
Calcium Hydroxyapatite: A Highly Stable and Selective Solid Catalyst for Glycerol Polymerization




Univ. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181—UCCS—Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(10), 1247; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101247
Submission received: 28 September 2021
/
Revised: 11 October 2021
/
Accepted: 12 October 2021
/
Published: 17 October 2021
(This article belongs to the Special Issue Catalytic Valorization of Glycerol: Strategies and Perspectives)
Abstract
:Calcium-based catalysts are of high interest for glycerol polymerization due to their high catalytic activity and large availability. However, their poor stability under reaction conditions is an issue. In the present study, we investigated the stability and catalytic activity of Ca-hydroxyapatites (HAps) as one of the most abundant Ca-source in nature. A stochiometric, Ca-deficient and Ca-rich HAps were synthesized and tested as catalysts in the glycerol polymerization reaction. Deficient and stochiometric HAps exhibited a remarkable 100% selectivity to triglycerol at 15% of glycerol conversion at 245 °C after 8 h of reaction in the presence of 0.5 mol.% of catalyst. Moreover, under the same reaction conditions, Ca-rich HAp showed a high selectivity (88%) to di- and triglycerol at a glycerol conversion of 27%. Most importantly, these catalysts were unexpectedly stable towards leaching under the reaction conditions based on the ICP-OES results. However, based on the catalytic tests and characterization analysis performed by XRD, XPS, IR, TGA-DSC and ICP-OES, we found that HAps can be deactivated by the presence of the reaction products themselves, i.e., water and polymers.
1. Introduction
Polyglycerols (PGs) are very interesting polyols with a wide range of structures and applications, particularly in the cosmetics, biomedical, and food sectors. PGs are water-soluble, biocompatible, and highly functional materials. Currently, 52% of the PGs market demand is occupied by the short-chain PGs, including PG2 and PG3 (PG2-3) [1].
In the last few years, attention has been paid to alkaline-based catalysts such as earth metal oxides [2,3,4], Ca/La mixed oxides [5,6] and alkaline-modified zeolite [7,8] due to their higher selectivity to PGs compared to acid catalysts [5,9,10,11]. Among the alkaline catalysts, CaO-based catalysts such as CaO [2,12], supported CaO on CNF [3], dolomite (CaO-MgO mixed oxides) [4], and calcined eggshell [13] have shown high catalytic performances with no acrolein formation [9]. Moreover, considering the wide availability of Ca, its low cost, and its absence of toxicity in the case of catalyst leaching into the media, Ca-based catalysts can be especially considered promising for glycerol polymerization reactions [9,12]. However, such catalysts usually exhibit a moderate selectivity to PG2-3. For instance, Kirby et al. [3] reported a 45% selectivity to PG2-3 at full glycerol conversion in the presence of CaO/CNF. Barros et al. [4] also reported 55% selectivity to PG2-3 at glycerol conversion of 90% in the presence of dolomite (CaCO3·MgCO3).
Furthermore, the heterogenous catalysts were generally unstable under polymerization reaction conditions, which induced totally or partially homogeneously-catalyzed conditions due to the activity of leached species. For Ca-based catalysts, this homogenous contribution has been attributed by many groups to the action of colloidal Ca(OH)2 and Ca2+ [2,7,13,14,15]. For instance, Barros et al. [13] reported that 49% of solid CaO-egg shell became homogeneous (i.e., dissolved in the reaction medium under the form of Ca2+) after 24 h of reaction. Thus, the homogeneous contribution in the PG reaction is still an unsolved problem.
Many types of Ca-rich compounds, particularly those derived from organic wastes such as waste fish scales and animal bones, have been successfully used as catalysts [16,17,18,19]. In fact, the mineral phase of bones consists of non-stoichiometric calcium hydroxyapatite (HAp). Hydroxyapatites are widely used in biomedical applications due to their similarity - from a crystallographic point of view and in terms of chemical composition, with human hard tissues such as bones and teeth [20]. Furthermore, due to their interesting properties such as high thermal stability, very limited water solubility, and insolubility in alkaline solutions [21], HAps have also found many applications in catalysis [16,17,18,19,22,23,24], protein separation [25], and in waste-water and soil treatment [21]. Hence, we decided to study heterogeneous catalysts based on Ca hydroxyapatite for the glycerol polymerization reaction due to these advantages.
Calcium hydroxyapatite can be present in various forms;
- I.
- Stochiometric HAp with formula Ca10(PO4)6(OH)2 with a Ca/P molar ratio of 1.67.
- II.
- Deficient HAp (non-stoichiometric) hydroxyapatites with Ca/P < 1.67, which are generally represented by the formula Ca10-x(PO4)6-x(HPO4)x(OH)2-x with 0 < x < 1. Outside this compositional range, the compounds can change to multiphasic mixtures including β-tricalcium phosphate (β-TCP) with a formula of Ca3(PO4)2 with Ca/P ratio of 1.5; octacalcium phosphate (OCP) (Ca8H2(PO4)6⋅xH2O) with a Ca/P ratio of 1.33, calcium hydrogen phosphate (CaHPO4) with a Ca/P ratio of 1 and calcium pyrophosphate (DCP) (β-Ca2P2O7) with a Ca/P ratio of 1 [26,27,28].
- III.
- Over-stoichiometric HAp or rich HAp (HAp-R) with a Ca/P molar ratio above 1.67. Generally, the excess of Ca in HAp-R could lead to the formation of CaO or Ca(OH)2 on the HAp surface [26].
Among the various HAp groups mentioned above, biological HAp has been studied for transesterification reactions by several groups [17,19]. For instance, Farooq et al. [17] studied HAp derived from waste chicken bones as a heterogeneous catalyst utilized in the transesterification reaction of waste cooking oil for biodiesel production. They reported a biodiesel yield of ca. 89% at 5 wt.% of catalyst loading, at a temperature of 65 °C, and a reaction time of 4 h. Furthermore, the catalyst was reused for four cycles with a biodiesel yield of 79%, where the recovered catalyst was calcined after each reaction. The performance decrease observed after the deactivation of the catalyst explained each cycle by deposition of unreacted oil, biodiesel, or glycerol on the active sites at the surface of the catalyst.
With respect to these results, HAp could be a suitable catalyst for glycerol polymerization, a liquid phase condensation reaction at high temperatures. To the best of our knowledge, no study has been reported so far on the catalytic activity of HAps in the glycerol polymerization reaction. Therefore, based on the literature review, we decided to develop new catalysts based on calcium hydroxyapatite, which were assumed to act heterogeneously in the glycerol polymerization reaction.
Thus, we developed several Ca-based HAps catalysts with various Ca/P ratios and studied their catalytic performances on the glycerol polymerization reaction in this work. Based on the catalyst characterizations and the catalytic activities, the catalyst deactivation mechanism has also been studied.
2. Results
2.1. Catalytic Performances
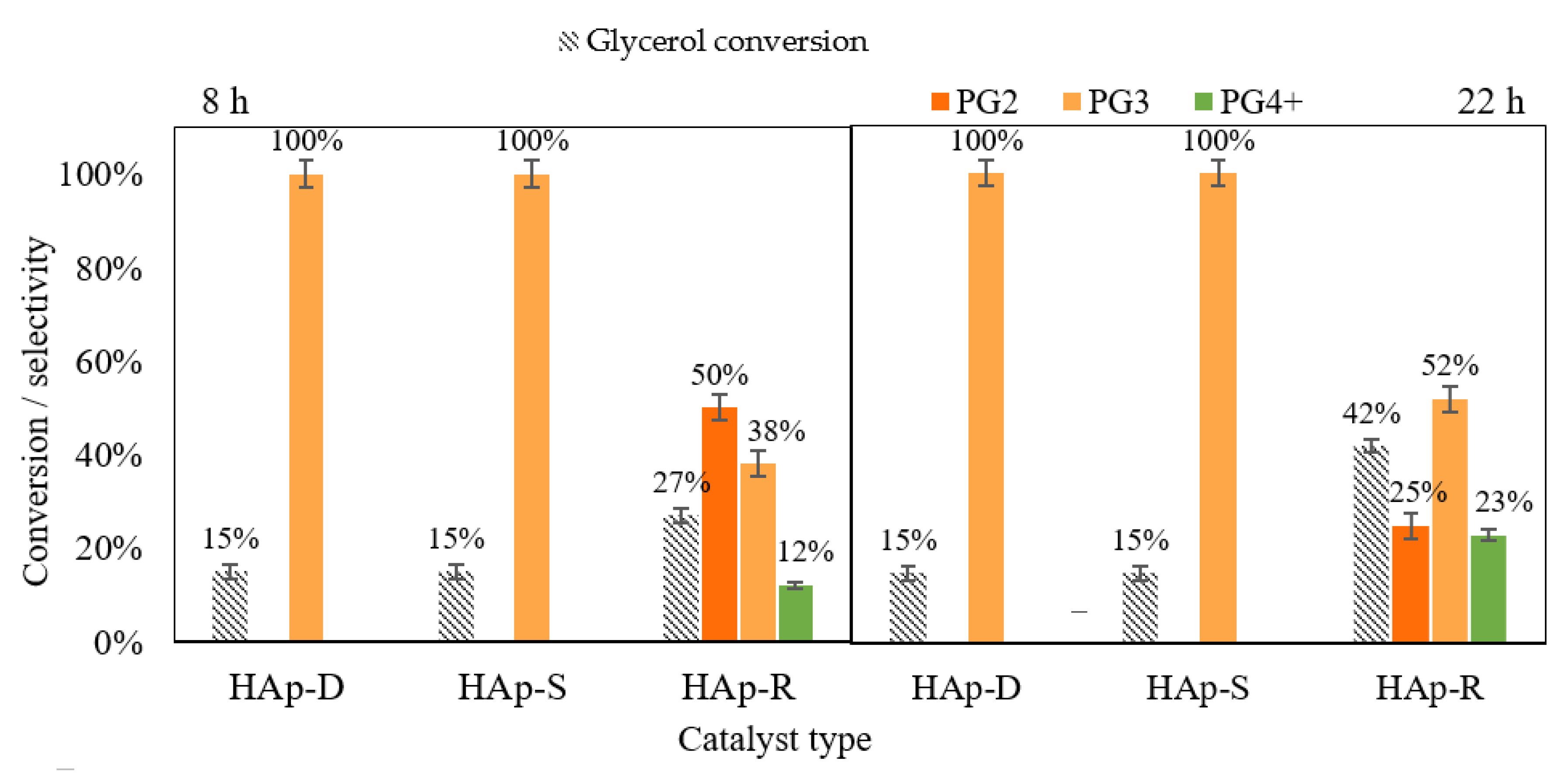
Herein, three Ca-based HAp catalysts have been tested: a stoichiometric HAp (HAp-S) with a theoretical Ca/P molar ratio of 1.67, a deficient HAp (HAp-D) with a theoretical Ca/P molar ratio of 1.45 and a rich-HAp (HAp-R) with a theoretical Ca/P molar ratio of 1.94. The catalytic performances of these three catalysts were evaluated at 245 °C in the presence of 0.5 mol.% of catalyst for 8 h reaction, as shown in Figure 1 (left).
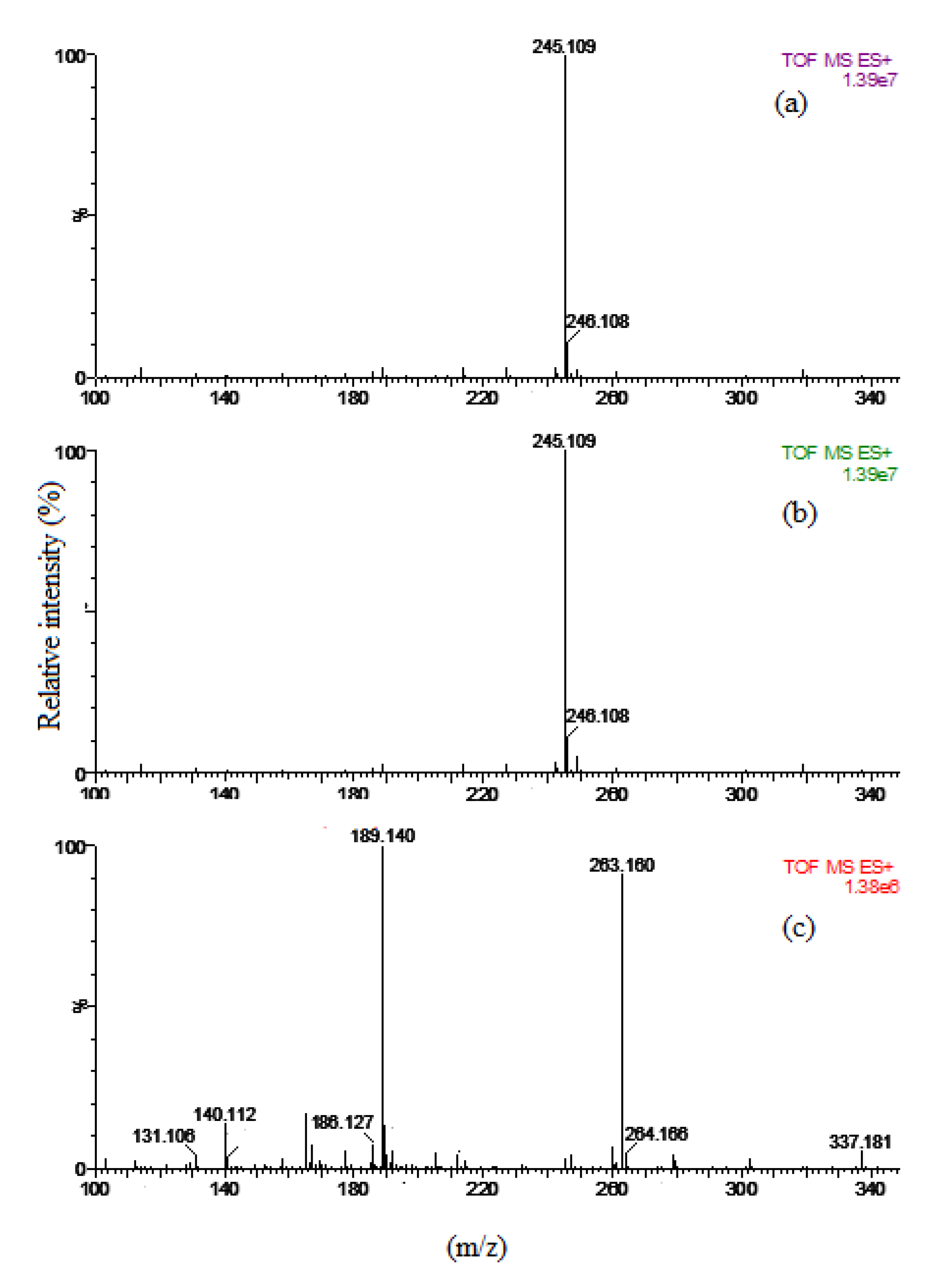
The results showed that HAp-S and HAp-D exhibited the same catalytic performances: 15% glycerol conversion with a full selectivity to PG3. In fact, these catalysts are highly selective to one molecule, which has been identified to be cyclic-PG3 by ESI-MS (Figure 2a,b). Surprisingly, the conversion of HAp-S and HAp-D did not increase when the reaction time was prolonged to 22 h (Figure 1 right).
For the Ca-rich hydroxyapatite (HAp-R), a glycerol conversion of 27% with high selectivity to PG2-3 (88%) was also observed after 8 h of reaction. Moreover, in contrast to HAp-S, the glycerol conversion increased to 42% with 77% of PG2-3 selectivity when the reaction was carried out for 22 h under the same reaction conditions (Figure 1 right). This increase in catalytic performances might also be attributed to the Ca leaching (0.37 mg/g) into the reaction medium, which could cause a partially homogeneous reaction. Furthermore, the HAp-R was also highly selective to linear PG2 and PG3 (as shown in Figure 2c), where the peaks at 189.1 Da and 263.1 Da correspond to linear PG2 and PG3, respectively.
When HAp-D and HAp-S were used as catalysts, low amounts of catalyst leached to the respective reaction media after 8 h of reaction (namely, 1.8% for HAp-D and 0.5% for HAp-S as shown in Table 1). Thus, since no increase in glycerol conversion nor PGs selectivity was observed after 22 h of reaction time (Figure 1), there is no strong evidence indicating that the catalytic performances were related to any homogeneous catalysis provoked by leached species.
The quantity of calcium leached in the reaction medium (Table 1) was also very low in the presence of HAp-R (3.4%) in the same reaction conditions. In contrast, CaO, as a typical Ca-based catalyst, became totally soluble after 1 h of reaction. Thus, HAp-based catalysts are highly stable compared to CaO.
The catalytic results obtained so far showed that among several HAp-based catalysts, the most promising one is HAp-S, both in terms of selectivity to PG3 and stability (very low Ca leaching). Hence, hereafter, the recycling of this catalyst was studied.
2.2. Activity of Recycled Catalyst
The catalytic performance of spent catalysts obtained from HAp-S was assessed upon recovery/reutilization under the same reaction conditions. The spent catalyst was washed with two different solvents: (1) water, labeled HAp-W or (2) EtOH, labeled HAp-Et.
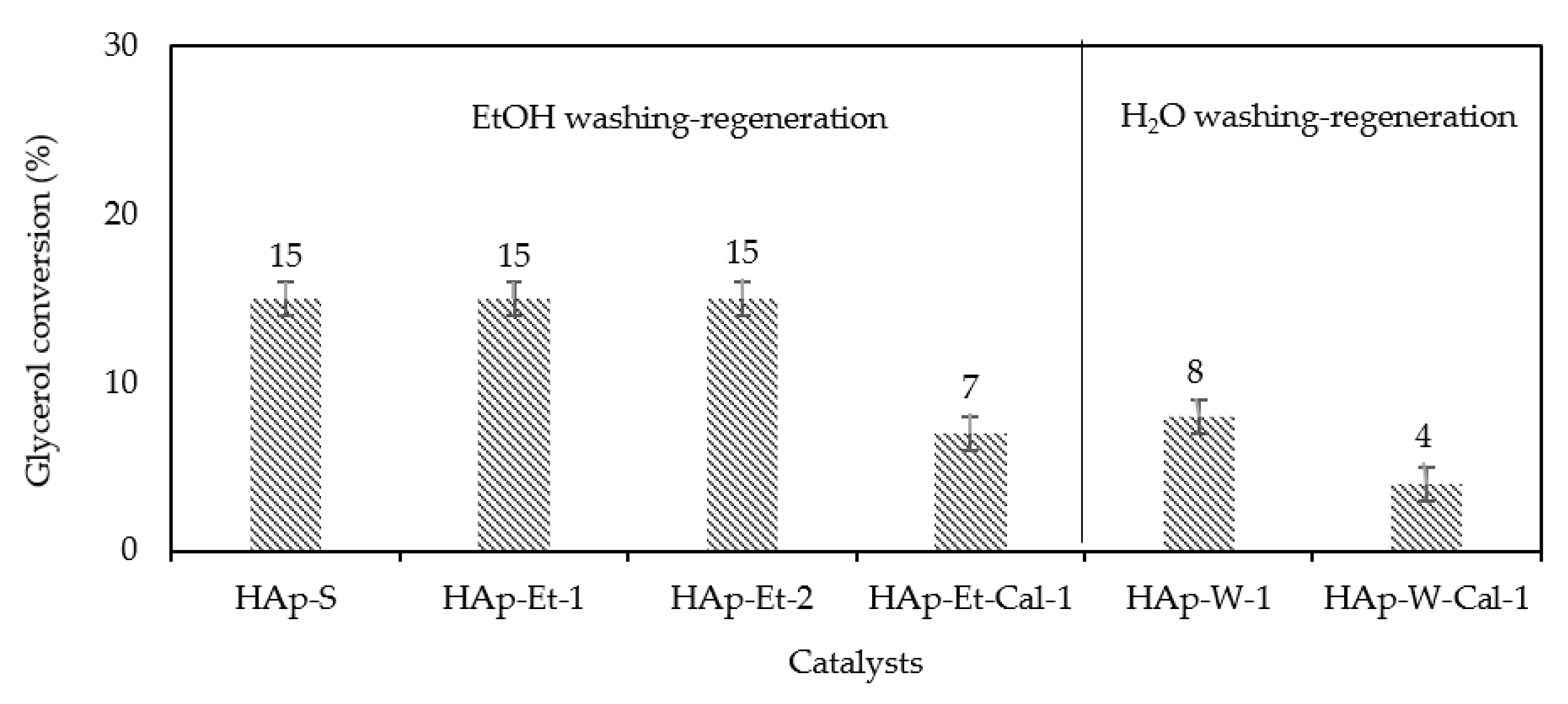
Using ethanol to wash the used catalyst, it was shown that the recovered catalyst from the 1st run reaction, HAp-Et-1, had a glycerol conversion of 15%, similar to that of the fresh HAp-S catalyst. Moreover, the spent catalysts, HAp-Et-1 and HAp-Et-2, were successfully reused in the 2nd and 3rd run reactions, without loss in activity (Figure 3). Most importantly, the reused catalyst (HAp-Et-1 and HAp-Et-2) still exhibited 100% selectivity to PG3.
Surprisingly, when the HAp-Et-1 was recalcined prior to the 2nd run reaction, the glycerol conversion decreased to 7% (HAp-Et-Cal-1). Recalcination of spent catalysts was carried out under static air, which probably caused a change in HAp stoichiometry. This could happen by forming carbonated groups due to the presence of O2 or CO2 in the oven atmosphere. This will be further discussed in Section 3.1.
Unlike HAp-Et-1, the used catalyst washed with water (HAp-W-1) showed a lower glycerol conversion of 8% with a 100% selectivity to PG3 compared to initial HAp-S (15% of conversion) (Figure 3). In this case, the decrease in catalytic performance might be due to the formation of a hydrated layer on the catalyst surface that causes a loss of catalyst activity. Moreover, when the recovered catalyst was recalcined prior to the reaction (HAp-W-Cal -1), lower performances than HAp-W-1 were observed (4% conversion vs. 8%), probably because the hydrated layer was removed but CO2 of the calcination atmosphere created carbonated species. Hence, like in the case of HAp-Et-Cal-1, the catalyst recalcination caused some deactivation.
To understand the deactivation phenomenon after water washing, further characterization of spent catalysts was needed, which will be further discussed.
2.3. Catalysts Characterization
HAp-D, HAp-S, and HAp-R were characterized using several techniques, including BET, XRD, XPS, IR, and TGA to study the influence of the Ca/P ratio on the bulk and surface properties of the catalysts.
2.3.1. Surface Characterization and Elemental Analysis
The specific surface area (SSA) of HAp-D, HAp-S, and Hap-R were measured using BET (Table 2). The HAp-S with 70.4 m2/g developed a higher SSA than HAp-D and HAp-R with 19.4 m2/g and 13.8 m2/g, respectively.
Moreover, the ICP analysis of the catalysts showed that the atomic Ca/P ratios of bulk HAp-S, HAp-D, and HAp-R were 1.66, 1.62, and 1.78, respectively, with ca. 36 wt.%, 36 wt.%, and 38 wt.% of calcium, respectively (Table 2). The difference between the experimental and the theoretical Ca/P atomic ratios for HAp-D (Table 2) suggests that no other apatite such as β-tricalcium phosphate, octacalcium phosphate, or calcium pyrophosphate was formed.
In addition, C analysis showed the presence of some carbon in HAp-D, HAp-S, and HAp-R. The presence of carbon suggests that some atmospheric CO2 was absorbed during the synthesis process, as already reported in the literature [26,27,29]. However, since the HAps were calcinated at 700 °C, this result suggests that only a partial release of CO2 could occur at this temperature level.
For the used catalysts washed with EtOH, the BET analysis showed a decrease in the surface area (36.6 m2/g for HAp-Et-1 and 38 m2/g for HAp-Et-2) compared to HAp-S (73.7 m2/g). The C content was also increased for HAp-Et-1 (1.96 wt.%) and HAp-Et-2 (2.38 wt.%) compared to HAp-S (0.11 wt.%) as shown in Table 2. These results suggest that the carbon deposition caused a decrease in the specific surface area of the recovered catalysts. However, it had no effect on its performance. Moreover, the recalcination of catalyst partially removed the C in HAp-Et-Cal-1, as expected. However, the higher amount of carbon in HAp-W-1 compared to HAp-Et-1 (2.13 wt.% vs 1.96 wt.%) confirmed that ethanol was more efficient than water to remove adsorbed species.
2.3.2. XRD
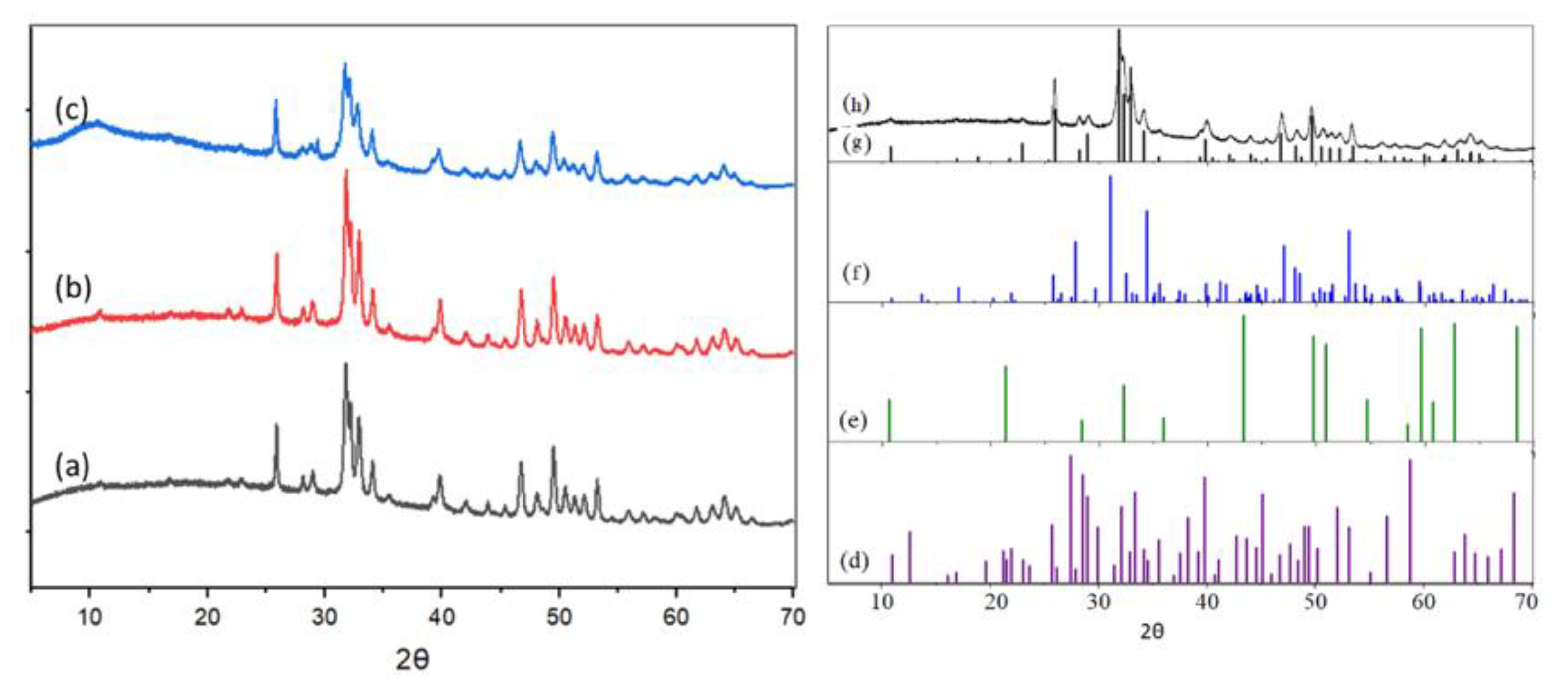
X-ray diffractograms of HAp-S, HAp-D, and HAp-R are presented in Figure 4(a–c). The characteristic peaks of calcium hydroxyapatite (PDF 04-014-8416) were observed for all these HAps.
Moreover, no peak was assigned to β-tricalcium phosphate (β-TCP) (PDF 00-055-0898); octacalcium phosphate (OCP) (PDF 00-044-0778), nor to calcium pyrophosphate (DCP) (PDF 00-044-0762) were observed (Figure 4 right). In addition, for HAp-R, no other crystalline phase, namely CaO with 2θ at 32.2°, 37.4° and 53.9° (PDF 01-070-4066) or Ca hydroxide with 2θ at 18.14°, 28.7°, 34.2°, or 50.8° (PDF-00-087-0673) were observed. However, it has to be noted that the main XRD peak for CaO at 2θ = 32.2° or Ca(OH)2 at 34.2°, could be hidden by intense peaks of HAp-R between 2θ = 31° and 35°.
Thus, it is impossible to distinguish the stoichiometric and non-stoichiometric HAps by XRD, as reported in the literature [27,30].
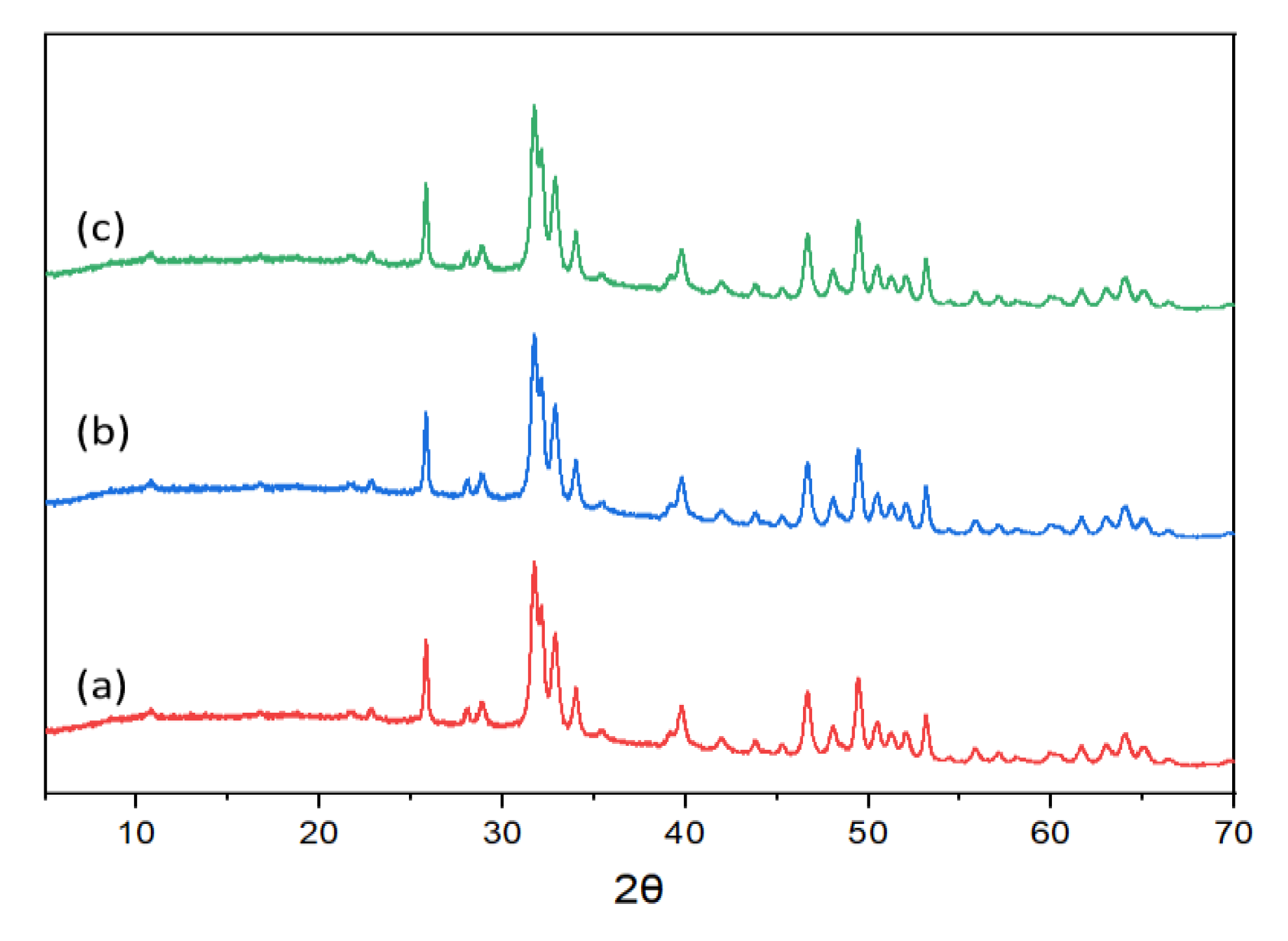
For the spent catalysts, either washed with water or ethanol, the characteristic peaks of calcium hydroxyapatite (PDF 04-014-8416) were also observed (Figure 5). The XRD results revealed that the crystalline structure of recovered HAps, including HAp-Et-1, HAp-W-1, and HAp-Et-Cal-1 remained unchanged after reaction.
Further, these catalysts were also analyzed by the IR technique to study the carbon bonds, carbon-carbon, and carbon-oxygen bonds, on the spent catalysts.
2.3.3. IR Analysis
The structures of the HAps were further studied by IR analysis. For HAp-S, HAp-D, and HAp-R, the IR spectra confirmed the presence of OH and phosphate groups.
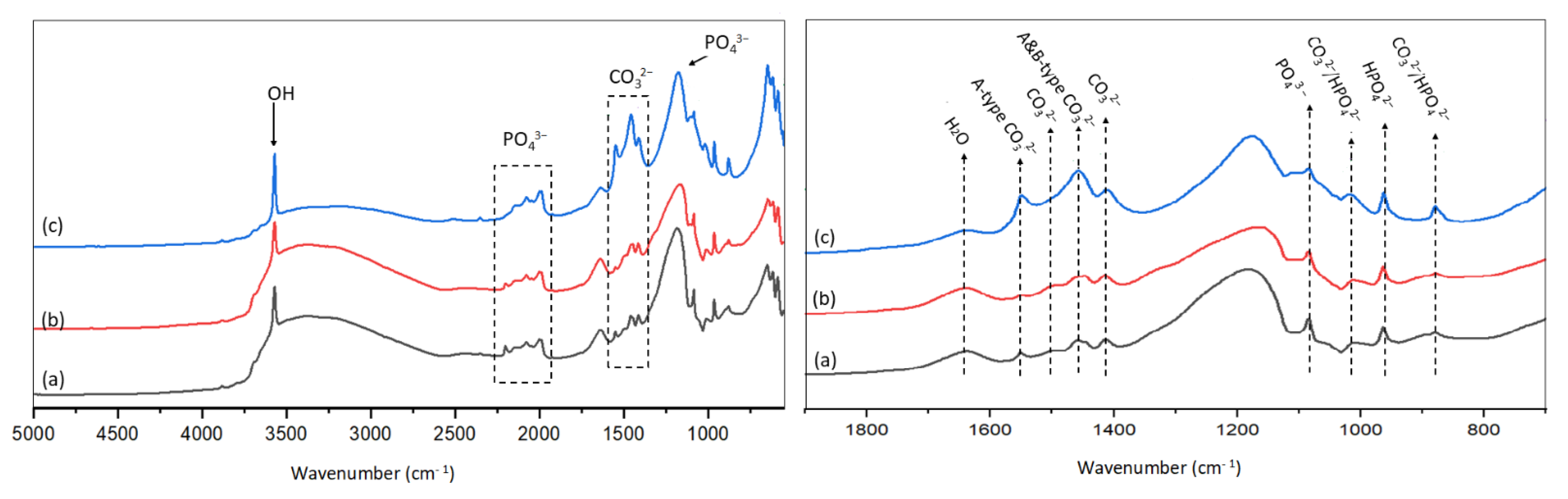
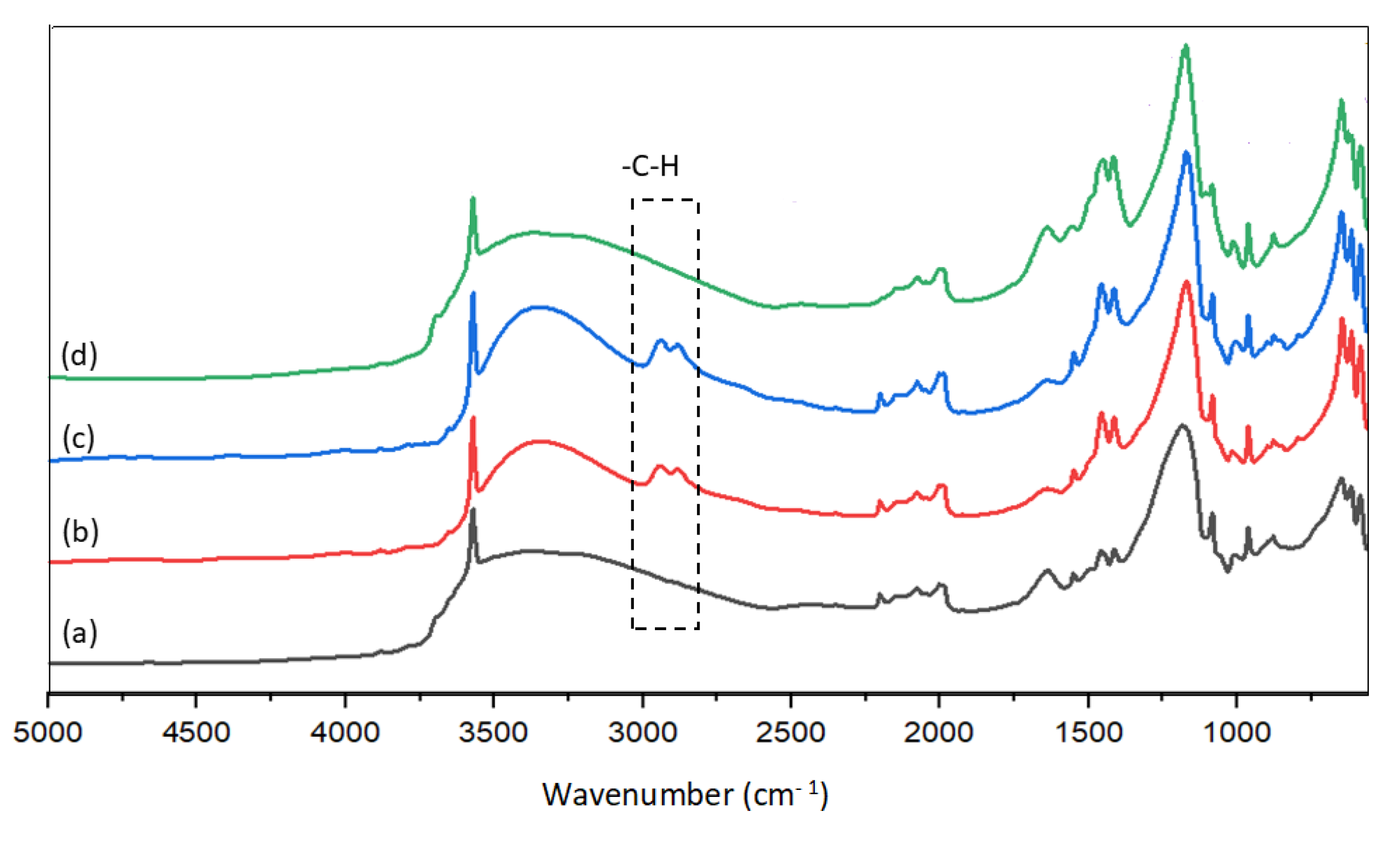
As shown in Figure 6a–c, the peak at 3572 cm−1 is assigned to the OH groups in HAp structures; and the peaks at 652 cm−1, 1084 cm−1, 1173 cm−1, and four peaks in the region of 1960 to 2220 cm−1 are assigned to the vibration of PO43− in HAps [27,29,31].
Furthermore, the bands in the regions of 1370 cm−1 and 1580 cm−1 with four peaks at 1413 cm−1, 1456 cm−1 (A and B-type), 1500 cm−1, and 1550 cm−1 (A-type) were attributed to carbonate groups [27,29,32]. The IR spectra for HAp-D and HAp-S (Figure 6 d–f) showed the presence of carbonated groups in these catalysts, where the intensity of two peaks at 1413 cm−1 and 1456 cm−1 were lower compared to HAp-R (Figure 6f). The presence of CO32− in non-carbonated HAps, is consistent with the XPS results (Section 2.3.3) and ICP results (Table 2).
Moreover, three peaks at 878 cm−1, 963 cm−1, and 1017 cm−1 assigned to HPO42− [27,29,33] were observed for all HAp samples (Figure 6e–f). For the deficient HAp, the presence of HPO42− was expected. However, for other HAps, the presence of HPO42− can be explained by A and B-type carbonate substitution.
In conclusion, regardless of the Ca/P molar ratio in HAps, they all contain PO43−, HPO42−, CO32− and OH- groups.
The recovered catalysts, including HAp-Et-1, HAp-W-1, and HAp-W-Cal-1, were also analyzed using IR (Figure 7b–d). Similarly to HAp-S as the precursor catalyst (Figure 6a), the peaks assigned to OH−, to phosphate groups, and carbonate groups (between 1377 cm−1 and 1580 cm−1) were also observed for all the recovered HAps (See Section 2.3.3). However, the intensity of peaks at 1413 cm−1, 1456 cm−1 (A and B-type) attributed to carbonate significantly increased for the spent catalysts compared to HAp-S, in the following order: HAp-S << HAp-Et-1 < HAp-W-1 < HAp-W-Cal-1.
Besides, two peaks observed at 2880 cm−1 and 2940 cm−1 (Figure 7b-c) are attributed to the -C-H bond on spent HAps. -CH bonds could form due to the presence of glycerol and/or PGs in the reaction, which is consistent with the decreased specific surface area (Table 2). These peaks disappeared after recalcination of the spent catalyst (HAp-Et-Cal-1) (Figure 7d).
Furthermore, the peak at 3400 cm−1 (3000–3500 cm−1), attributed to the H2O, became more intense for the non-calcined spent catalysts (Figure 7b–c), which suggests that the latter became hydrated during the reaction.
2.3.4. XPS
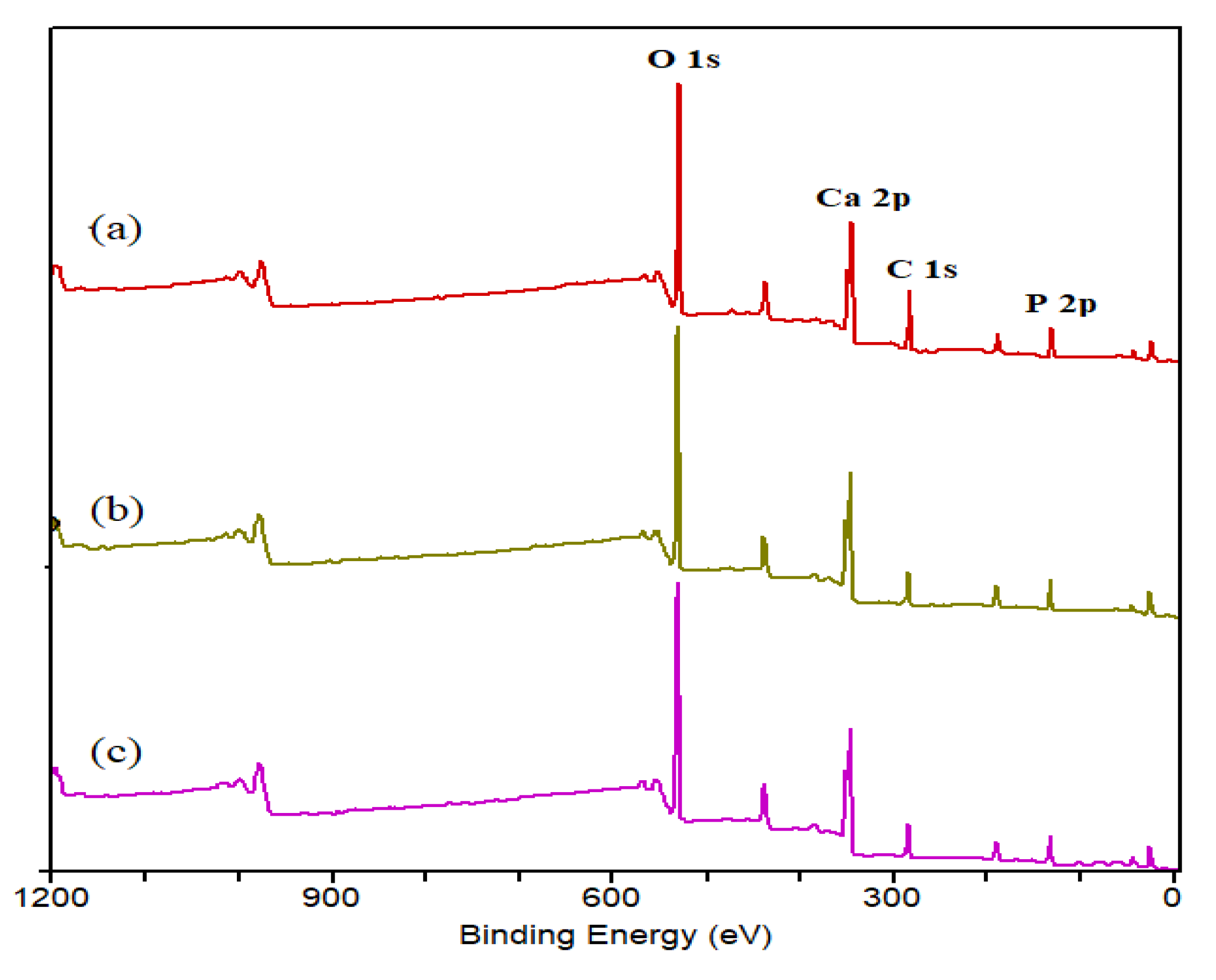
XPS analyzed the HAp-D, HAp-S and HAp-R samples to determine their surface compositions, Ca/P molar ratio, and oxidation state of the elements. The XPS spectra for these catalysts, showed in Figure 8, indicate the presence of Ca, P, O, and C elements, as was expected.
The peaks at 284.8 eV, 285.8 eV, and 288.6 eV corresponding to C 1s are attributed to the organic C (C-C), Ca-OH-C (C-O), and CO3, respectively (not shown), where the peak at 284.8 eV was used to calibrate the spectra. A considerable concentration of carbon contamination on the HAps’ surface can originate from the vacuum of the XPS chamber, which is generally known as “adventitious carbon” [34].
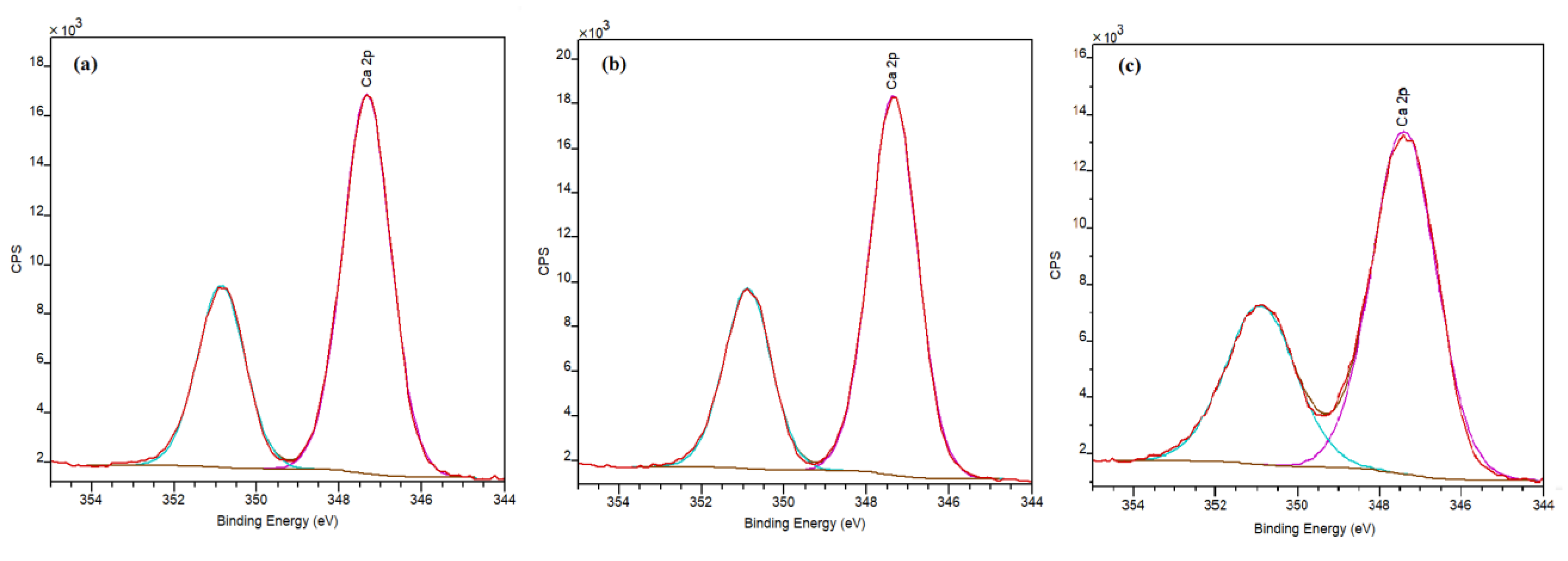
As shown in Figure 9, for HAp-D and HAp-S, a well-defined doublet with two components Ca 2p1/2 (347.4 eV) and Ca 2p3/2 (350.9 eV) is observed, which is attributed to Ca involved in bonds characteristic of HAps [30,35]. However, the Ca 2p spectrum of HAp-R suggests that the environment of Ca ions on the surface of HAp-R is not similar to that in HAp-S and HAp-D.
For all HAps, the O 1s envelope (not shown) was fitted with two peaks at 531.1 eV and 532.6 eV, attributed to O-Ca and O-P (or O-H) in HAps structure, respectively [35,36]. It should be noted that the peak assigned to O-C cannot be distinguished in the fitted peaks; however, the presence of carbonate peak (288.6 eV) already confirmed the presence of carbonate on the HAp surface.
Furthermore, the atomic percentages of Ca, P, O, and C are presented in Table 3. These ratios represent the atomic proportion on the surface layer of approximately 10 nm of HAps’ surface. The Ca/P molar ratio for HAp-D, HAp-S, and HAp-R were 1.4, 1.36, and 1.48, respectively. The lower Ca/P ratios obtained on the surface compared to Ca/P ratios obtained for the bulk HAps by ICP (Table 2) can be due to the carbonate adsorption on the surface, which caused a change in stoichiometries. This was in agreement with previous studies [31,36] and indicated that Ca species are less exposed on the surface.
2.3.5. Thermal Analysis
To study their thermal behaviors, TGA was performed on the non-calcined catalysts, including HAp-D, HAp-S, and HAp-D.
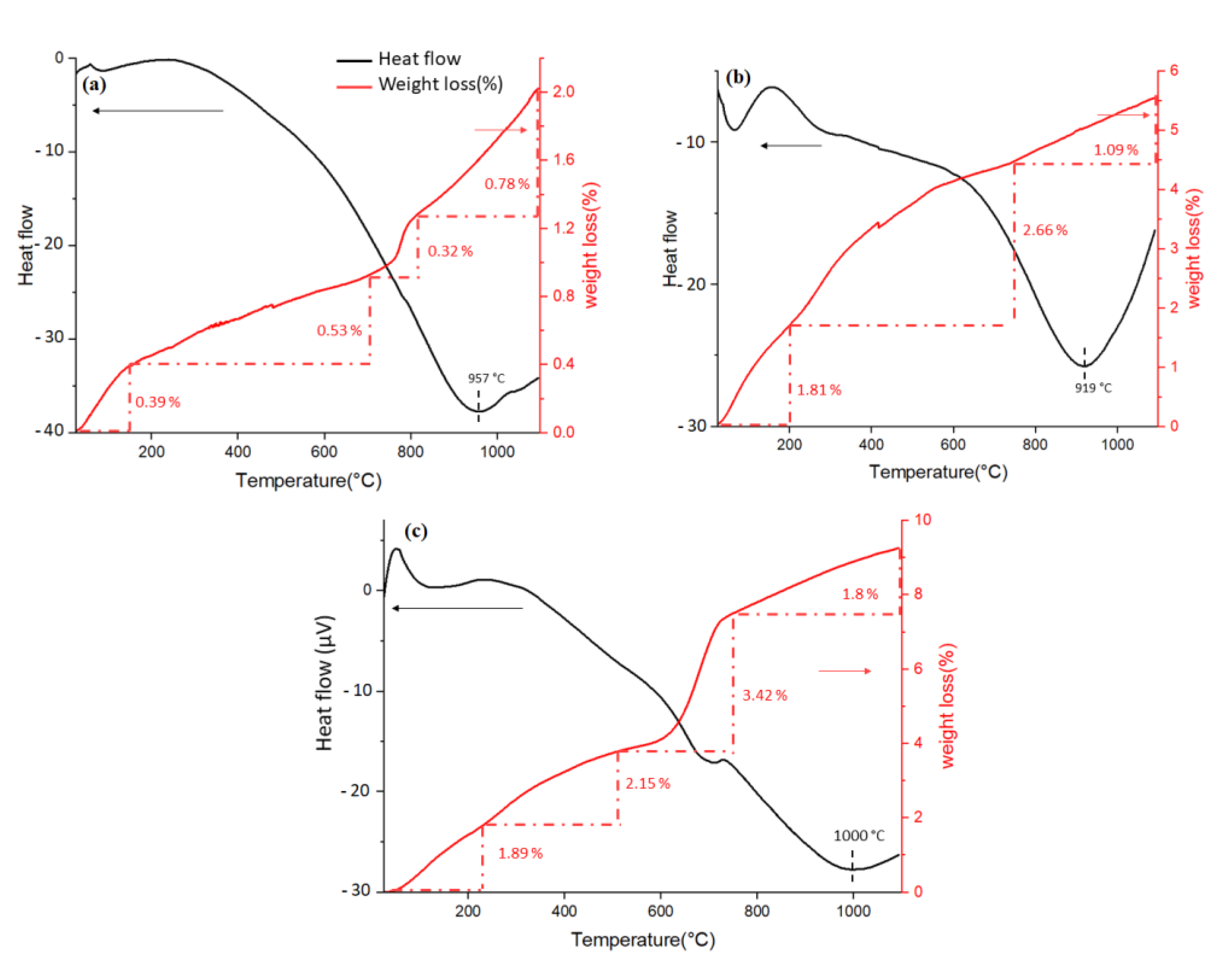
The TGA curve for HAp-D showed a total weight loss of 2.02 wt.% between room temperature (RT) and 1100 °C. The first weight loss from RT to 140 °C can be attributed to the release of H2O from the sample surface, followed by two steps of mass loss of 0.53 wt.% and 0.32 wt.% up to 800 °C (Figure 10a). The final mass loss of 0.78 wt.% occurred between 800 °C and 1100 °C, where an endothermic peak at 957 °C associated with this weight loss was also observed in the DSC curve.
Similarly, the TGA curve for HAp-S showed a three steps weight loss with a total loss of 5.56 wt.% (Figure 10b). The first weight loss (1.81 wt.%) from RT to 214 °C, followed by a further weight loss of 2.66 wt.% up to 743 °C, suggests the release of H2O and CO2 from the sample surface. The final weight loss of 1.09 wt.% with a corresponding endothermic peak at 919 °C was recorded between 743 °C and 1100 °C.
For HAp-R, the weight loss was observed in 4 steps. The first two mass losses of 1.89 wt.% and 2.15 wt.% were recorded between RT and 587 °C, corresponding to the desorption of H2O and CO2 from the surface. The further weight loss of 3.42 wt.% was recorded between 587 °C and 745 °C. An endothermic peak at 705 °C, which corresponds to this mass loss, suggests that decarboxylation of HAp-R might have occurred at this temperature range. Finally, HAp-R lost 9.26 wt.% when heated up to 1100 °C, as shown in Figure 10c.
For these samples, the large, broad endothermic peak at the range of 900 to 1100 °C suggests a phase transition of HAps to other apatites such as OCP, DCP, and TCP, meaning that the chosen calcination temperature (700 °C) was optimal.
3. Discussion
The results of catalytic tests showed that HAp-D and HAp-S exhibited an excellent selectivity to PG3 (100%), where the glycerol conversion was 15%. The same catalytic activity for these two catalysts could be due to their similar Ca/P bulk molar ratios as measured by ICP-OES (Table 2) or the similar Ca surface content for HAp-S and HAp-D (ca. 36 wt.%) as obtained by XPS (Table 3). The relatively low glycerol conversion of 15% can be due to the blockage of the active sites; otherwise, the conversion would still increase with reaction time.
More interestingly, these catalysts also showed good stability during the glycerol polymerization reaction, where 98% and 99.5% of HAp-D and HAp-S, respectively, remained as solid in the reaction media after 8 h of reaction. However, the similar catalytic performances after 8 and 22 h reaction for the catalysts (Figure 1) suggested that the catalysts deactivated during the reaction.
On the other hand, a higher glycerol conversion was obtained in the presence of HAp-R (27%), in the same reaction conditions, where the catalyst showed good selectivity to linear PG2-3 of 88%. An increase in the glycerol conversion (42%) with a selectivity of 77% to PG2-3, after 22 h reaction, suggested that increased catalytic performance could influence Ca leaching. However, 96.5% of the catalyst was still solid based on the ICP results.
So far, HAp-D and HAp-S showed better stability compared to HAp-R, while HAp-R exhibited a higher glycerol conversion in the same reaction condition, where the three catalysts were highly selective to PG2-3 and highly stable compared to CaO.
The results of recycling experiments also showed that used HAp-S washed by EtOH (HAp-Et) was successfully reused in the reaction, where it exhibited a glycerol conversion of 15% after 3rd run, similar to fresh HAp-S (15%). In contrast, HAp-W lost its activity when reused for the 2nd run of reaction (conversion of 7% vs. 15%). These results suggested that the catalyst was deactivated upon hydration.
Furthermore, the IR and C analyses confirmed that the catalysts after reaction became carbonized with the formation of CO32− and also contained -CH groups. In addition, IR analysis showed that the -CH groups were removed after recalcination of the catalyst (HAp-Et-Cal-1). This result is consistent with C analysis, where the C content decreased from 1.96 to 0.37 wt.% in HAp-Et -1 and HAp-Et-Cal-1, respectively.
However, the catalytic results proved that -CH groups did not affect catalytic activity when the regenerated catalyst was reused without any further thermal treatment. However, -CH groups could convert to CO32− under calcination treatment and decrease the activity of the catalyst (HAp-Et-Cal).
As a short conclusion for this part, the XRD patterns for HAps, including HAp-S, HAp-D, and HAp-R confirmed the formation of calcium hydroxyapatite structures, where no other crystalline phase was observed. The XRD results were in good agreement with IR analysis, where the IR spectra exhibited the characteristic peaks of hydroxyapatite, including hydroxyl and phosphate groups for all HAps. In addition to that, the presence of CO32− and HPO42− were also observed in IR spectra. The IR analysis agreed with C analysis, where it showed the presence of carbon in HAps.
In addition, the XPS spectra for HAp-D, HAp-S, and HAp-R, confirmed the presence of less Ca exposed on the surface of the catalysts compared to the bulk. This can be due to the presence of various groups, including CO32− and HPO42− observed by IR, which changed the surface stoichiometries.
Moreover, the thermal analysis showed 2 to 10 wt.% of weight loss for these catalysts, which could be attributed to the release of H2O and CO2. The TGA results were also in good agreement with C analysis, where it confirmed that calcination at 700 °C partially removed the carbonates in the HAps’ structures. Thus, since the results of catalyst characterization confirmed the presence of unexpected carbonates in HAps, its effect on the catalyst activity must be studied.
3.1. Deactivation Mechanisms
Herein, based on the catalytic performances results and the catalyst characterizations, we focus on the deactivation of the HAps.
In brief, the HAps can be deactivated due to the following reasons:
- i.
- Physical deposition of glycerol and/or reaction products/polymers;
- ii.
- H2O interaction with Ca2+ on the surface;
- iii.
- Carbonate formation.
These three hypotheses were examined by performing the following experiments.
3.1.1. Physical Deactivation
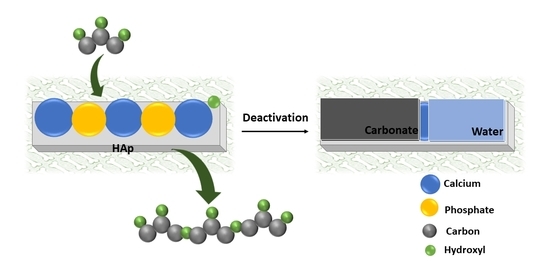
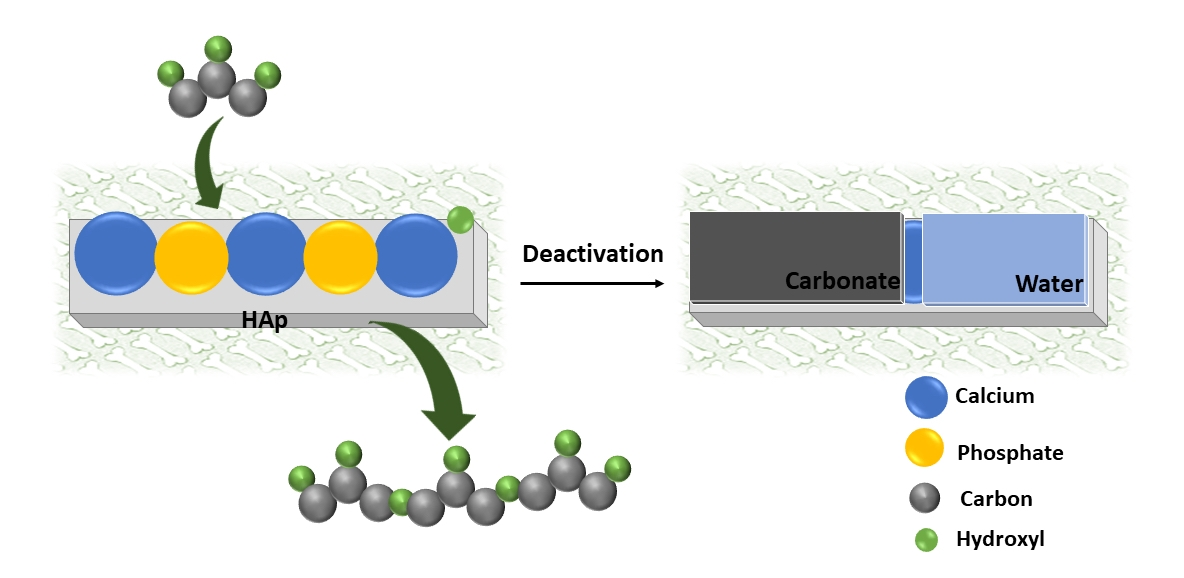
As aforementioned in Section 2.1, HAp-D and HAp-S were deactivated after 8 h of reaction. Here, we assumed that the formed PGs could poison the catalyst surface by physical interaction with the catalyst. This hypothesis was examined by performing a reaction with HAp-S as the catalyst in the presence of 3 wt.% of a PG3 standard. In this case, no glycerol conversion was observed. This result proved that the catalyst’s surface was poisoned by PGs (as illustrated in Scheme 1).
However, as previously observed, when EtOH regenerated the spent catalyst, the PGs layer was washed out, and the catalyst recovered its activity (see Section 2.2).
3.1.2. Water Deactivation
This hypothesis of water deactivation was assessed by performing a reaction in the presence of 0.5 wt.% of extra water in the reaction medium. The results revealed that the catalytic activity dropped to 5% of glycerol conversion (vs. 15%). Moreover, when H2O was used to regenerate the spent catalyst (see Section 2.2), the catalyst’s surface became more hydrated, as shown by the IR analysis (Figure 7), which caused a drop in the activity.
3.1.3. Carbonate Formation
In addition to physical deactivations, the catalyst can also be deactivated by carbonate formation. This hypothesis was examined by performing a reaction with HAp-S as the catalyst in the presence of CO2 atmosphere. In this case, very low glycerol conversion (6%) was observed. This result proved that the catalyst was deactivated due to the carbonate formation on the surface. In fact, the carbonation probably changed the Ca nature through CaCO3 formation and caused a reduction in catalytic performances. We also previously reported that CaCO3 had a low conversion compare to other catalysts [12].
Moreover, the -CH bonds on the spent catalysts (Figure 7b,c) can also deactivate the catalysts upon conversion to carbonate during calcination (CaCO3 formation). For instance, the catalytic performances of HAp-Et decreased when it was recalcined under static air (HAp-Et-Cal), where -CH bonds were removed after thermal treatment and converted to CO3. This phenomenon was confirmed by IR analysis (Figure 7d), where the intensity of carbonate peaks was increased by removing the -CH peaks.
4. Materials and Methods
4.1. Materials
CaO and Ca(OH)2 were purchased from Alfa Aesar. Glycerol (99.5% purity), Ca(NO3)2.4H2O, (NH4)H2PO4, NH4OH (25–30% solution), ethanol (99.5% purity), and were purchased from Sigma-Aldrich. All the chemicals were used as such without any pre-treatment or purification.
4.2. Catalyst Preparation
Stoichiometric hydroxyapatite (HAp-S) with a theoretical formula of Ca₁₀(PO₄)₆(OH)₂ was synthesized by adding dropwise 60 mL of an aqueous solution containing 0.033 mol (7.882 g) of Ca(NO3)2.4H2O to 200 mL of a 0.0198 mol (2.273 g) of NH4H2PO4 solution placed under stirring at 70 °C. The pH of the solution was adjusted to 10 prior to mixing by adding a 25–30% NH4OH solution. The formed precipitate was centrifuged and washed with milli-Q H2O several times before drying at 80 °C for 48 h. Then, the obtained white solid was calcined at 700 °C with a ramp of 10 °C/min under static air for 4 h.
Deficient hydroxyapatite (HAp-D) with a theoretical formula of Ca8.7(PO4)4.7(HPO4)1.3(OH)0.7 was synthesized by a method similar to the HAp-S by adding dropwise 60 mL of an aqueous solution containing 0.033 mol (7.882 g) of Ca(NO3)2.4H2O to 200 mL of a 0.023 mol (2.64 g) of (NH4)H2PO4 solution.
Ca-rich hydroxyapatite (HAp-R) with the theoretical formula of Ca11.62(PO4)6(OH)2.62 was synthesized by an impregnation method similar to methods described previously in the literature [41,42]. To synthesize HAp-R, 4.9 g of Ca(OH)2 (0.066 mmol) were mixed in 145 mL of milli-Q H2O as a solvent, where the pH of the initial solution was 12.9. Then, 2.32 mL of an 85 % of H3PO4 solution (0.0341 mmol) were added very slowly to adjust the pH to 4. The slurry mixture was heated at 80 °C for 48 h, before washing with milli-Q water for several times and drying at 80 °C for 72 h before calcination at 700 °C with a ramp of 10 °C/min for 4 h.
Calcined calcium oxide (CaO) was freshly prepared by performing a high-temperature treatment of calcium nitrate tetrahydrate (Ca(NO3)2.4H2O) under static air in a calcination oven, where the nitrate salt was heated to 700 °C with a ramp of 10 °C/min for 2 h.
4.3. Catalytic Test
Then, 218 mmol (20 g) of pure glycerol were placed in a 100 mL Schlenk tube in the presence of 0.5 mol.% of catalyst (based on the number of moles of glycerol), including CaO, HAp-S, HAp-D, and HAp-R. The reaction took place at 245 °C under N2 atmosphere at a stirring rate of 800 rpm. After 8 h of reaction, the catalyst was separated from the reaction medium by centrifugation and washed with ethanol or water. The collected catalysts were characterized after drying under static air at 80 °C for 4 h.
4.4. Glycerol and Polyglycerol Analyses
The analysis of glycerol was performed using high-performance liquid-phase chromatography (HPLC) equipped with a refractive index (RI) detector. A Bio-Rad (Des Plaines, IL, USA) column (300 mm × 7.8 mm) was used, and the eluent was a 5 mM H2SO4 solution fed at 0.5 mL/min flowrate. The glycerol conversion was calculated using Equation (1):
PGs analyses were conducted by direct infusion Electrospray Ionization Mass Spectrometry (ESI-MS) in a Waters ESI-MS/MS SYNAPT G2-Si HDMS (High Definition Mass Spectrometer) (USA). ESI was set in the positive ion mode, the heated capillary temperature was 300 °C, the spray voltage 3 kV and the capillary voltage 60 V.
Although quantitative analysis required a pure PG standard for each product, whereas the available standards were a mixture of PGs, a relative PG selectivity was chosen to evaluate the catalysts’ performances.
Relative PG selectivity was defined as the ratio of the intensities of the ESI-MS peaks of the di- and tri- glycerol’s (PG2-3) or the tetra-, penta- and hexa-glycerol’s (PG4+) divided by the sum of the intensities of all the PGs peaks detected, as shown in Equations (2) and (3):
4.5. Characterization of Catalyst
4.5.1. BET
The Brunauer–Emmett–Teller (BET) model serves as the basis for the measurement of the specific surface area of materials. The samples were outgassed at 130 °C in vacuum for 3 h before plotting the N2 physisorption isotherms at liquid nitrogen temperature on a TriStar II Plus gas adsorption analyzer (Micromeritics, Norcross, GA, USA). The specific surface areas were evaluated with the BET model over the P/P0 = 0.05–0.30 range.
4.5.2. XRD
X-ray diffraction patterns of the solid catalysts were recorded on a D8 Discover X-ray Diffractometer from Bruker using an X-Ray tube with the Cu (Kα) radiation (λ = 1.54060 Å). The diffraction angle 2θ was taken in the 10–70° range with steps of 0.01° per second.
4.5.3. XPS
XPS spectra were collected on an Axis UltraDLD Kratos (U.K) spectrometer using the monochromatic Al Kα radiation (hν = 1486.6 eV) as the excitation source. The calibration of the XPS spectra was made using the carbon C1s reference peak at 284.8 eV.
4.5.4. Infrared Spectroscopy (IR)
Infrared spectroscopy (IR) spectra were recorded on a Fourier transform infrared spectrometer (FTIR), Tensor 37-HTS-XT from Bruker (Mannheim, Germany). An MCT (Mercury-Cadmium-Telluride) photoelectric detector cooled by liquid N2 was used in reflection mode. The spectra were recorded by accumulating 16 scans with a resolution of 4 cm−1.
4.5.5. Thermal Analyses
Thermo Gravimetric (TG) and Differential Scanning Calorimetry (DSC) were carried out using a Setaram Labsys instrument (France). Continuous heating was applied from 25 to 1100 °C with a heating rate of 10 °C min−1 under 30 ml/min nitrogen flow.
4.5.6. Inductively Coupled Plasma-optic Emission Spectroscopy (ICP-OES)
These analyses were performed on a 720-ES ICP-OES from Agilent (USA) with axially viewing and simultaneous CCD detection. For liquid samples from reaction media, 20 mg of liquid samples were prepared by diluting them with 4 mL of milli-Q water. Prior to the analysis of the solid samples, an acid digestion method was applied for the total dissolution of the catalysts before analysis. 10 mg of each sample were digested with 2 mL of aqua regia (0.5 mL HNO3 + 1.5 mL HCl) and mixed under sonification for 15 min before being kept overnight at ambient temperature before analysis.
4.5.7. Carbon Analysis
The carbon amount of HAps catalysts was determined using a Thermo Scientific (Waltham, MA, USA) FlashSmart automated analyzer. The samples were weighted in tin containers and introduced into the combustion reactor. The reactor operated with dynamic flash combustion of the sample at 950 °C, where the C was detected as CO2. The resultant gases were separated on a packed column heated at 60 °C in an oven and detected by a thermal conductivity detector (TCD).
5. Conclusions
In this paper, the catalytic performances of three calcium-based hydroxyapatites: one stoichiometric, one deficient, and one rich HAps with different theoretical Ca/P molar ratios were assessed for glycerol polymerization. The catalytic tests showed that HAp-D and HAp-S exhibited similar performances, 15% of conversion with a total selectivity to PG3 (100%) at 245 °C after 8 h of reaction. HAp-R also exhibited a very good selectivity to linear PG2-3 (88%) in the same reaction conditions but with a slightly higher activity (27% glycerol conversion instead of 15%).
The same catalytic performances obtained for HAp-D and HAp-S were explained by their similar bulk and surface atomic Ca/P ratio, as observed by ICP and XPS. The higher catalytic activity of HAp-R was explained by a higher atomic Ca/P ratio than HAp-S and also by the higher amount of Ca leached into the reaction medium (homogeneous contribution). However, this leaching was very low compared to what is observed for CaO.
Further, these catalysts were characterized by XRD, IR, XPS, TGA-DSC, and ICP-OES. As said above, the elemental analysis showed that HAp-D and HAp-S had a close bulk Ca/P molar ratio, 1.62 and 1.66, respectively, while HAp-R had a higher Ca/P molar ratio of 1.78. However, XRD analysis confirmed that these catalysts had the crystallized structure of Ca hydroxyapatite, despite the difference in their theoretical Ca/P ratios. Furthermore, XPS analysis showed the Ca/P ratios on the surface of the catalysts were lower than the bulk ones. Hence, less Ca was exposed at the surface of catalysts. Moreover, IR analysis brought new insight into the HAps’ structure. The IR analysis confirmed that the HAps contain CO32− and HPO42−, PO43− and OH− groups in their structure. The intensity of carbonate groups in IR was in good agreement with elemental analysis, which was HAp-R (0.8 wt.%) >> HAp-S (0.111 wt.%) ~ HAp-D (0.108 wt.%). The thermal analysis also confirmed that the release of CO2 gradually continued up to 1100 °C for these catalysts.
Thus far, based on the results, we proposed and examined the three following hypotheses that could explain the catalyst deactivation: (i) Physical deposition of PGs, (ii) water deactivation, and (iii) carbonate formation. Finally, the results confirmed that HAps were mostly deactivated by the presence of products, i.e., “PGs” and water formed by the polymerization reaction.
Thus, it can be concluded that the HAp calcium-based catalysts discussed in this paper are promising solid catalysts for the glycerol polymerization reaction, regarding their high selectivity to PG2-3 and their stability under the reaction conditions.
Author Contributions
N.E. wrote the paper; S.P., F.D. and B.K. led the study, gave advice on the interpretation of results, and revised the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Chevreul Institute (FR 2638), Centrale Lille Institut, Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation, Région Hauts-de-France and FEDER are acknowledged for supporting and funding partially this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Polyglycerol Market Size. 2019. Available online: https://www.grandviewresearch.com/industry-analysis/polyglycerol-market (accessed on 5 May 2020).
- Ruppert, A.; Meeldijk, J.D.; Kuipers, B.W.M.; Erné, B.H.; Weckhuysen, B.M. Glycerol Etherification over Highly Active CaO-Based Materials: New Mechanistic Aspects and Related Colloidal Particle Formation. Chem.-A Eur. J. 2008, 14, 2016–2024. [Google Scholar] [CrossRef] [Green Version]
- Kirby, F.; Nieuwelink, A.-E.; Kuipers, B.W.M.; Kaiser, A.; Bruijnincx, P.C.A.; Weckhuysen, B.M. CaO as Drop-In Colloidal Catalysts for the Synthesis of Higher Polyglycerols. Chem.-A Eur. J. 2015, 21, 5101–5109. [Google Scholar] [CrossRef] [Green Version]
- Barros, F.J.S.; Cecilia, J.A.; Moreno-Tost, R.; De Oliveira, M.F.; Rodríguez-Castellón, E.; Luna, F.M.T.; Vieira, R.S. Glycerol Oligomerization Using Low Cost Dolomite Catalyst. Waste Biomass Valorization 2018, 11, 1499–1512. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z. Selective Etherification of Glycerol over Heterogeneous Mixed Oxide Catalyst: Optimization of Reaction Parameters. Chem. Eng. Sci. 2013, 1, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.T. Heterogeneously catalyzed etherification of glycerol to diglycerol over calcium–lanthanum oxide supported on MCM-41: A heterogeneous basic catalyst. Appl. Catal. A Gen. 2014, 479, 76–86. [Google Scholar] [CrossRef]
- Krisnandi, Y.K.; Eckelt, R.; Schneider, M.; Martin, A.; Richter, M. Glycerol Upgrading over Zeolites by Batch-Reactor Liquid-Phase Oligomerization: Heterogeneous versus Homogeneous Reaction. ChemSusChem 2008, 1, 835–844. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z.; Ahmad, M.; Sultana, S. Performance of lithium modified zeolite Y catalyst in solvent-free conversion of glycerol to polyglycerols. J. Taibah Univ. Sci. 2014, 8, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Ebadipour, N.; Paul, S.; Katryniok, B.; Dumeignil, F. Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review. Catalysts 2020, 10, 1021. [Google Scholar] [CrossRef]
- Sayoud, N.; Vigier, K.D.O.; Cucu, T.; De Meulenaer, B.; Fan, Z.; Lai, J.; Clacens, J.-M.; Liebens, A.; Jérôme, F. Homogeneously-acid catalyzed oligomerization of glycerol. Green Chem. 2015, 17, 4307–4314. [Google Scholar] [CrossRef]
- Medeiros, M.A.; Araujo, M.H.; Augusti, R.; De Oliveira, L.C.A.; Lago, R.M. Acid-catalyzed oligomerization of glycerol investigated by electrospray ionization mass spectrometry. J. Braz. Chem. Soc. 2009, 20, 1667–1673. [Google Scholar] [CrossRef]
- Pour, N.E.; Dumeignil, F.; Katryniok, B.; Delevoye, L.; Revel, B.; Paul, S. Investigating the active phase of Ca-based glycerol polymerization catalysts: On the importance of calcium glycerolate. Mol. Catal. 2021, 507, 111571. [Google Scholar] [CrossRef]
- Barros, F.; Moreno-Tost, R.; Cecilia, J.; Ledesma-Muñoz, A.; de Oliveira, L.; Luna, F.M.T.; Vieira, R. Glycerol oligomers production by etherification using calcined eggshell as catalyst. Mol. Catal. 2017, 433, 282–290. [Google Scholar] [CrossRef]
- Nieuwelink, A.-E. CaO/CNF for the Oligomerization of Glycerol. Master’s Thesis, Department of Chemistry, Utrecht University, Utrecht, The Netherland, 2015. [Google Scholar]
- Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Selective etherification of glycerol to polyglycerols over impregnated basic MCM-41 type mesoporous catalysts. Appl. Catal. A Gen. 2002, 227, 181–190. [Google Scholar] [CrossRef]
- Chinglenthoiba, C.; Das, A.; Vandana, S. Enhanced biodiesel production from waste cooking palm oil, with NaOH-loaded Calcined fish bones as the catalyst. Environ. Sci. Pollut. Res. 2020, 27, 15925–15930. [Google Scholar] [CrossRef]
- Farooq, M.; Ramli, A.; Naeem, A. Biodiesel production from low FFA waste cooking oil using heterogeneous catalyst derived from chicken bones. Renew. Energy 2015, 76, 362–368. [Google Scholar] [CrossRef]
- Gupta, J.; Agarwal, M.; Dalai, A.K. Marble slurry derived hydroxyapatite as heterogeneous catalyst for biodiesel production from soybean oil. Can. J. Chem. Eng. 2018, 96, 1873–1880. [Google Scholar] [CrossRef]
- Yan, B.; Zhang, Y.; Chen, G.; Shan, R.; Ma, W.; Liu, C. The utilization of hydroxyapatite-supported CaO-CeO2 catalyst for biodiesel production. Energy Convers. Manag. 2016, 130, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Ma, P.X. Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering. Biomaterials 2004, 25, 4749–4757. [Google Scholar] [CrossRef]
- Ibrahim, M.; Labaki, M.; Giraudon, J.-M.; Lamonier, J.-F. Hydroxyapatite, a multifunctional material for air, water and soil pollution control: A review. J. Hazard. Mater. 2019, 383, 121139. [Google Scholar] [CrossRef]
- Li, X.; Sun, L.; Zou, W.; Cao, P.; Chen, Z.; Tang, C.; Dong, L. Efficient Conversion of Bio-Lactic Acid to 2,3-Pentanedione on Cesium-Doped Hydroxyapatite Catalysts with Balanced Acid-Base Sites. ChemCatChem 2017, 9, 4621–4627. [Google Scholar] [CrossRef]
- Matsumura, Y.; Moffat, J.B. Catalytic oxidative coupling of methane over hydroxyapatite modified with lead. Catal. Lett. 1993, 17, 197–204. [Google Scholar] [CrossRef]
- Silvester, L.; Lamonier, J.-F.; Lamonier, C.; Capron, M.; Vannier, R.-N.; Mamede, A.-S.; Dumeignil, F. Guerbet Reaction over Strontium-Substituted Hydroxyapatite Catalysts Prepared at Various (Ca+Sr)/P Ratios. ChemCatChem 2016, 9, 2250–2261. [Google Scholar] [CrossRef]
- Ajish, P.; Alex, X. Removal of Protein Aggregates from Biopharmaceutical Preparations Using Calcium Phosphate Salts. WO2011156073A1, 2011. [Google Scholar]
- Raynaud, S.; Champion, E.; Bernache-Assollant, D.; Thomas, P. Calcium phosphate apatites with variable Ca/P atomic ratio I. Synthesis, characterisation and thermal stability of powders. Biomaterials 2001, 23, 1065–1072. [Google Scholar] [CrossRef]
- Diallo-Garcia, S. Hydroxyapatites, un Système Basique Atypique Modulable par la Synthèse: Vers L’identification Des sites actifs. Ph.D. Thesis, Université Pierre-et-Marie-Curie, Paris, France, 2012. [Google Scholar]
- Ruiz-Aguilar, C.; Olivares, U.; Aguilar-Reyes, E.A.; López-Juárez, R.; Alfonso, I. Characterization of β-tricalcium phosphate powders synthesized by sol–gel and mechanosynthesis. Boletín Soc. Española Cerámica Vidr. 2018, 57, 213–220. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Silvester, L.; Lamonier, J.-F.; Vannier, R.-N.; Lamonier, C.; Capron, M.; Mamede, A.-S.; Pourpoint, F.; Gervasini, A.; Dumeignil, F. Structural, textural and acid–base properties of carbonate-containing hydroxyapatites. J. Mater. Chem. A 2014, 2, 11073–11090. [Google Scholar] [CrossRef] [Green Version]
- Silvester, L. Synthesis of Higher Alcohols from Ethanol over Hydroxyapatite-Based Catalysts; Lille University: Lille, France, 2013. [Google Scholar]
- Mohammad, N.F.; Amiruddin, N.L.; Saleh, S.S.M.; Taib, M.A.A.; Nasir, N.F.M. Effect of swelling agent on pore properties of mesoporous carbonated hydroxyapatite. J. Phys. Conf. Ser. 2019, 1372. [Google Scholar] [CrossRef] [Green Version]
- Von Euw, S.; Wang, Y.; Laurent, G.; Drouet, C.; Babonneau, F.; Nassif, N.; Azaïs, T. Bone mineral: New insights into its chemical composition. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.J.; Biesinger, M.C.; McIntyre, N.S. Interactions of CO2 and CO at fractional atmosphere pressures with iron and iron oxide surfaces: One possible mechanism for surface contamination? Surf. Interface Anal. 2002, 33, 299–305. [Google Scholar] [CrossRef]
- Gomes, G.C.; Borghi, F.; Ospina, R.; López, E.; Borges, F.; Mello, A. Nd:YAG (532 nm) pulsed laser deposition produces crystalline hydroxyapatite thin coatings at room temperature. Surf. Coatings Technol. 2017, 329, 174–183. [Google Scholar] [CrossRef]
- Sheikh, L.; Tripathy, S.; Nayar, S. Biomimetic matrix mediated room temperature synthesis and characterization of nano-hydroxyapatite towards targeted drug delivery. RSC Adv. 2016, 6, 62556–62571. [Google Scholar] [CrossRef]
- Bolis, V.; Busco, C.; Martra, G.; Bertinetti, L.; Sakhno, Y.; Ugliengo, P.; Chiatti, F.; Corno, M.; Roveri, N. Coordination chemistry of Ca sites at the surface of nanosized hydroxyapatite: Interaction with H 2 O and CO. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 1313–1336. [Google Scholar] [CrossRef] [Green Version]
- Prakash, M.; Lemaire, T.; Caruel, M.; Lewerenz, M.; De Leeuw, N.; Di Tommaso, D.; Naili, S. Anisotropic diffusion of water molecules in hydroxyapatite nanopores. Phys. Chem. Miner. 2017, 44, 509–519. [Google Scholar] [CrossRef]
- Corno, M.; Rimola, A.; Bolis, V.; Ugliengo, P. Hydroxyapatite as a key biomaterial: Quantum-mechanical simulation of its surfaces in interaction with biomolecules. Phys. Chem. Chem. Phys. 2010, 12, 6309–6329. [Google Scholar] [CrossRef]
- Tanaka, H.; Chikazawa, M.; Kandori, K.; Ishikawa, T. Influence of thermal treatment on the structure of calcium hydroxyapatite. Phys. Chem. Chem. Phys. 2000, 2, 2647–2650. [Google Scholar] [CrossRef]
- Verwilghen, C.; Rio, S.; Nzihou, A.; Gauthier, D.; Flamant, G.; Sharrock, P.J. Preparation of high specific surface area hydroxyapatite for environmental applications. J. Mater. Sci. 2007, 42, 6062–6066. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ye, X.; Wang, H.; Zhu, M.; Wang, B.; Yan, H. The influence of pH and temperature on the morphology of hydroxyapatite synthesized by hydrothermal method. Ceram. Int. 2003, 29, 629–633. [Google Scholar] [CrossRef]
Figure 1.
Catalytic activity in the glycerol polymerization reaction of HAp-D, HAp-S, and HAp-R at 245 °C after 8 h of reaction (left) and after 22 h (right).
Figure 1.
Catalytic activity in the glycerol polymerization reaction of HAp-D, HAp-S, and HAp-R at 245 °C after 8 h of reaction (left) and after 22 h (right).

Figure 2.
ESI-MS spectra of reaction media when (a) HAp-D, (b) HAp-S, and (c) HAp-R were used as the catalyst at 245 °C after 8 h of reaction.
Figure 2.
ESI-MS spectra of reaction media when (a) HAp-D, (b) HAp-S, and (c) HAp-R were used as the catalyst at 245 °C after 8 h of reaction.

Figure 3.
The catalytic activity of fresh HAp-S and recovered catalysts with washing using EtOH or H2O.
Figure 3.
The catalytic activity of fresh HAp-S and recovered catalysts with washing using EtOH or H2O.

Figure 4.
XRD diffractograms of (a) HAp-D, (b) HAp-S and (c) HAp-R (left); and the standard PDF cards for (d) HAp-S, (e) calcium hydroxyapatite, (f) β-TCP, (g) OCP and (h) DCP (right).
Figure 4.
XRD diffractograms of (a) HAp-D, (b) HAp-S and (c) HAp-R (left); and the standard PDF cards for (d) HAp-S, (e) calcium hydroxyapatite, (f) β-TCP, (g) OCP and (h) DCP (right).

Figure 5.
XRD patterns of (a) HAp-Et-1, (b) HAp-W-1, and (c) HAp-Et-Cal-1.

Figure 6.
IR spectra for (a) HAp-S, (b) HAp-D, and (c) HAp-R (left) and zooms of the IR spectra for (d) HAp-S, (e) HAp-D and (f) HAp-R (right).
Figure 6.
IR spectra for (a) HAp-S, (b) HAp-D, and (c) HAp-R (left) and zooms of the IR spectra for (d) HAp-S, (e) HAp-D and (f) HAp-R (right).

Figure 7.
IR spectra for (a) HAp-S before the reaction and spent catalysts (b) HAp-Et-1, (c) HAp-W-1, and (d) HAp-Et-Cal-1.
Figure 7.
IR spectra for (a) HAp-S before the reaction and spent catalysts (b) HAp-Et-1, (c) HAp-W-1, and (d) HAp-Et-Cal-1.

Figure 8.
XPS spectra of (a) HAp-D, (b) HAp-S, and (c) HAp-R.

Figure 9.
XPS of Ca 2p spectra for (a) HAp-D, (b) HAp-S, and (c) HAp-R.

Figure 10.
TGA-DSC profiles for non-calcined (a) HAp-D, (b) HAp-S and (c) HAp-R.

Scheme 1.
Schematic of HAp’ surface before reaction (left) and after reaction upon poisoning by PGs, water, and carbonate (right).
Scheme 1.
Schematic of HAp’ surface before reaction (left) and after reaction upon poisoning by PGs, water, and carbonate (right).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalytic performances in glycerol polymerization reaction in the presence of 0.5 mol% of CaO and different HAps and percentages of leached Ca based on the ICP-OES when 0.5 mol.% of catalysts were used at 245 °C for 8 h.
Table 1.
Catalytic performances in glycerol polymerization reaction in the presence of 0.5 mol% of CaO and different HAps and percentages of leached Ca based on the ICP-OES when 0.5 mol.% of catalysts were used at 245 °C for 8 h.
| Catalyst | Glycerol Conversion (%) | PG2 Selectivity (%) | PG3 Selectivity (%) | PG4+ Selectivity (%) | Leached Ca. (%) |
|---|---|---|---|---|---|
| CaO | 36 | 8 | 22 | 70 | 100 |
| HAp-D | 15 | 0 | 100 | 0 | 1.8 |
| HAp-S | 15 | 0 | 100 | 0 | 0.5 |
| HAp-R | 27 | 50 | 38 | 12 | 3.4 |
Table 2.
Chemical compositions and surface areas of fresh and reused HAps.
| Catalyst | Name | BET Specific Surface Area (m2/g) | Theoretical Ca/P | Ca/P Atomic Ratio (ICP) | Ca (wt.%) | P (wt.%) | C (wt.%) |
|---|---|---|---|---|---|---|---|
| Fresh catalysts | HAp-D | 19.4 | 1.45 | 1.62 | 36.16 | 17.33 | 0.108 |
| HAp-S | 70.4 | 1.67 | 1.66 | 36.07 | 16.84 | 0.111 | |
| HAp-R | 13.8 | 1.94 | 1.78 | 38.44 | 17.39 | 0.8 | |
| Reused catalysts | HAp-W-1 | 38.3 | 1.67 | n.d * | n.d | n.d | 2.13 |
| HAp-Et-1 | 36.6 | 1.67 | n.d | n.d | n.d | 1.96 | |
| HAp-Et-2 | 38 | 1.67 | n.d | n.d | n.d | 2.38 | |
| HAp-Et-Cal-1 | 60 | 1.67 | n.d | n.d | n.d | 0.37 |
*n.d.: Not determined.
Table 3.
Surface chemical compositions (atomic %) of HAp-D, HAp-S, and HAp-R by XPS analysis.
| Catalyst | C 1s (%) | O 1s (%) | Ca 2p (%) | P 2p (%) | Ca/P (XPS) |
|---|---|---|---|---|---|
| HAp-D | 35 | 43.8 | 12.4 | 8.8 | 1.40 |
| HAp-S | 21.7 | 51.4 | 15.5 | 11.4 | 1.36 |
| HAp-R | 22.9 | 50.9 | 15.6 | 10.6 | 1.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ebadipour, N.; Paul, S.; Katryniok, B.; Dumeignil, F. Calcium Hydroxyapatite: A Highly Stable and Selective Solid Catalyst for Glycerol Polymerization. Catalysts 2021, 11, 1247. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101247
AMA Style
Ebadipour N, Paul S, Katryniok B, Dumeignil F. Calcium Hydroxyapatite: A Highly Stable and Selective Solid Catalyst for Glycerol Polymerization. Catalysts. 2021; 11(10):1247. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101247
Chicago/Turabian StyleEbadipour, Negisa, Sébastien Paul, Benjamin Katryniok, and Franck Dumeignil. 2021. "Calcium Hydroxyapatite: A Highly Stable and Selective Solid Catalyst for Glycerol Polymerization" Catalysts 11, no. 10: 1247. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101247
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.