy [8,9,10,11]. In this route, indeed, the electricity is converted in a secondary chemical energy carrier that can be used on demand [12,13,14,15,16]. The combustion of H2 in the presence of O2, in devices like fuel cells, formally produces electricity and H2O, a green product.
On the other hand, the H2 is not present on earth and it can be obtained/stored from/in organic compounds, like methanol and formic acid (FA) [17,18,19]. In particular, since its chemical-physical properties and its involvement in chemical industries and biomass production, the FA is believed a promising species for the hydrogen economy [1,2,20,21]. The dehydrogenation of FA, which generates CO2 and H2, (Scheme 1) is usually mediated by metal-containing catalysts.
In detail, the most widely adopted species contain Pd, Pt, Ir, Rh and Ru (see refs [22,23,24,25,26] as recent examples), with turnover frequencies (TOFs) variating from 102 to 106 h−1 [27]. Since all these species contain precious metals, nowadays one of the main goals is synthesizing complexes containing equally efficient metal ions belonging to the first row of transition series, which are notoriously cheaper and more abundant in the Earth’s crust.
Promising solutions can arise by Fe-based catalysts showing turnover values comparable to those containing precious metals, but still demanding an improvement of their catalytic efficiency [28,29,30]. In addition, these species require the presence of a general base and Lewis acid, like Li+, to explicate the catalysis [29].
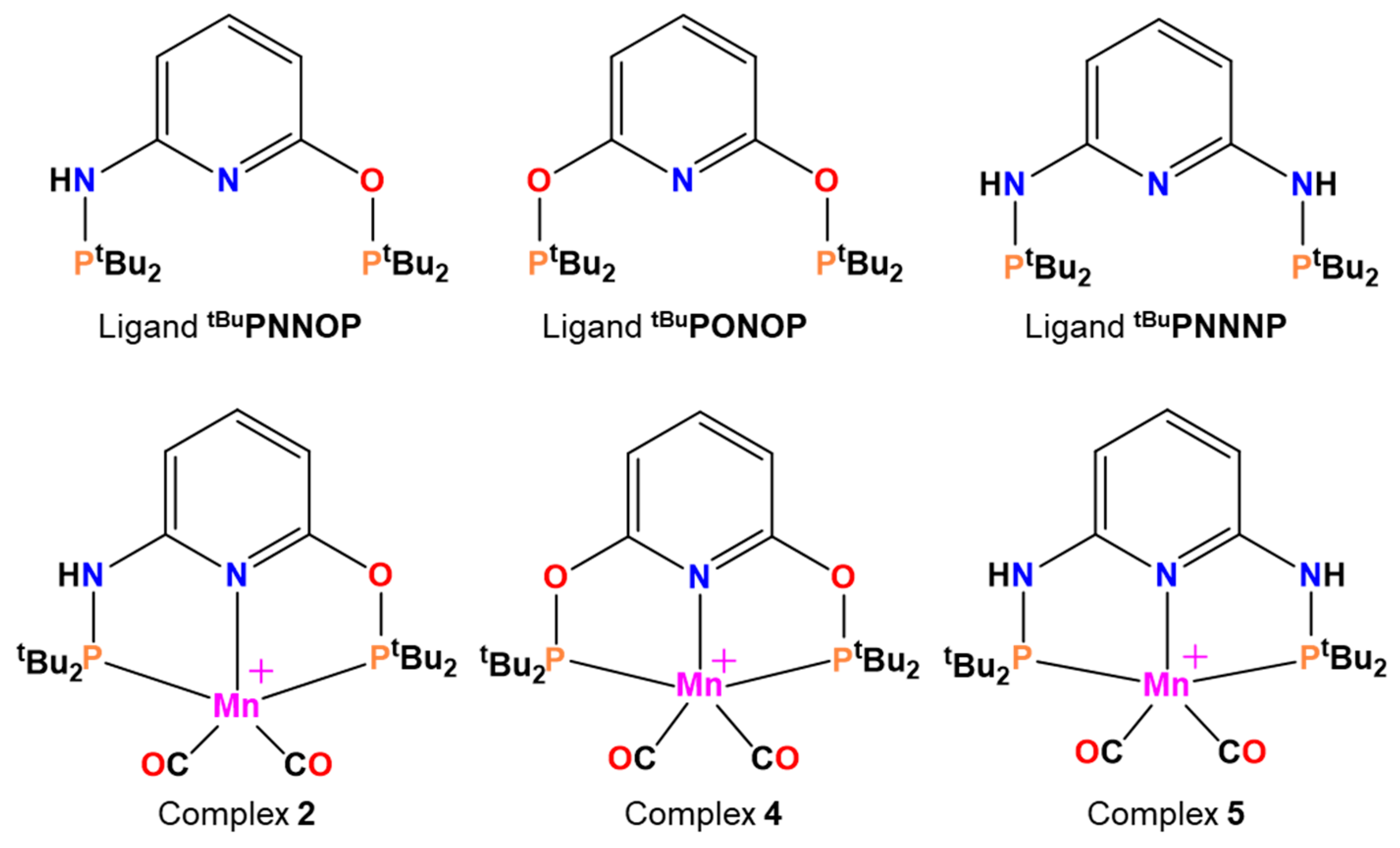
Very recently, for a new synthesized complex of Mn(I)-based complex, fast catalyzed dehydrogenation of formic acid has been described [31]. The organometallic compound, having the structure [(tbuPNNOPMn(CO)2]+ (2), with tbuPNNOP = 2,6-(di-tert-butylphosphinito)(di-tert-butylphosphinamine)pyridine), exhibited substantially higher catalytic activity with respect to the unique known Mn-dependent complex acting on the formic acid [32], with TOFs improved to the value of 8500 h−1 [31]. The reaction requires the presence of a proton-acceptor system, but intriguingly the catalysis was not affected by the nature of the base [31].
In addition, the contribution of the hybrid tbuPNNOP ligand has been highlighted, since attempts with tbuPONOP (tBuPONOP =2,6-bis(di-tert-butylphosphinito)pyridine, 4) and tbuPNNNP (tBuPNNNP = 2,6-bis (di-tert-butylphosphinamine)pyridine, 5), two ligands tested on a previous Ru-based organometallic complex and depicted in Scheme 2 [33], did not provide remarkable catalytic activity [31].
Encouraged by the results’ novelty, which bring Mn into the family of earth-abundant metals catalyzing FA dehydrogenation [31], a detailed mechanistic study on the catalysis mediated by 2 in the framework of density functional theory (DFT) has been performed, with the aim of shedding light on the possible reaction mechanism and to characterize the intermediates and transitions states intercepted along the related potential energy surface (PES).
The adducts with formic acid of two other manganese ligands (complex 4, buPNNOP and complex 5, BuPNNNP of Scheme 2) have been considered. In addition, different solvents have been taken into account to evaluate their effect on the rate limiting step.
We hope that our results on the detailed catalytic mechanism of 2 can increase the knowledge of such a promising complex, helping the rationalization of experiments design of more efficient catalysts.
2. Results and Discussion
Before proceeding with the mechanistic study, it was mandatory to determine the spin multiplicity. For this purpose, optimizations of 2 catalyst in different spin states (2S + 1 = 1, 3, 5) have been performed. The singlet state (2S + 1 = 1) resulted in being the most stable one in agreement with d6 configuration of Mn(I) species, as analogously determined in a recent study concerning the Mn(I)-mediated catalysis of organic nitriles to amides [34], while the triplet and quintet lay at 12.1 kcal/mol and 18.7 kcal/mol, respectively, with respect to the singlet spin state.
Small spin contamination (<6%, see Table S1), monitored as described in other works [35,36], has been observed with <S2> equal to 2.10 and 6.29 for triplet and quintet (2.00 and 6.00 after annihilation, respectively) and the single state has been selected for mechanistic investigation. Consistently with the X-ray structure of substrate-free complex (CCDC 1848774) [31], in the case of singlet state, good agreement with crystallographic distances between Mn(I) and atoms of its coordination sphere were found, with small deviation (<0.05 Å) compared with the experimental counterpart (see Figure S1). In the case of higher spin states, an increasing shift with respect to the X-ray structure has been observed (see Figure S1), confirming that the singlet spin state represents the suitable species for the study of catalytic mechanism.
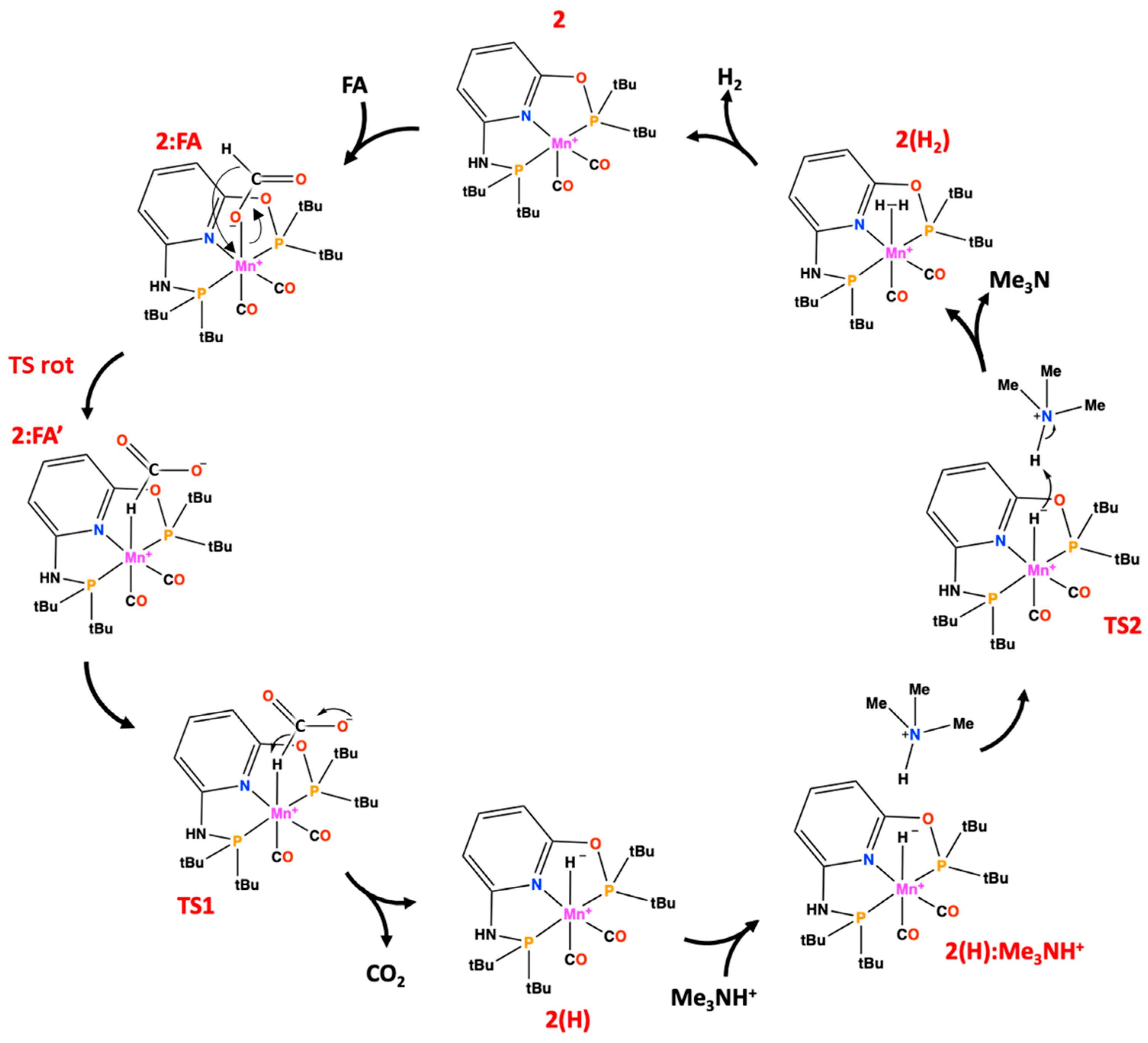
The proposed reaction mechanism is reported in Scheme 3 and the related free energy profile is presented in Figure 1. After the formation of the complex-FA− adduct, the rearrangement of the substrate takes place, in which the bond with the metal switches from the oxygen to the hydrogen of the substrate. Successively, the reaction mechanism can be divided in two phases, as follows:
[(tbuPNNOPMn(CO)2]+ + HCOO− → [(tbuPNNOPMn(H)(CO)2] + CO2
[(tbuPNNOPMn(H)(CO)2] + Me3NH+ → [(tbuPNNOPMn(CO)2]+ + H2 + Me3N
In the first (Equation (1)), the CO2 (the first product) is released with concerted formation of H-Mn bond. In the second phase (Equation (2)), due to the presence of Me3NH+ species, the H2 (the second product) is formed and subsequently released. Every phase consisted of a multistep process. The final mechanism included the formic acid in its deprotonated form (FA) produced by the trimethylamine acting as proton acceptor, since all the attempts performed on neutral species failed.
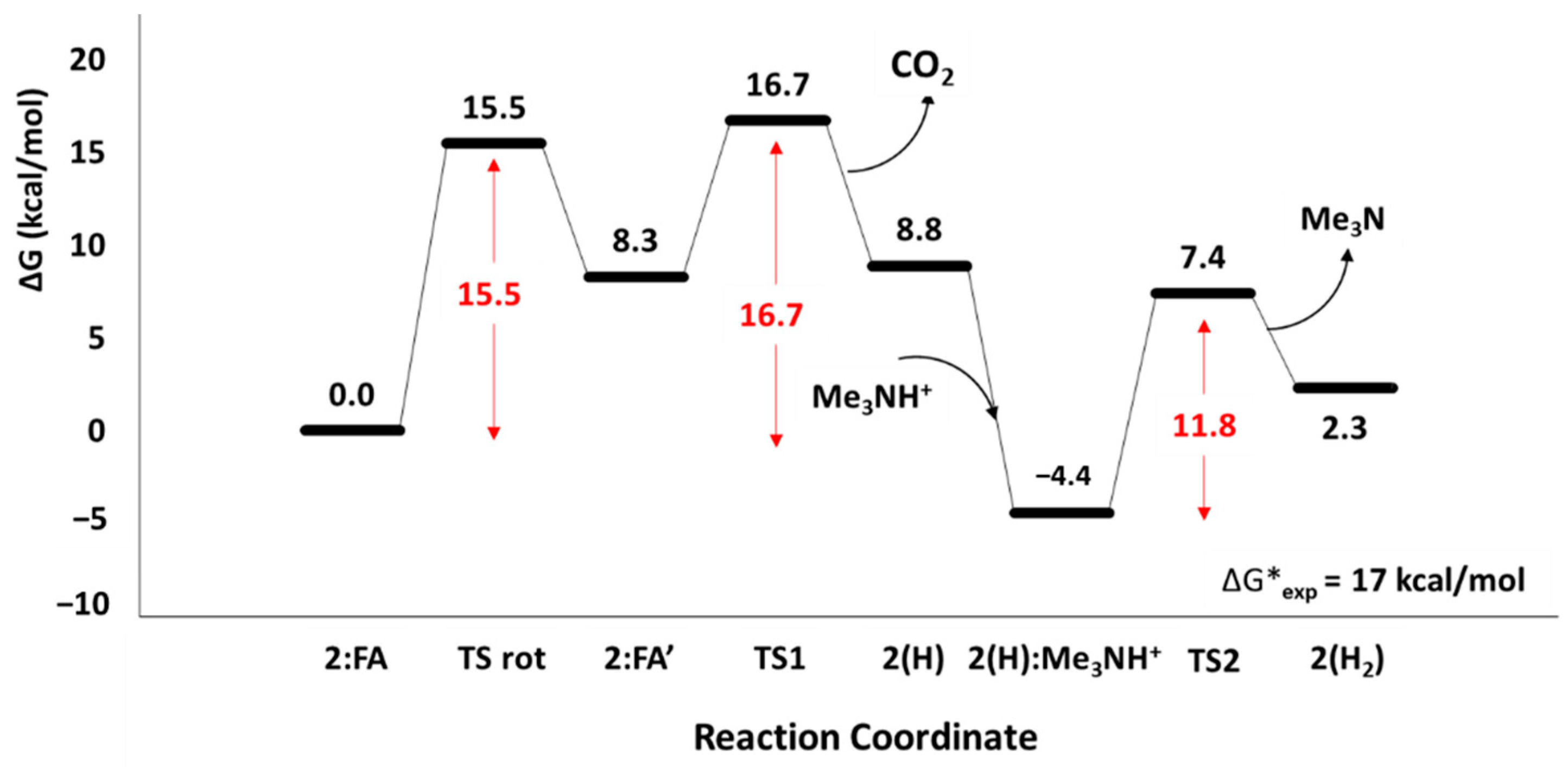
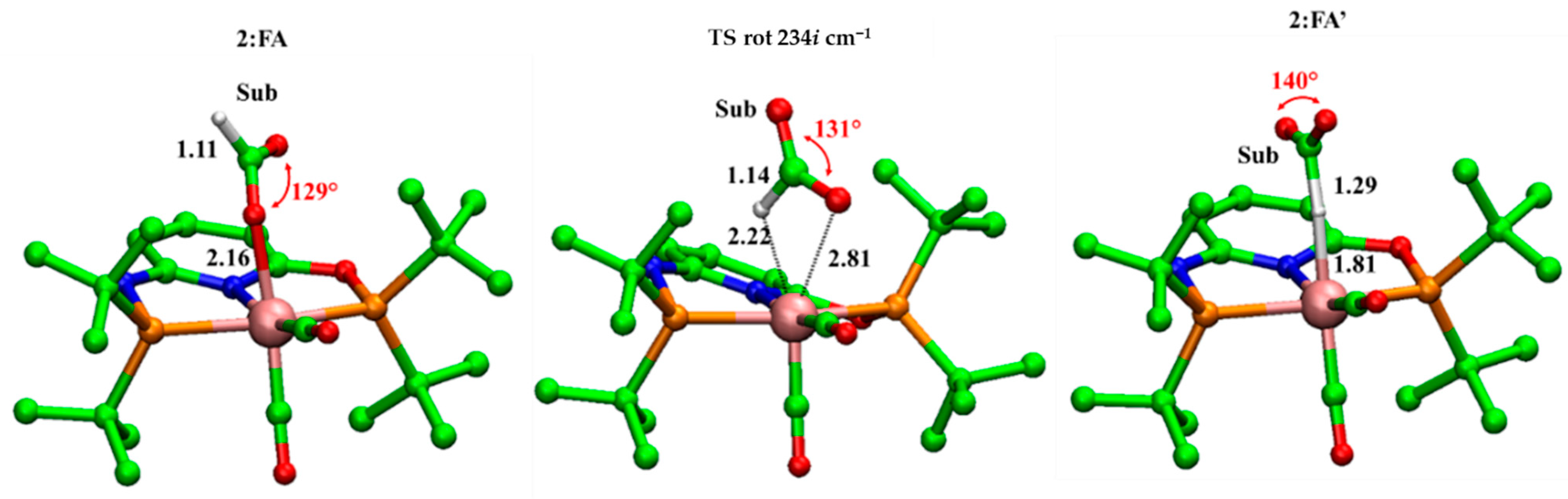
The first part of the study is related to the formation of adduct between complex 2 and formate (FA). Two different conformers have been characterized with the formate bound to the Mn(I) in both Mn-O− (2:FA) and Mn-H (2:FA’) fashions as shown in the optimized structures illustrated in Figure 2. The 2:FA’ is higher in energy (8.3 kcal/mol) with respect to 2:FA, but, in spite of this, it resulted in being more reactive with the C-H distance of 1.29 Å and the OĈO angle of 140°, if compared to the corresponding ones in 2:FA (1.11 Å and 129°, respectively).
The interconversion between the Mn-O− and Mn-H takes place with an energy barrier of 15.5 kcal/mol across the transition state TS rot (see Figure 1). In the corresponding optimized geometry (see Figure 2), the formate acts as a bidentate ligand owing to the forming Mn-H 2.22 Å and the breaking Mn-O-bonds 2.81 Å, thus offering an epta-coordinated manganese. DFT calculations on the dehydrogenation of formic acid promoted by a pincer-supported iron catalyst evidenced analogous behavior proposing an energy barrier of 21.4 kcal/mol for the switching from the Fe-O− to the Fe-H conformer [29].
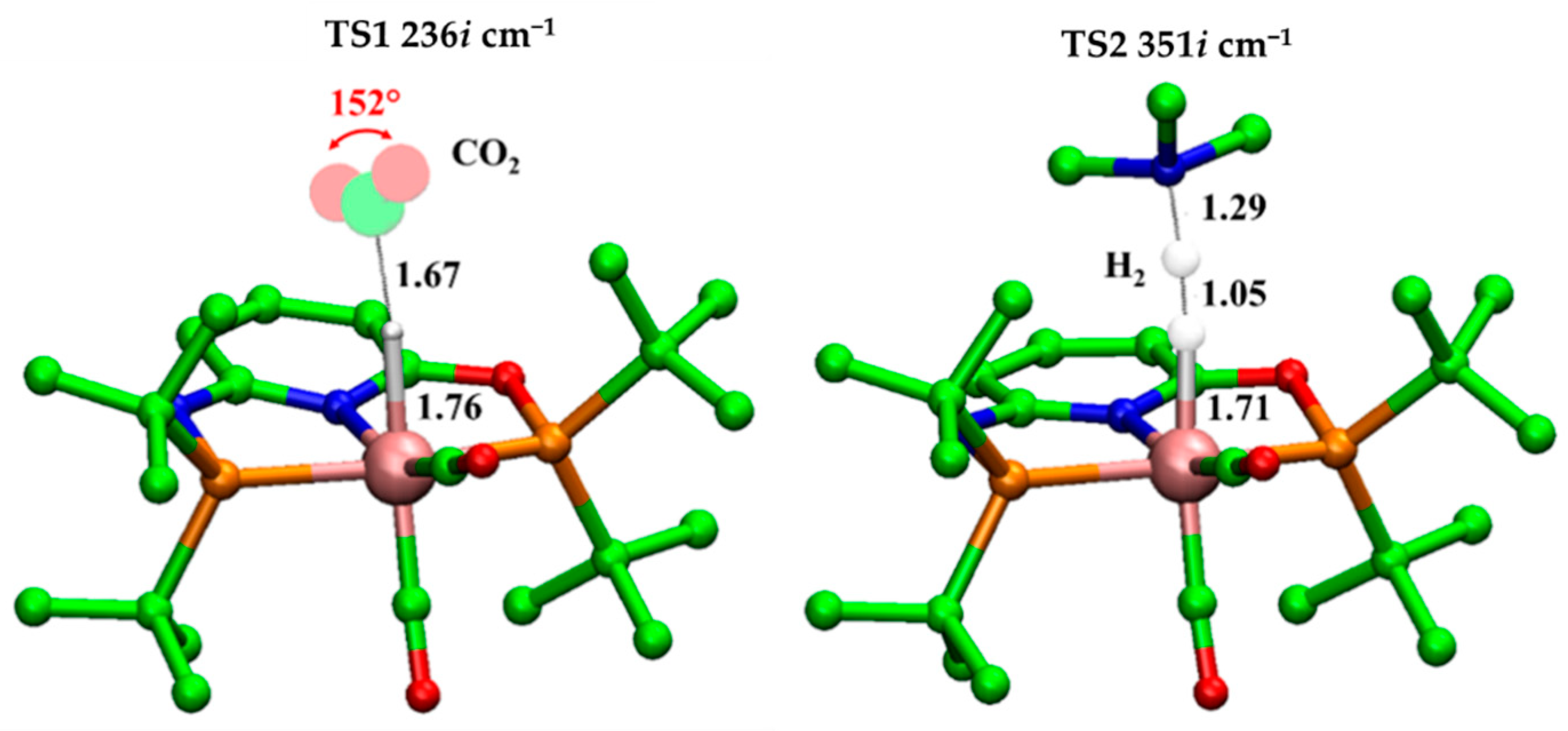
Starting from 2:FA’, the CO2 release takes place by overcoming the barrier of 8.4 kcal/mol represented by TS1 in Figure 3. In particular, the H lies at 1.76 Å from the Mn(I) and at 1.67 Å from the C, while the value of OĈO angle (152°) increased preparing for the next carbon dioxide release. The related imaginary frequency (236i cm−1) concerns the C-H stretching.
In the intermediate 2(H) the H-Mn bond resulted in being definitely formed, as confirmed by the distance of 1.60 Å (see Figure S2). From this species starts the second phase of the dehydrogenation of formic acid leading to the formation of H2. To do this, a protonated species, most likely the conjugated acid of the base involved in the catalysis, must come into play acting as proton donor. As confirmed by experimental evidences [31], its presence is fundamental for the advancing of the reaction, being deputed to the deprotonation of the formic acid. Moreover, Tondreau et al. in their kinetic studies advised that “the identity of the base does not significantly alter catalytic results” [31], so in our investigation the protonated triethylamine used in the experiment has been replaced by the protonated trimethylamine (Me3NH+ or TMA) in order to save computational cost.
The 2(H):Me3NH+ species lying at 4.4 kcal/mol below 2:FA- is the result of the addition of the protonated base and represents the starting point of the next H2 release step. The presence of the Me3NH+ induces a small elongation of the Mn-H bond (1.65 Å) if compared with the value of 1.60 Å in 2(H) species (see Figure S2) indicating its early activation.
Indeed, the proton shift, which leads to the formation of the H2 product, is described in the TS2, where the forming Hbase-H bond assumes a value of 1.05 Å vs. that of 1.45 Å found in 2(H):Me3NH+ of Figure S2 while the Mn-H results to be 1.71 Å. These findings evidence the important role of base also in the formation of the second product, since its nature as a proton-donor species. TS2 lies at 11.8 kcal/mol related to the 2(H):Me3NH+ intermediate but at 7.4 kcal/mol with respect to 2:FA (see Figure 1). In the 2(H2) species, the molecular hydrogen is directly linked to the Mn(I). The H2 σ bond length is 0.77 Å and the distance of H2 from metallic center is equal to 1.91 Å, see Figure S2.
The TS1 describing the release of CO2 represents the highest relative barrier, presenting an energy 1.2 and 9.3 kcal/mol higher than those of the TSrot and TS2, respectively. The value of 16.7 kcal/mol, in addition, is in good agreement with the available kinetic data proposing a kcat value of 2.4 s−1, converted to a ΔG# = 17 kcal/mol adopting Eyring’s equation [31].
As can be also noted from the analysis of the energy profile, the kinetic of reversible reaction, proceeding from 2(H2) → 2:FA is slower. Indeed, for this reaction an energy barrier of 21.1 kcal/mol resulted for the step 2(H):Me3NH+-TS1, 4.4 kcal/mol higher than that calculated for the 2:FA → 2(H2) pathway.
We also attempted to rationalize the effect of the pincer ligands 4 and 5 of Scheme 2 on the catalysis, focusing on their affinity (ΔHaff) for the substrate and to the fundamental formation of complex:substrate adduct that represents a crucial step for the proceeding of catalysis, as follows:
[XMn(CO)2]+ + HCOO− → [(XMn(HCOO-)(CO)2] X = tbuPNNOP, tbuPONOP, tbuPNNNP
ΔHaff = H[(XMn(HCOO−)(CO)2] − H[XMn(CO)2]+ − HHCOO−
In the case of the complex 2 the adduct 2:FA is more stabilized in energy (−35.1 kcal/mol respect to the separated reactants), contrarily to the complexes 4:FA and 5:FA (16.6 and 22.0 kcal/mol). Based on these results, the increasing energy request for the formation of adduct (complex 2 > complex 4 > complex 5) well reproduces the experimental observations [31].
A possible explanation for this behavior arises from the analysis of the charge distribution in the three 2:FA, 4:FA and 5:FA adducts, reported in Figure S3.
In particular, an evident effect concerns the Mn(I) center that exhibits a more negative charge in presence of tbuPNNNP (−0.08 |e|) and of tbuPONOP (−0.05 |e|) than in presence of the tbuPNNOP. This could consequently affect the coordination of the negatively charged HCOO− with a reduced catalytic activity.
The ligand tbuPNNOP proposes the right balance between electron withdrawing and electron donating effect, represented respectively by the O and N atoms, to the pyridine ring. Moreover, from the maps of the electrostatic potential of the three ligands depicted in Figure S4 emerges as the presence of two oxygens in tbuPONOP leads to a marked decrease of negative charge on the aromatic ring contrarily to what occurs in tbuPNNNP, this behavior can have repercussions on their catalytic activity.
Given the important role played in homogeneous catalysis by the solvent, with the aim of investigating its effect on the dehydrogenation of formic acid assisted by the 2 catalyst, different implicit solvents have been tested on the CO2 release step (TS1), considering dielectric constant with gradually increasing values to simulate apolar, polar aprotic and polar protic solvents extensively used in organometallic reactions [37]. The results are collected in Figure 4.
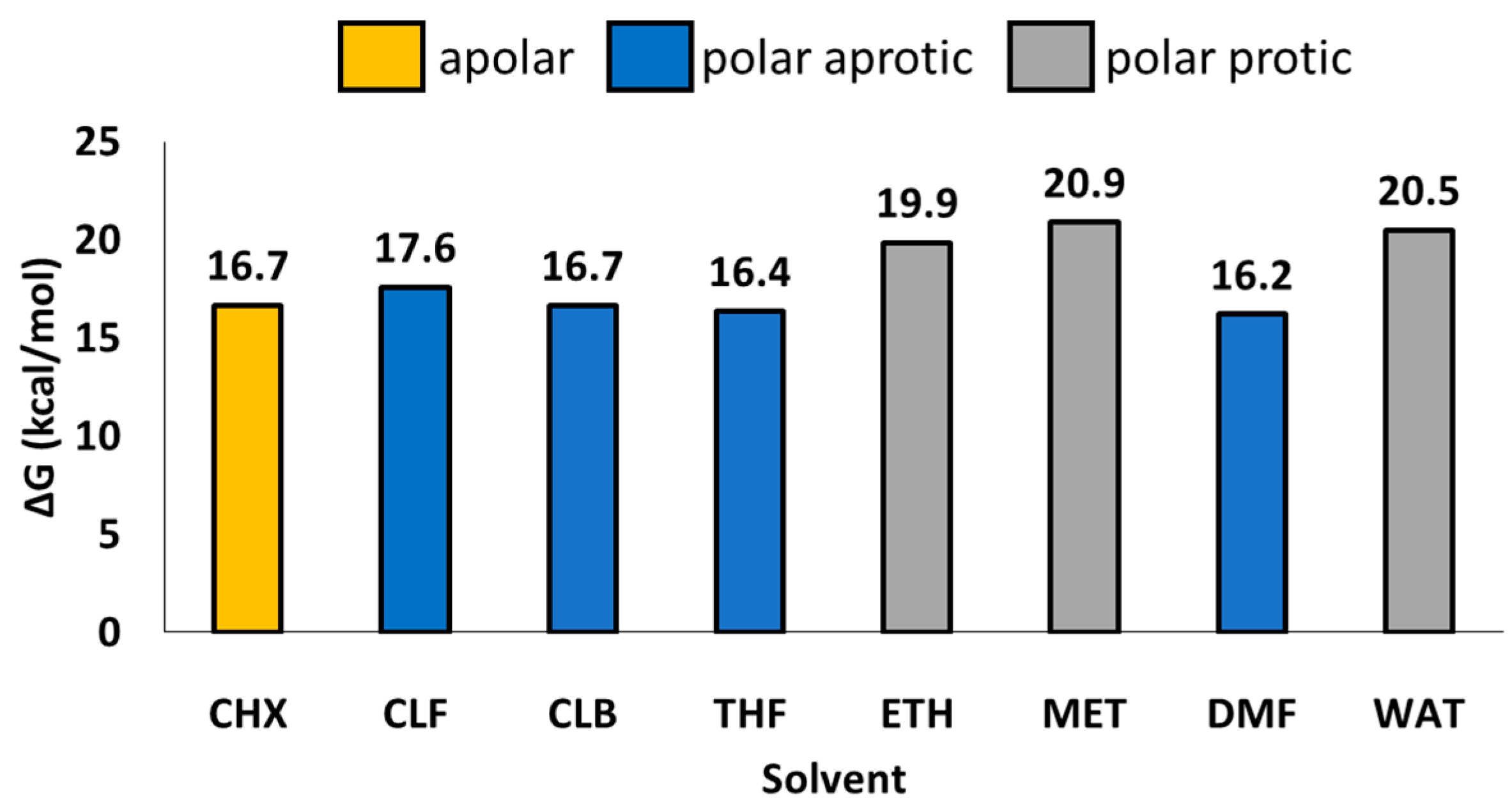
The use of polar aprotic solvent with lower dielectric constant, respect to chlorobenzene (ε = 5.7), like chloroform (ε = 4.7), results in a destabilization of the energy barrier (+0.9 kcal/mol), while an increased ε, like in the case of tetrahydrofuran (ε = 7.4) and dimethylformammide (ε = 37.2) gives rise to lower energy barriers (0.3 kcal/mol and 0.5 kcal/mol, respectively).
An interesting behavior has been observed in the case of protic polar solvents. In the case of ethanol (ε = 24.8), methanol (ε = 32.6) and water (ε = 78.3), a remarkable increment of TS1 barrier resulted, with values of 3.2 kcal/mol, 4.2 kcal/mol and 3.8 kcal/mol, respectively, indicating that the choice of operative conditions can affect the efficiency of catalytic mechanism. Finally, for the apolar solvent cyclohexane (ε = 2.0), no relevant variations occurred.
3. Materials and Methods
All the calculations have been carried out with the Gaussian09 D.01 software package [38]. The available x-ray data (CCDC 1848774) has been selected as starting structure for the mechanistic study [31]. The geometries have been fully optimized without any physical constraints, adopting B3LYP functional [39,40] and 6-31+G(d,p) basis set for C, H, N, O and P atoms. For the Mn, instead, the SDD pseudopotential [41] and its related basis set has been chosen. In addition, the dispersions according to Grimme’s scheme (D3-BJ) have been included in all the computations [42]. The strategy here presented has been successfully described and adopted in previous works on catalytic systems containing first row transition metals [34,43,44,45,46,47]. Given the lack of information on the multiplicity of the catalytic system, preliminary investigation has been carried out considering different spin states, (2S + 1 = 1, 3, 5).
The potential energy surfaces have been explored via relaxed linear transit scan and the nature of intercepted minima and maxima has been confirmed through thermochemical analysis (one and no imaginary frequency for intermediates and transition states, respectively). To include the effect of solvent, the SMD implicit solvation method [48] has been assumed, selecting the chlorobenzene as medium (ε = 5.7) to better reproduce the experimental conditions [31]. In order to investigate the effect to the catalysis, cyclohexane (ε = 2.0) chloroform (ε = 4.7), chlorobenzene (ε = 5.7), Tetrahydrofuran (ε = 7.4), ethanol (ε = 24.8), methanol (ε = 32.6), dimethylformammide (ε = 37.2) and water (ε = 78.3) implicit solvents have been additionally tested.
The final energies (ΔG) reported on the PESs additionally include the most accurate electronic energy obtained from single point energy calculations employing the more extended 6-311+G(2d,2p) basis set, and the Gibbs free energy corrections, extrapolated from thermochemical analysis.
4. Conclusions
On the basis of a detailed computational investigation performed in solvent and taking into account the experimental information, a catalytic cycle for the dehydrogenation of formic acid promoted by a recently synthetized Mn(I)-dependent complex [(tBuPNNOP)Mn(CO)2]+ as catalyst has been proposed. The process consists of two elementary steps: release of CO2 and production of H2. The proposed reaction mechanism well fits the experimental behaviors.
The CO2 release represents the rate determining state requiring 16.7 kcal/mol.
The pincer ligand with its steric hindrance ensures the occurrence of the catalytic process confirmed by the scaffold stability on all the intercepted stationary points.
Our calculations highlight that the presence of a base is fundamental for the dehydrogenation of formic acid, not only in the deprotonation of acid, but also during the formation of the second product, acting as proton-donor species.
The behavior of other pincer ligands (with tbuPONOP (tBuPONOP = 2,6-bis(di-tert-butylphosphinito)pyridine 4 and tbuPNNNP (tBuPNNNP = 2,6-bis (di-tert-butylphosphinamine)pyridine, 5) has been rationalized in terms of a different electrostatic distribution induced on the pyridine ring coordinated to Mn(I) by nitrogen.
The effect of the solvent on the catalytic process has been evaluated by taking into account apolar, polar aprotic and polar protic solvents simulated by opportune dielectric constant values.
We believe that the results from the present investigation can stimulate further experimental works in the design of new more efficient catalysts devoted to H2 production.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/11/1/141/s1, Figure S1. On the left, superposition of substrate-free complex fixing singlet, triplet (green) and quintet (gray) state. On the right, comparison between optimized and experimental observed relevant geometrical parameters. Figure S2. B3LYP-D3/6-31+G(d,p) optimized geometries of 2(H), 2(H):Me3NH+ and 2(H2) stationary points. Relevant distances (in black) are in Å. Figure S3. Charge distribution in NBO population of relevant atoms. Figure S4. Map of electrostatic potential plotted for the three different ligands. Table S1. Calculated spin contamination for triplet and quintet spin states. Cartesian coordinates.
Author Contributions
T.M. and M.P. performed the calculations and analyzed the results. T.M. and M.P wrote the manuscript and contributed to the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
We thank the Università degli Studi della Calabria-Dipartimento di Chimica e Tecnologie Chimiche (CTC) for the financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mellmann, A.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material—Development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, G.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Jacobson, M.Z. Review of solutions to global warming, air pollution, and energy security. Energy Environ. Sci. 2009, 2, 148–173. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V. The Hydrogen Issue. ChemSusChem 2011, 4, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Princiotta, F.T. Global Climate Change and the Mitigation Challenge. In Global Climate Change—The Technology Challenge; Princiotta, F.T., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp. 1–50. [Google Scholar]
- Armaroli, N.; Balzani, V. The future of energy supply: Challenges and opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Ortenzi, F.; Pede, G.; Ramadhas, A.S. Alternative Fuels for Transportation; Ramadhas, A.S., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 243–293. [Google Scholar]
- Abuadala, A.; Dincer, I. A review on biomass-based hydrogen production and potential applications. Int. J. Energy Res. 2012, 36, 415–455. [Google Scholar] [CrossRef]
- Kırtay, E. Recent advances in production of hydrogen from biomass. Energy Convers. Manag. 2011, 52, 1778–1789. [Google Scholar] [CrossRef]
- Christopher, K.; Dimitrios, R. A review on exergy comparison of hydrogen production methods from renewable energy sources. Energy Environ. Sci. 2012, 5, 6640–6651. [Google Scholar] [CrossRef]
- Moriarty, P.; Honnery, D. A hydrogen standard for future energy accounting? Int. J. Hydrog. Energy 2010, 35, 12374–12380. [Google Scholar] [CrossRef]
- Hobein, B.; Krueger, R. Hydrogen and Fuel Cells—Fundamentals, Technologies and Applications; Stolten, D., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 377–393. [Google Scholar]
- Schüth, F. Chemical Compounds for Energy Storage. Chem. Ing. Tech. 2011, 83, 1984–1993. [Google Scholar] [CrossRef]
- Makowski, P.; Thomas, A.; Kuhn, P.; Goettmann, F. Organic materials for hydrogenstorage applications: From physisorption on organic solids to chemisorption in organic molecules. Energy Environ. Sci. 2009, 2, 480–490. [Google Scholar] [CrossRef]
- Dalebrook, A.F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Hydrogen storage: Beyond conventional methods. Chem. Commun. 2013, 49, 8735–8751. [Google Scholar] [CrossRef]
- Li, S.-L.; Xu, Q. Metal–organic frameworks as platforms for clean energy. Energy Environ. Sci. 2013, 6, 1656–1683. [Google Scholar] [CrossRef]
- Rodrıguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grutzmacher, H. A homogeneous transition metal complex for clean hydrogen production from methanol-water mixtures. Nat. Chem. 2013, 5, 342–347. [Google Scholar] [CrossRef]
- Nielsen, N.; Alberico, E.; Baumann, W.; Drexler, H.-J.; Junge, H.; Gladiali, S.; Beller, M. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—Recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Malacea, R.; Poli, R.; Manoury, E. Asymmetric hydrosilylation, transfer hydrogenation and hydrogenation of ketones catalyzed by iridium complexes. Coord. Chem. Rev. 2010, 254, 729–752. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef]
- King, R.B.; Bhattacharyya, N.K. Catalytic reactions of formate 4. A nitrite-promoted rhodium (III) catalyst for hydrogen generation from formic acid in aqueous solution. Inorg. Chim. Acta 1995, 237, 65–69. [Google Scholar] [CrossRef]
- Marcinkowski, M.D.; Liu, J.; Murphy, C.J.; Liriano, M.L.; Wasio, N.A.; Lucci, F.R.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Selective Formic Acid Dehydrogenation on Pt-Cu Single-Atom Alloys. ACS Catal. 2017, 7, 413–420. [Google Scholar] [CrossRef]
- Barrett, S.M.; Slattery, S.A.; Miller, A.J.M. Photochemical Formic Acid Dehydrogenation by Iridium Complexes: Understanding Mechanism and Overcoming Deactivation. ACS Catal. 2015, 5, 6320–6327. [Google Scholar] [CrossRef]
- Fink, C.; Laurenczy, G. A Precious Catalyst: Rhodium-Catalyzed Formic Acid Dehydrogenation in Water. Eur. J. Inorg. Chem. 2019, 2019, 2381–2387. [Google Scholar] [CrossRef]
- Frenklah, A.; Treigerman, Z.; Sasson, Y.; Kozuch, S. Formic Acid Dehydrogenation by Ruthenium Catalyst—Computational and Kinetic Analysis with the Energy Span Model. Eur. J. Org. Chem. 2019, 2019, 591–597. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen Energy Future with Formic Acid: A Renewable Chemical Hydrogen Storage System. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Zhang, Y.; MacIntosh, A.D.; Wong, J.L.; Bielinski, E.A.; Williard, P.G.; Mercado, B.Q.; Hazari, N.; Bernskoetter, W.H. Iron catalyzed CO2 hydrogenation to formate enhanced by Lewis acid co-catalysts. Chem. Sci. 2015, 6, 4291–4299. [Google Scholar] [CrossRef] [Green Version]
- Bielinski, E.A.; Lagaditis, P.O.; Zhang, Y.; Mercado, B.Q.; Würtele, C.; Bernskoetter, W.H.; Hazari, N.; Schneider, S. Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer Supported Iron Catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Hazari, N. Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res. 2017, 50, 1049–1058. [Google Scholar] [CrossRef]
- Anderson, N.H.; Boncella, J.M.; Tondreau, A.M. Manganese-Mediated Formic Acid Dehydrogenation. Chem. Eur. J. 2019, 25, 10557–10560. [Google Scholar] [CrossRef] [PubMed]
- Andérez-Fernández, M.; Vogt, L.K.; Fischer, S.; Zhou, W.; Jiao, H.; Garbe, M.; Elangovan, S.; Junge, K.; Junge, H.; Ludwig, R.; et al. A Stable Manganese Pincer Catalyst for the Selective Dehydrogenation of Methanol. Angew. Chem. Int. Ed. 2017, 56, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.H.; Boncella, J.M.; Tondreau, A.M. Reactivity of Silanes with (tBuPONOP) Ruthenium Dichloride: Facile Synthesis of Chloro-Silyl Ruthenium Compounds and Formic Acid Decomposition. Chem. Eur. J. 2017, 23, 13617–13622. [Google Scholar] [CrossRef]
- Prejanò, M.; Alberto, M.E.; Russo, N.; Marino, T. Hydration of Aromatic Nitriles Catalyzed by Mn-OH Complexes: A Rationalization from Quantum Chemical Investigations. Organometallics 2020, 39, 3352–3361. [Google Scholar] [CrossRef]
- Parise, A.; Muraca, M.C.; Russo, N.; Toscano, M.; Marino, T. The Generation of the Oxidant Agent of a Mononuclear Nonheme Fe(II) Biomimetic Complex by Oxidative Decarboxylation. A DFT Investigation. Molecules 2020, 25, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, T.; Fortino, M.G.; Russo, N.; Toscano, M.; Alberto, M.E. Computational Mechanistic Insights on the NO Oxidation Reaction Catalyzed by Non-Heme Biomimetic Cr-N-Tetramethylated Cyclam Complexes. Int. J. Mol. Sci. 2019, 20, 3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, V. Solvent effects on the reactivities of organometallic compounds. Coord. Chem. Rev. 1976, 18, 225–255. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 2009; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Andrae, D.; Hussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Alberto, M.E. A trispyrazolylborato iron cysteinato complex efficiently mimics the cysteine dioxygenation process: Mechanistic insights. Chem. Commun. 2015, 51, 8369–8372. [Google Scholar] [CrossRef]
- Mazzone, G.; Marino, T.; Piazzetta, P.; Ponte, F.; Prejanò, M.; Sicilia, E.; Toscano, M. Quantum mechanical DFT elucidation of CO2 catalytic conversion mechanisms: Three examples. Int. J. Quantum Chem. 2018, 118, e25572. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.N.; Himo, F.; Maseras, F.; Perrin, L. Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis. ACS Catal. 2019, 9, 6803–6813. [Google Scholar] [CrossRef]
- Marino, T.; Ponte, F.; Mazzone, G.; Sicilia, E.; Toscano, M.; Russo, N. The ability of a zinc pyrrolidine complex to catalyze the synthesis of cyclic carbonates from carbon dioxide and epoxides: A mechanistic theoretical investigation. Dalton Trans. 2018, 46, 9030–9035. [Google Scholar] [CrossRef] [PubMed]
- Piazzetta, P.; Marino, T.; Russo, N.; Salahub, D.R. Direct hydrogenation of carbon dioxide by an artificial reductase obtained by substituting rhodium for zinc in the carbonic anhydrase catalytic center. A mechanistic study. ACS Catal. 2015, 5, 5397–5409. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chim. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
Scheme 1.
Conversion of formic acid in CO2 and H2.

Scheme 2.
The three ligands (on the top) and the three related complexes (on the bottom). The name of the complexes has been retained according to the experimental work [31].
Scheme 2.
The three ligands (on the top) and the three related complexes (on the bottom). The name of the complexes has been retained according to the experimental work [31].

Scheme 3.
The investigated mechanism for the FA to CO2 conversion catalyzed by 2.

Figure 1.
Free energy profile of the FA to CO2 conversion catalyzed by complex 2 at the B3LYP-D3/6-311+G(2d,2p)/SDD/Chlorobenzene level of theory. Energy barriers are depicted in red.
Figure 1.
Free energy profile of the FA to CO2 conversion catalyzed by complex 2 at the B3LYP-D3/6-311+G(2d,2p)/SDD/Chlorobenzene level of theory. Energy barriers are depicted in red.

Figure 2.
B3LYP-D3/6-31+G(d,p) optimized geometries of 2:FA, TS rot and 2:FA’. Distances implicated in chemical event (black) are in Å. Imaginary frequencies are also reported. For clarity, hydrogens are not shown.
Figure 2.
B3LYP-D3/6-31+G(d,p) optimized geometries of 2:FA, TS rot and 2:FA’. Distances implicated in chemical event (black) are in Å. Imaginary frequencies are also reported. For clarity, hydrogens are not shown.

Figure 3.
B3LYP-D3/6-31+G(d,p) optimized geometries of TS1 and TS2. Relevant distances (in black) are in Å. Imaginary frequencies are also indicated. For clarity, hydrogens were not represented.
Figure 3.
B3LYP-D3/6-31+G(d,p) optimized geometries of TS1 and TS2. Relevant distances (in black) are in Å. Imaginary frequencies are also indicated. For clarity, hydrogens were not represented.

Figure 4.
Variation of TS1 barrier as function of implicit solvents: Cyclohexane (CHX) ε = 2.0; Chloroform (CLF) ε = 4.7; CLB (Chlorobenzene) ε = 5.7; THF (Tetrahydrofuran) ε = 7.4; ETH (Ethanol) ε = 24.8; MET (Methanol) ε = 32.6; DMF (Dimethylformamide) ε = 37.2; WAT (Water) ε = 78.3.
Figure 4.
Variation of TS1 barrier as function of implicit solvents: Cyclohexane (CHX) ε = 2.0; Chloroform (CLF) ε = 4.7; CLB (Chlorobenzene) ε = 5.7; THF (Tetrahydrofuran) ε = 7.4; ETH (Ethanol) ε = 24.8; MET (Methanol) ε = 32.6; DMF (Dimethylformamide) ε = 37.2; WAT (Water) ε = 78.3.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marino, T.; Prejanò, M. Dehydrogenation of Formic Acid to CO2 and H2 by Manganese(I)–Complex: Theoretical Insights for Green and Sustainable Route. Catalysts 2021, 11, 141. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010141
AMA Style
Marino T, Prejanò M. Dehydrogenation of Formic Acid to CO2 and H2 by Manganese(I)–Complex: Theoretical Insights for Green and Sustainable Route. Catalysts. 2021; 11(1):141. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010141
Chicago/Turabian StyleMarino, Tiziana, and Mario Prejanò. 2021. "Dehydrogenation of Formic Acid to CO2 and H2 by Manganese(I)–Complex: Theoretical Insights for Green and Sustainable Route" Catalysts 11, no. 1: 141. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010141
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.