
Apiose-Relevant Glycosidases


Laboratory of Biocatalysis and Organic Synthesis, Institute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, SK-845 38 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(10), 1251; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101251
Submission received: 15 September 2021
/
Revised: 15 October 2021
/
Accepted: 16 October 2021
/
Published: 18 October 2021
(This article belongs to the Special Issue New Potential Applications of Enzymes in Biocatalysis and Next Challenges for a Green Chemistry)
Abstract
:Apiose is a branched pentose naturally occurring either as a component of the plant cell wall polysaccharides or as a sugar moiety present in numerous plant secondary metabolites such as flavonoid and phenylethanoid glycosides, substrates in plant defense systems or as glycosylated aroma precursors. The enzymes catalyzing hydrolysis of such apiosylated substances (mainly glycosidases specific towards apiose or acuminose) have promising applications not only in hydrolysis (flavor development), but potentially also in the synthesis of apiosides and apioglucosides with pharmaceutical relevance. This review summarizes the actual knowledge of glycosidases recognizing apiose and their potential application in biocatalysis.
1. Introduction
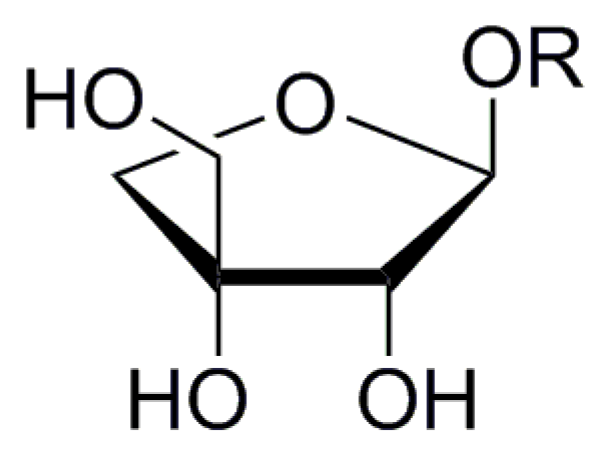
Apiose is one of the best studied branched sugars of plant origin. Despite its recognition as a “rare sugar”, apiose could be found in all higher plants, although never occurring as a free sugar and with some exceptions found always as D-apiofuranoside (3-C-(hydroxymethyl)-d-erythrofuranoside, Figure 1).
An excellent review focused on apiose, including its natural occurrence and secondary metabolites containing apiose, was released in 2016 [1]. Apiose represents an important component of rhamnogalacturonan II, a minor constituent of plant cell walls which is widespread among all vascular plants. The role of apiose in rhamnogalacturonan is bridging its two chains through apiose-borate ester bonds [2]. Another type of cell wall pectic polysaccharide containing apiose is apiogalacturonan, typical for aquatic monocots, which can represent several percent of the whole dry plant mass [3,4]. On the other side, D-apiose occurs also in nearly 1200 secondary metabolites (glycosides) found in more than 530 plant species from 106 families of ferns, gymnosperms, angiosperms (most often Asteraceae, Fabaceae, Lamiaceae), lichens and exceptionally also in fungi. Several of these glycosides are attractive for the scale of their beneficial health properties and for their role in flavor release in food and beverage production. Aglycones of these glycosides belong to various phenolics, terpenes, terpenoids, saponins, cyanogenic nitriles, alcohols or lactones [1]. As a component of glycosides, apiose can be bound either directly to the aglycone, or it is incorporated to the sugar chain of the glycone, where it can be linked to D-glucose, D-galactose, L-rhamnose or D-xylose [1]. At least one instance of C-linked apiose was identified as luteolin 6-C-apioside-8-C-glucoside [5].
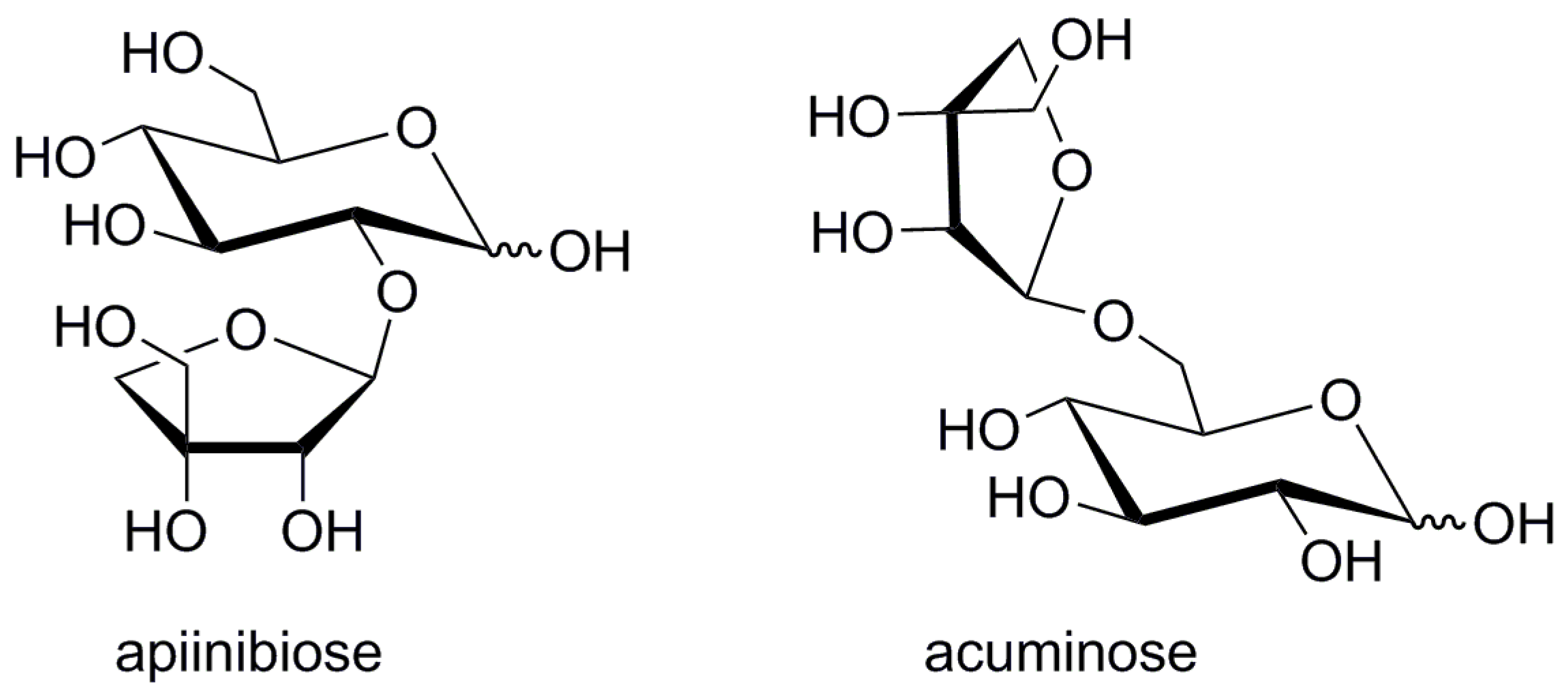
High levels of apiin (apigenin 7-O-apiinibioside) are present in parsley and celery [6]. The glycone of apiin-β-apiinibiose (β-d-apiofuranosyl-(1→2)-β-d-glucopyranose, Figure 2) is a typical disaccharide motif present also in plant glycosides such as isoliquiritin apioside [7], luteosides A-C [8] or cucurbitosides A-M [9,10]. β-Acuminose (β-d-apiofuranosyl-(1→6)-β-d-glucopyranose, Figure 2) is another abundant apiose-derived glycone [11]. This disaccharide differs from apiinibiose in its positional arrangement of apiose and glucose units. Besides its occurrence in various glycosides such as acuminosylated flavonoids [12,13], stilbenes [14] and coumarins [15,16], it is also a constituent of substrates of glycosidase-driven plant alarm and defense systems [17,18] as well as an odor releasing glucosidase mechanism in grapes [11].
Within our research on chemoenzymatic preparation of bioactive sugar derivatives, we are highly interested in enzymatic synthesis of apiose-containing glycosides. The purpose of this brief review is therefore to summarize the knowledge on glycosidases specific to apiose-containing saccharides and glycosides and to evaluate their potential in synthesis of apiosaccharides and apioglycosides.
2. General Remarks on Glucosidases
The main role of glycosidases in nature is obviously the hydrolysis of glycosidic bond in glycosides, oligosaccharides or polysaccharides. Besides simple apiosides, the apioglycosides are more complex molecules consisting of several sugars forming an oligosaccharide bound to the aglycone by glycosidic bond on its reducing end. A significant number of flavor precursors of tea, wine and juices are for example constituted by glycosylated odor-active alcohols [19,20,21] that become volatile after releasing the sugar moieties by acidic or enzymatic hydrolysis. Isolation and characterization of substances such as geranyl acuminoside [11] have consequently led to systematic studies of enzymatic hydrolysis of diglycosidic flavor precursors containing apiose [22,23,24].
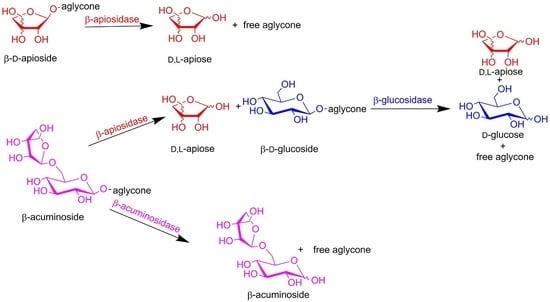
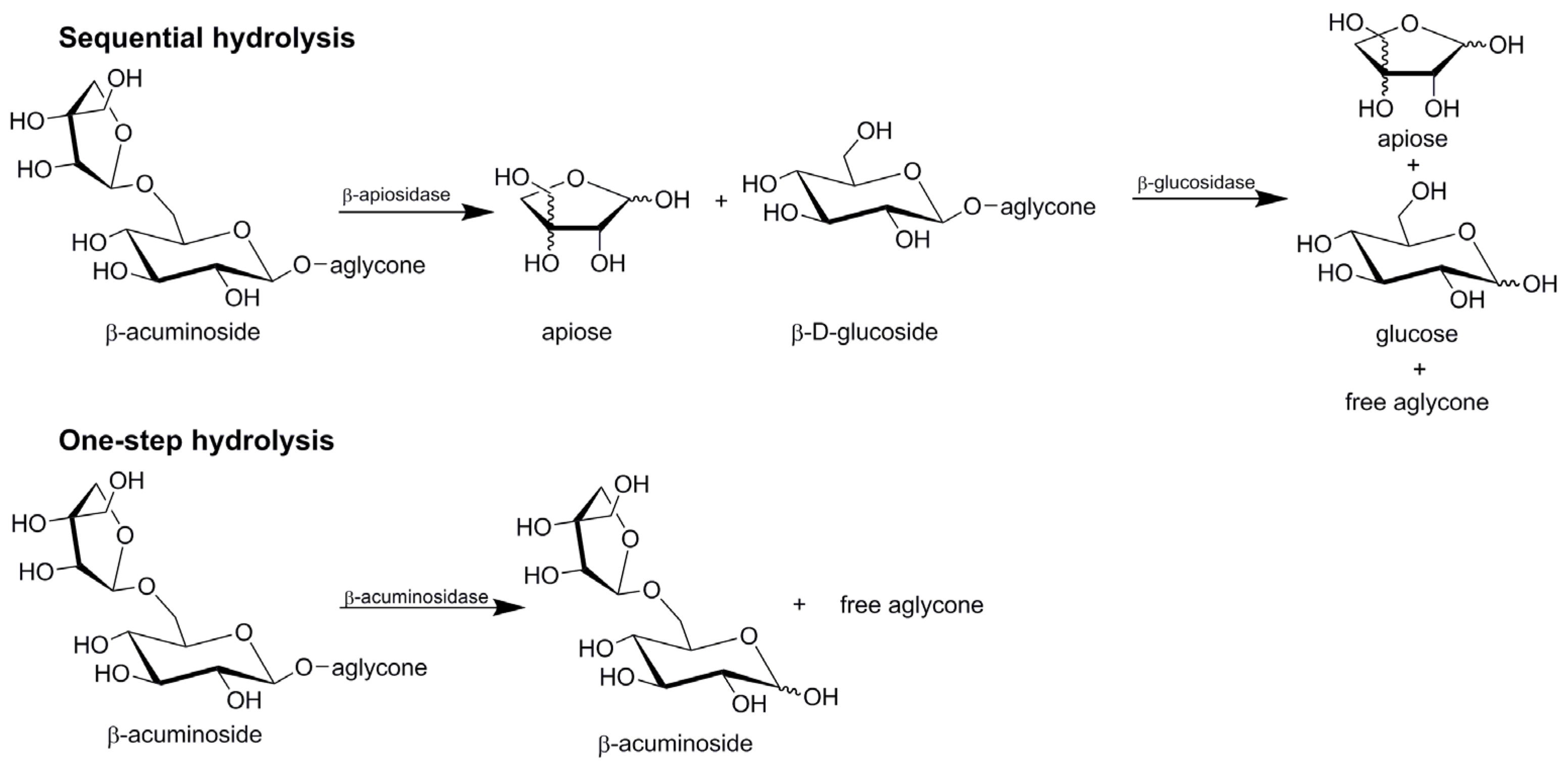
The enzymatic hydrolysis of diglycosides (acuminosides, primeverosides, rutinosides or vicianosides) may generally proceed by two distinct mechanisms (Figure 3). In the sequential mode, β-apiosidase, β-xylosidase, α-l-rhamnosidase or α-l-arabinopyranosidase) initially cleaves the intersugar bond between the terminal sugar and β-d-glucoside. In the second step, a β-glucosidase catalyzes the hydrolysis of the resulting β-d-glucoside and liberates the corresponding aglycone and glucose. In the one-step mode, the diglycosidase catalyzes direct cleavage of the glycosidic linkage between the aglycone and the disaccharide.
Despite their natural role as destroyers of glycosidic bond, glycosidases can also be used in the synthesis of oligosaccharides and glycosides. Chemical synthesis of glycosides requires laborious steps and noxious catalysts to acquire a specific glycoside linkage, while the enzymes can attain this goal straightforward due to the advantages of their stereo- and chemo-selectivity. On the other hand, reaction regioselectivity can remain as a problem if glycosidases are used to the synthesis of oligosaccharides and oligoglycosides.
The enzymatic glycosylation catalyzed by glycosidases can be simply realized by two methods, reverse hydrolysis and transglycosylation. Reverse hydrolysis is a thermodynamically controlled process in which a free saccharide reacts with present alcohol until an equilibrium is reached between starting substances and the target glycoside [25]. Syntheses of glycosides realized through reverse hydrolysis usually suffer from long reaction times and low product yields. Higher concentrations of substrates required for reverse hydrolysis result in complications during product purification and limit the use of this process to inexpensive starting sugars. Moreover, the enzymes used for hydrolysis must be free of glycosidases with the opposite anomer specificity to prevent formation of undesired product anomers. On the other side, transglycosylation is a kinetically controlled process employing partial transferase activity of glycosidases [25]. The enzyme catalyzes transfer of the glycosyl unit from the non-reducing end of oligosaccharide, polysaccharide or other glycoside to the alcohol or other acceptor (saccharide, amine, thiol, etc.). Compared to reverse hydrolysis, the transglycosylation is faster, requires lower substrate concentrations and gives usually higher yields. Anomeric purity of product is controlled by the choice of the substrate, so contamination of biocatalyst by glycosidase with the opposite anomer specificity does not give rise to formation of undesired products. Secondary product hydrolysis must, however, be controlled through monitoring of the reaction progress. The level of transferase activity is strongly varying among glycosidases and is limited only to glycosidases operating through the retention reaction mechanism since the inverting glycosidases do not catalyze transglycosylation.
3. Glycosidases in Processing Apiose-containing Saccharides and Glycosides
3.1. β-d-apiofuranosidase (β-apiosidase)
3.1.1. Sources
Glycosidase activity towards β-d-apiofuranosides was detected in three related filamentous fungi Aspergillus niger, A. oryzae and AO 10207 [26] and A. aculeatus [27] in various commercial enzyme preparations (Table 1) [22,24,26,27] or in aspergilli AN 55465, AN 12648, AO 12559 [26] and AN CBS 554.65 [28] after their incubation with apiin as the inductor. Previous studies detected β-apiosidase activity only in fungal enzymes dedicated for use in winemaking [22,24], but the activity has been recently found also in some fungal lipase and cellulase preparations [27]. On the other side, many marketed crude pectinases and polygalacturonases of the same microbial origin (frequently used in winemaking) lack this activity. This indicates the non-constitutive [22,24,26,27] and inducible character of β-apiosidase [26,28].
The β-apiosidase-comprising commercial glycanases are produced mainly as pectinases or cellulases and contain other glycosidases as side activities. The apiosidase activity was not confirmed in various varieties of Vitis vinifera or yeast strains typically used for wine production [20,24,29]. Most recently, a new type of endoapiosidase was, however, identified in the rhamnogalacturonan II (RGII) degradome of Bacteroides thetaiotaomicron (PDB, 5MSY, EC 3.2.1.-), which revealed a new glycoside hydrolase family group GH140. The enzyme hydrolyses D-apiofuranosyl-β-(1→2)-d-galacturonopyranosyl linkage in RGII in cooperation with several different hydrolases and glycosidases responsible for depolymerisation of RGII [30]. This enzyme is available only from NZYTech, Lda. (Lisbon, Portugal).
3.1.2. Purification and Characterization of Apiosidases
Purification of β-apiosidase was for the first time reported in 1992 [26]. After inducing β-apiosidase production in Aspergillus strains and determining extracellular production of the enzyme, its crude preparation was separated by a combination of gel permeation chromatography on Ultrogel AcA 44 and ion-exchange chromatography on DEAE-Sepharose CL6B. The isolated β-apiosidase had the best stability at pH 7 and below 60 °C with rapid decrease in pH 3–3.6 to 50%. The β-apiosidase was later isolated from crude enzyme preparation Klerzyme 200 (nowadays known as Rapidase AR 2000) by the same procedure, the enzyme was, however, not completely void of α-rhamnosidase. Separation on Concanavalin A-Ultrogel AcA 22 column was also unsuccessful, as the enzyme was moreover attached to the gel [22]. Guo et al. [23] attempted to improve the purification of β-apiosidase from Klerzyme 200 by use a pre-separation through precipitation of proteins with 30–65% saturated ammonium sulphate and dialyzation before application on Ultrogel AcA 44 and DEAE-Sepharose CL-6B, they were, however, unable to eliminate an accompanying β-glucosidase activity. According to SDS-PAGE combined with the detection with 4MUmAp, the molecular weight of both enzymes was 120 kDa. The β-apiosidase eluted on ion-exchange HPLC in two peaks. SDS-PAGE analysis confirmed that the first peak consisted of fragments with 120 and 80 kDa, but only the band corresponding to Mw 120 kDa displayed the apiosidase activity. Repeated separation of fractions with β-apiosidase activity with a changed NaCl gradient resulted in a single peak at 280 nm. The final purification factor was 270. Table 2 summarizes molecular and catalytic properties of purified apiosidases.
Mastihuba et al. [31] studied the reaction mechanism of β-apiofuranosidase from Aspergillus aculeatus in commercial glycanase Viscozyme L and revealed that hydrolysis of pNP β-d-apiofuranoside proceeds with the inversion of the configuration of hydroxyl in the anomeric center. Since crystal structures and molecular biology of purified enzymes are still unavailable, further research work is required to fully understand the molecular mechanism of catalysis by this enzyme.
Endo-apiosidase from B. thetaiotaomicron was not purified from the original source, but was available as the recombinant protein in the mixture with other enzymes forming RGII degradome expressed in Escherichia coli. The recombinant proteins were purified using metal ion affinity chromatography (IMAC) using Talon (cobalt-based matrix) eluted with 100 mM imidazole. The separated endo-apiosidase was crystallized in 40% (v/v) 2-methyl-2,4-pentanediol, 0.2 M ammonium phosphate, and Tris, pH 8.5. [30]. The crystal structure revealed an (α/β)8-barrel catalytic domain and C-terminal β-sandwich domain. The retention mechanism of this enzyme was predicted (essential catalytic residues ASP187 and Glu284 are separated by approximately 5.5 Å, which is consistent with double-displacement mechanism with retention of anomeric configuration) and also experimentally confirmed. The +1 Subsite includes two arginine residues, which probably contribute to the binding of galacturonic acid through the formation of ion pairs (as supported by results from mutagenesis). Sequential involvement of endo-apiosidase in the degradation process also suggested a narrow −1 active site and +1 subsites preventing binding of apiofuranose linked to borate or galacturonic acid appended to the backbone. Alignment of the endo-apiosidase sequence (GenBank AAO76119.1) revealed affiliation to new glycoside hydrolase family GH140 [30].
3.1.3. Substrate Specificity and Assaying of β-Apiosidases
Extracellular β-apiofuranosidase from Aspergillus spp. induced by apiin had specificity towards the terminal apiose in terpenol acuminosides from grapes. Specificity of the purified β-apiosidase was tested towards the glycosidic extract from grape and according to the GS-MS analysis, acuminosides of nerol, geraniol and pyran linalool oxide were hydrolysed to apiose and the corresponding glucosides, while L-arabinosyl- or rhamnosylglucosides were not affected. Lower effectivity of hydrolysis towards apiosylglucoside of furan linalool oxide indicated some degree of aglycone specificity [24]. It should be stressed at this point that although apiin, an abundant apiosylated diglycosidic flavonoid was used to induce production of β-apiosidase, no publication has appeared reporting its hydrolysis by any apiose-relevant glycosidase.
Since there is no commercialized substrate for determination of β-apiosidase on the market and the natural substrates are difficult to obtain or are not economically available, crude grape extract containing the monoterpenyl glycosides was used for the assay as an alternative. The trifluoroacetylated reactants were analyzed by GC-MS [23,26]. Complete hydrolysis of geranyl and linalyl apiosylglucosides and apiin was also confirmed by TLC analysis [22,23].
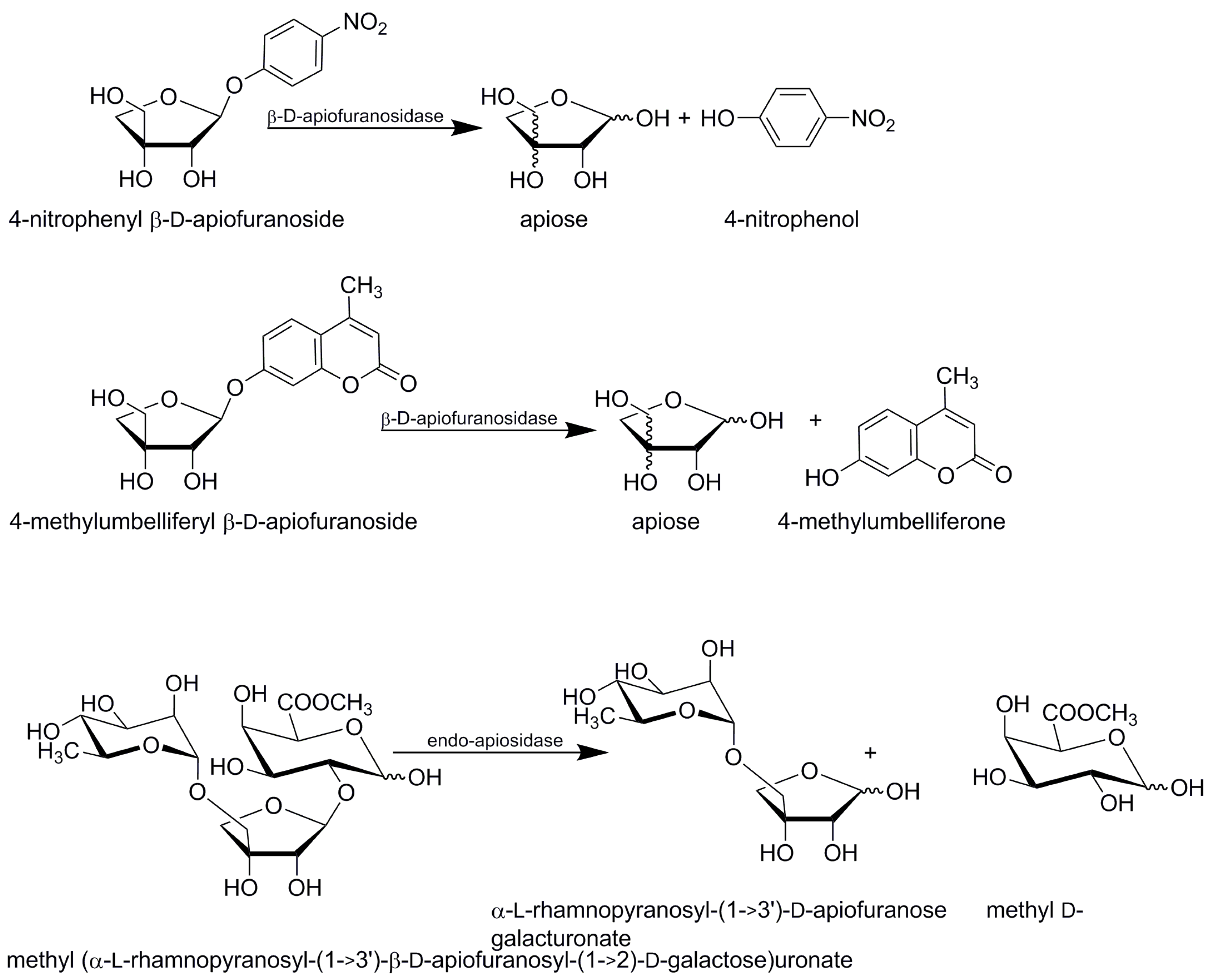
Many exo-acting β-apiosidases were able to hydrolyse 4-nitrophenyl (pNP) β-d-apiofuranoside in screenings. Synthetic pNP β-d-apiofuranoside was successfully used for spectrophotometric analysis of liberated nitrophenol (Figure 4), [22,23,26,27,31]. The assay protocols vary in details such as substrate concentration, type and pH of the buffer, the way of stopping of the substrate hydrolysis or reaction time. According to personal communication with Oenobrand (subsidiary of DSM), the company uses this method to assay β-apiosidase in their Rapidase AR 2000. Similarly, 4-methylumbelliferyl β-d-apiofuranoside (4Mum β-d-apiofuranoside) was used as a highly sensitive fluorogenic probe to simplify monitoring of β-apiosidase isolation and purification during electrophoretic separation [23] (Figure 4). The electrophoretic gel was incubated in a solution of 4Mum β-d-apiofuranoside to stain the protein with the target activity.
Endo-apiosidase from B. thetaiotaomicron liberates D-apiose chains A and B linked to the remaining D-galacturonic acids after depolymerising the RG II backbone. Its hydrolysing activity was also screened with 4-nitrophenyl glycosides and no activity was observed against other pNP glycosides (β-d-glucuronide, α-l-rhamnopyranoside, α-l-arabinofuranoside, β-d-arabinopyranoside, β-d-xylopyranoside or α-l-galactopyranoside) [30]. Activity of the endo-apiosidase from B. thetaiotaomicron was quantified through hydrolysis of trisaccharide methyl (α-l-rhamnopyranosyl-(1→3’)-β-d-apiofuranosyl-(1→2)-d-galactose) uronate monitored by NMR and LC-MS. NMR analysis revealed liberation of α-l-rhamnopyranosyl-(1→3’)-d-apiofuranose with the appearance of anomeric signals of apiose typical for residues with free reducing end fixed in the D-erythro configuration corresponding respectively to β- and α-anomer [30].
3.2. Acuminosidases
Diglycosidases, or disaccharide-specific glycosidases classified to GH 1 family [32] are typical by their plant origin, since they play a key role in the plant defense mechanism, liberating toxic aglycones or attractants from their diglycosylated precursors. This group of enzymes is supposed to be a case of endo-β-glucosidases, since they cleave the glycosidic bond between the aglycone and the glucose part at the reducing end of the disaccharide [33].
Diglycosidases involved in processing of apiose-containing disaccharides catalyze the cleavage of β-glycosidic bond at the reducing end of acuminose (β-d-apiofuranosyl-(1→6)-β-d-glucopyranose). They can be therefore generally supposed as acuminosidases, although various authors call them according to their natural substrate (e.g., furcatin hydrolase). The acuminosidases vary in terms of their substrate specificity and occur usually in plant tissues, being involved in degradation of various plant glycosides such as monoterpenyl and flavonoid glycosides.
3.2.1. Sources and Substrate Specificity of β-Acuminosidases
To date, no study reporting a microbial acuminosidase has appeared. All known acuminosidases are of plant origin and their discoveries are usually connected with studies of the fate of particular secondary metabolites in plants. The reason is probably in non-existence of an appropriate probe enabling detection and assay of acuminosidases. The studies of plant acuminosidases use the secondary metabolites as detection substrates (and every enzyme has usually strong affinity to one particular substrate), but prices of such natural acuminosides are prohibitive for extensive acuminose screenings among microbial glycosidases.
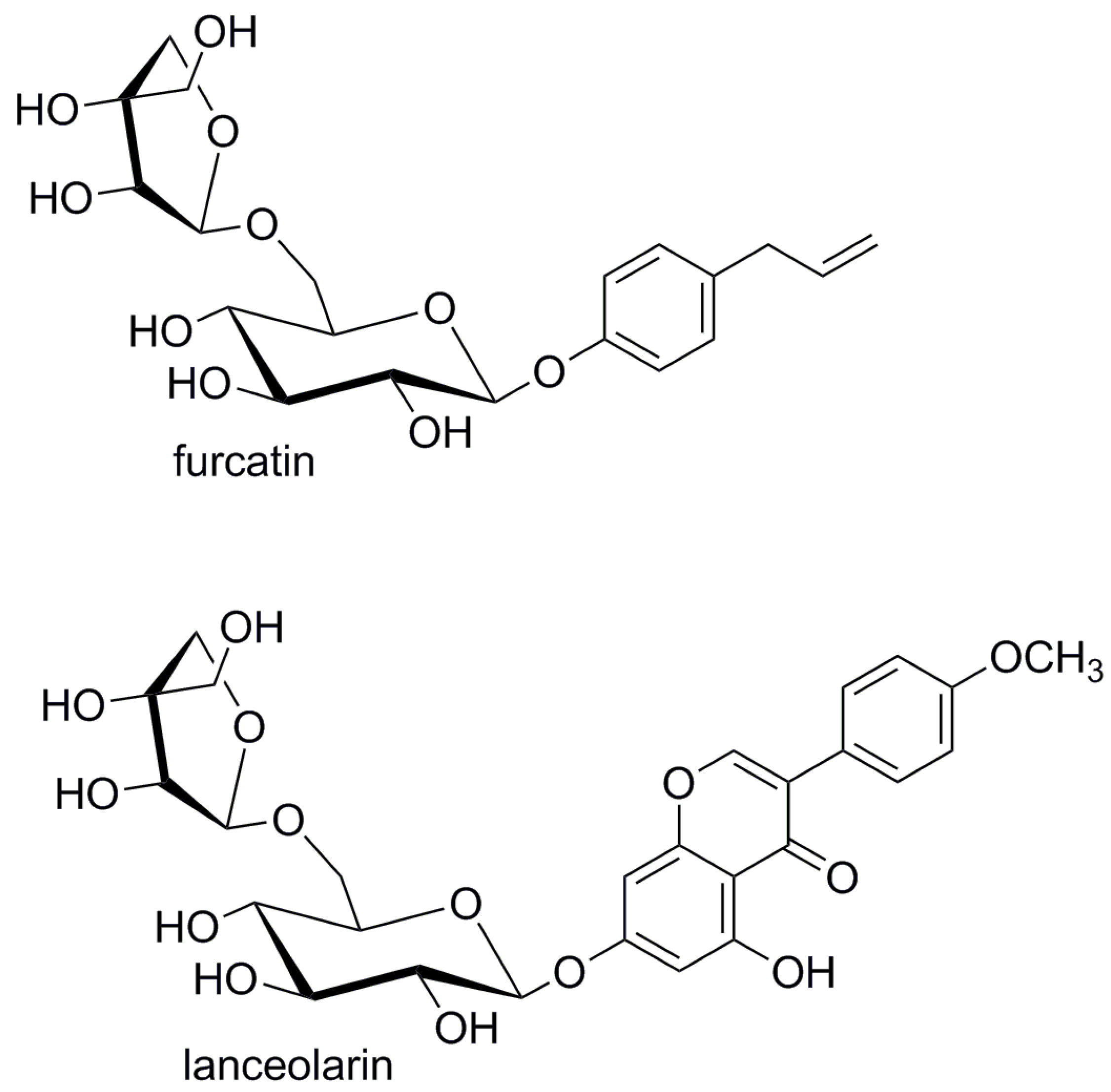
Diglycosidase furcatin hydrolase found in leaves of the ornamental bush Viburnum furcatum Blume [17,18] has a narrow specificity towards the aglycone. The purified enzyme hydrolyzed its natural substrate furcatin (4-allylphenyl acuminoside, Figure 5), being active also towards alkyl acuminosides, but the degree of their hydrolysis was remarkably low. Furcatin hydrolase did not act on rutin or apiin, suggesting that the enzyme accepts a β-d-apiose-(1→6)-glucoside and does not accept its L-rhamnose analogue [17]. Later analysis of the purified enzyme revealed low activity towards monoglucosides with the same aglycone. The activity of the enzyme towards 4-allylphenyl β-d-glucopyranoside and pNP β-d-glucopyranoside represented only 43% and 3% of its activity against furcatin, respectively, and the enzyme was inactive toward prunasin, 2-phenylethyl-β-d-glucopyranoside and pNP monoglycosides, including β-d-xylopyranoside, α-l-arabinopyranoside and β-d-galactopyranoside. An NMR study of furcatin hydrolysis revealed initial formation of β-acuminose confirming the retaining mechanism of furcatin hydrolase [18].
Endoglycosidase from grape berry skins (Vitis vinifera cv. Muscat of Alexandria) possessing β-glucosidase activity was found to hydrolyze monoterpenyl and 2-phenylethyl diglycosides including acuminosides and to yield the corresponding disaccharides (Table 3). On the other hand, the hydrolysis of peltatoside (quercetin 3-O-α-l-arabinopyranosyl-(1→6)-β-d-glucopyranoside yielded free monosaccharides. The enzyme therefore displays diglycosidase activity toward diglycosides with variable monosaccharide at the reducing end and possessing (1→6) intersugar linkage with the exception of peltatoside, where different aglycone or its glycosylation position may interfere with hydrolysis. The hydrolysing activity of grape berry skin endoglycosidase towards glycosides comprising tertiary alcohol (linalool) as aglycone was very low [20]. In their more recent review, the authors indicate the presence of this enzyme in various grapevine tissues, most abundantly in the berry skins. Its activity is increased during berry maturation [24].
Three glycosidases isolated from leaves, roots and hypocotyls of Cicer arietinum L. (chickpea) were specific toward isoflavonoids and flavonoids glycosylated in the position 7. The enzymes hydrolyzed biochanin A 7-O-β-acuminoside (lanceolarin, Figure 5) and released whole acuminose as the disaccharide, which was not further hydrolyzed. The enzymes have however higher affinity to isoflavonoid glucosides over the respective apioglucosides [34].
Chuankhayan et al. [13] reported isolation of isoflavonoid 7-O-β-acuminoside specific β-glycosidase from seeds of Dalbergia nigrescens Kurz. The enzyme released whole disaccharide from its natural substrates, 7-O-β-acuminosides of dalpatein and dalnigrein. The enzyme is selective towards the natural glycosides, displaying high affinity to β-glucosides daidzin and genistin and low hydrolytic activity towards pNP β-d-glucopyranoside and pNP β-d-fucoside. The enzyme has an 81% identity with a β-glucosidase (dalcochinase) from D. cochinchinensis, an enzyme effectively hydrolyzing dalcochinin β-d-glucopyranoside but possessing only low level of acuminosidase activity. Mutations of β-d-glycosidase from D. nigrescens provided a recombinant enzyme which hydrolysed natural acuminosides less efficiently than the native enzyme, regardless, it retained 400- and 5000-fold higher catalytic towards dalpatein and dalnigrein, respectively, when compared to hydrolysis of dalcochinin β-d-glucopyranoside. Mutations of substrate binding residues in dalcochinase increased its acuminosidase activity only in a limited extent [35].
3.2.2. Assay of Acuminosidases
Proof and estimation of diglycosidase activity is more complex than assaying monoglycosidases. Since many diglycosidases, especially those of plant origin, are strongly specific towards the structure of aglycon, synthetic substrates may not be effective as diglycosidase probes. Moreover, the assay requires several tests or structural characterization of the products to exclude interferences by co-occurring monoglycosidases. Flavonoids and isoflavonoids as natural diglycosidase substrates can be determined by HPLC [36] or GC/MS [20]. The structure of the liberated disaccharide is usually verified by TLC [13,18,20] paper chromatography [17] or by NMR [18].
The kinetic experiments with natural substrates can be carried out spectrophotometrically if there is sufficient difference between extinction coefficients of the substrate and the released aglycone [34,37]. Günata et al. [20] determined kinetic parameters of grape berry skin diglycosidase (having wide substrate specificity) on pNP rutinoside, while hydrolysis of other diglycosides to aglycone and disaccharide was identified by TLC and by GC-MS after derivatization (trifluoroacetylation). Ahn et al. [18] measured the furcatin hydrolase (acuminosidase) activity through estimation of liberated 4-allylphenol from furcatin by HPLC. The presence of free glucose and apiose was excluded by parallel TLC and MS analysis. Chuankhayan et al. [13] monitored the hydrolysis of dalpatein and dalnigrein acuminosides by diglycosidase from D. nigrescens with HPLC as release of aglycones while they used TLC to confirm liberation of whole acuminose.
3.2.3. Purification and Characterization of Acuminosidases
The acuminosidases have been isolated and purified by combination of standard procedures involving precipitation with ammonium sulphate or acetone, ion exchange chromatography and gel permeation. Some of the enzymes were expressed in bacterial or yeast hosts to obtain the target.
Furcatin hydrolase (FH) was for the first time isolated from fresh leaves of V. furcatum by fractional precipitation with ammonium sulphate [17]. The final extract after filtration and dialysis contained 4 mg protein/mL and was stable for several months at 3 °C. Ahn et al. [18] used precipitation of the leaf extract with acetone and ammonium sulphate followed by separation on CM-Toyopearl and DEAE-Sepharose, achieving 5.78-fold purification of FH (final yield 20.5%). The purified enzyme contained major protein with molecular weight 56 kDa, with minor contaminants having 60 and 45 kDa.
The same authors used cDNA cloning to amplify 18 cDNA fragments obtained with specific primers constructed according to highly conserved regions for plant β-glucosidases from family 1. Using pGEM-T vectors, three different glucosidases were identified including partial clone encoding putative β-glucosidase showing greatest homology to tea leaf β-primeverosidase. cDNA of FH consisted of 12 untranslated nucleotides at 5’end, coding sequence of 1617 nucleotides and 229 untranslated nucleotides including poly(A) tail at 3’end. Open reading frame encoded protein consisted of 538 amino acid residues with calculated molecular mass 60.625 Da. [18]. Recombinant FH was expressed as fused protein with GST (glutathione S-transferase) in Escherichia coli by expression vector pGEX-5X as 88-kDa protein and released by factor Xa cleavage as free FH. Expression of recombinant FH could be visualized using β-primeverosidase antibody, which recognized both fused and free FH. The purified recombinant protein showed acuminosidase activity towards furcatin, with no observation of formation of free monosaccharides [18].
Hosel and Barz [34] extracted β-glycosidase from various parts of Cicer arietinum L., pre-purified by precipitation with MnCl2 and ammonium sulphate and further separated the extract by combination of chromatography on Sephadex G-25, DEAE-cellulose, CM-Sephadex C-50 and Sephadex G-200. pNP β-d-glucopyranoside was used to monitor the purification process.
The endoglycosidase from grape berry skins is a membrane-bound protein and therefore the detergent (1% polyethylene glycol, Mw 4000 Da) was used to assist its extraction [20]. The enzyme was further separated on DEAE Sepharose CL-6B followed by FPLC gel filtration on a Protein-Pak SW with resulting purification factor 1071. A β-glucosidase however co-eluted with the isolated endoglycosidase and occurred in all fractions.
Glycosidase from Dalbergia nigrescens Kurz was extracted from homogenized pre-soaked seeds and purified by combination of precipitation steps with chromatography on DEAE-Sepharose and Sephacryl S-300. The enzyme had a subunit molecular weight of 62–63 kD and was probably a tetramer [13].
The same enzyme was cloned using cDNA from D. nigrescens seeds with PCR primers designed from the cDNA sequence of the related β-glucosidase from D. cochinchinensis. RNA isolated from immature and germinating seeds was used as a template. The sequence of the initial cDNA clones was further used to design two new specific primers, which were used to clone acuminosidase from D. nigrescens. The recombinant enzyme was expressed as a fusion protein with yeast α-factor prepropeptide in Pichia pastoris. Approximately 63 kDa protein was purified from desalted media by adsorption to cobalt chelating resin (IMAC), which represented the subunit of previously declared tetramer structure of the native enzyme [34]. Molecular and catalytic properties of purified acuminosidases are summarized in Table 4.
3.2.4. Computational and Structural Analysis of Known Diglycosidases and Comparison with Endo-Apiosidase
Furcatin hydrolase (FH) isolated from the fresh leaves of V. furcatum [17] was stable above pH 5 and below 50 °C. In a more recent study [18], different tissues of V. furcatum were analyzed for the presence of FH (young and fully expanded leaf, immature and mature fruit and stem) by RT-PCR. FH transcript was detected only in leaves regardless of their age. The amino acid sequence of FH (Uniprot, Q75W17) was also analysed and compared to databases to compare with related glycosidases. Based on the presence of highly conserved sequence motifs (NEP sequence motif with Glu residue as acid-base catalyst; ITENG sequence with Glu residue as catalytic nucleophile, but with exception of Asn replaced by Cys; residues involved in binding of glycone (glucose) moiety conserved at Gln88, His192, Asn237, Glu238, Glu501, and Trp502), FH was classified as a family 1 glycosyl hydrolase. Comparison of amino acid sequence of FH with primeverosidase and more related glycosidases with high sequence identity and conserved regions suggested that diglycosidases interact with glucose unit of the substrate in a similar manner as monoglycosidases. Phylogenetic tree analysis revealed the most related structure suitable for homology modelling (PDB: 1CBG) and also suggested that FH and primeverosidase from Camellia sinensis evolved from a common ancestor of plant monoglycosidases unlike vicianin hydrolase (Vicia angustifolia), which was far from these diglycosidases, but close to other monoglycosidases [38]. D. nigrescence and C. arientinum β-glycosidases with acuminosidase activity discussed in this review occur at a more distant branch together with few other cyanogenic and non-cyanogenic β-glucosidases. According to our verification of BLAST (basic local alignment search tool) data, the most related proteins to FH are two tobacco “furcatin hydrolase-like” proteins predicted by automated computational analysis using gene prediction method: Gnomon, from genomic sequences of Nicotiana tomentosiformis (NW_008931554.1) and Nicotiana tabacum (NW_015877216.1). Figure 6 shows a phylogenetic tree constructed from the mentioned sequences after Clustal W alignment.
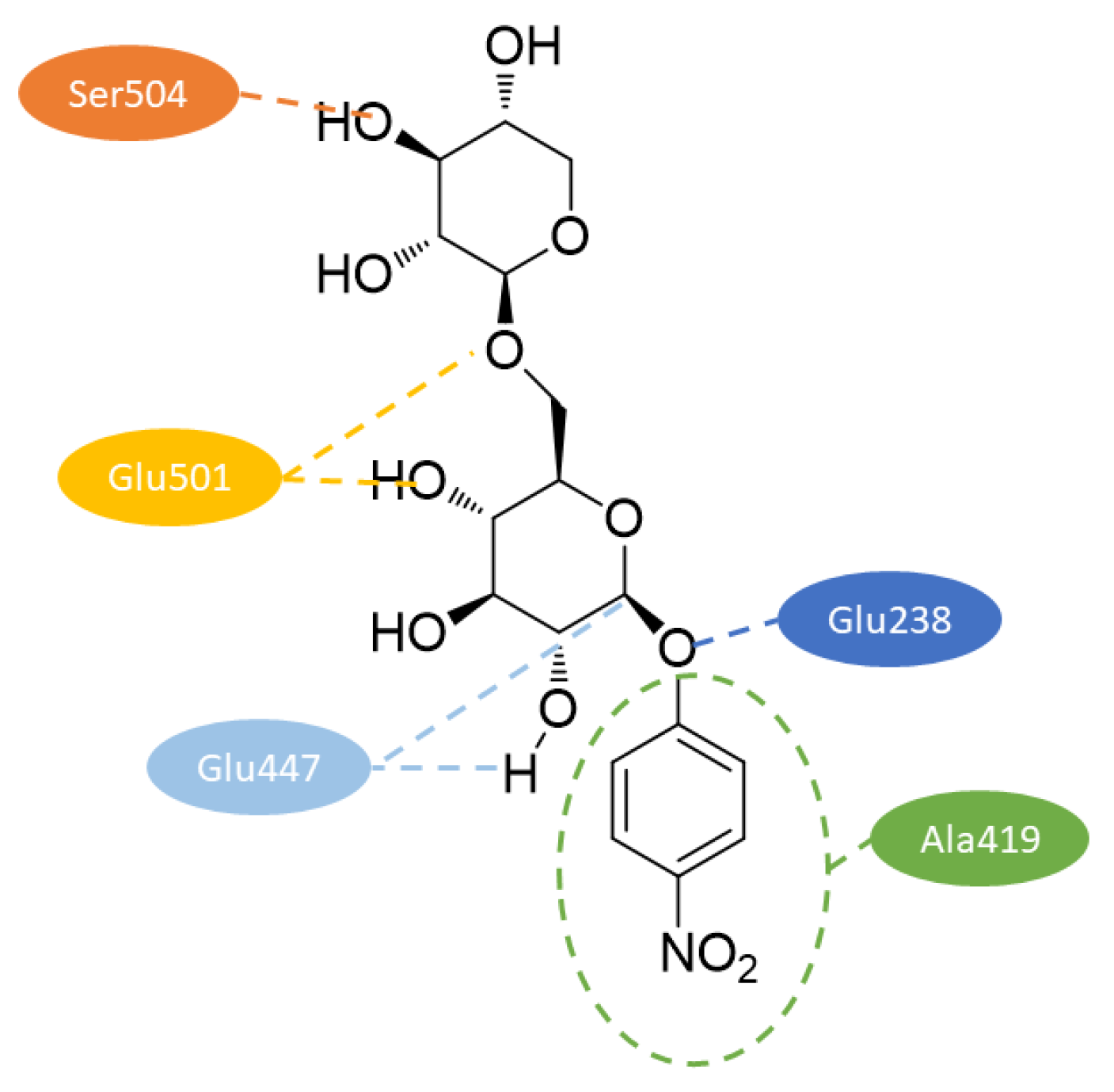
Model structure including pNP β-primeveroside in substrate binding mode formed a typical TIM barrel fold. Two regions with no corresponding equivalent in 1CBG represented two loops, which were far from the substrate, so probably without catalytic significance. Figure 7 represents interactions with pNP β-primeveroside in the catalytic pocket. Carboxylate groups of catalytic residues Glu238 and Glu447 were localized in a position suitable for catalysis through the retaining catalytic mechanism. The oxygen of Glu447 carboxylate, which acts as a catalytic nucleophile, was close enough to C1 and hydrogen of O2 in the glucose unit of the substrate. The oxygen in the carboxylate of Glu238 that acted as the acid/base catalyst was near the glycosidic oxygen between the aglycone and the glycone. In order to compare substrate specificity of mono and diglycosidases, the close surroundings of the substrate in the model structure were analyzed. Among 19 selected residues, Ala419 and Ser504 were further analyzed, because they were specifically conserved within FH and primeverosidase. Ala419 was near to entrance of the slot close to aglycone of the substrate and in alignment with other glycosidases was substituted by conserved Trp which aromatic residue anchors aglycone at the entrance. Ser504 was at the bottom of the catalytic hole and close enough to O3 of xylose as external saccharide, the same position in alignment was not conserved among monoglycosidases. Site directed mutagenesis of these two residues resulted in remarkable changes in hydrolytic activity. The replacement of Ala419 by Trp as residue with larger side chain cause reduction in activity towards furcatin, pNP-β-primeveroside, p-allylphenyl (pAP) β-glucoside and pNP β-glucoside, while replacement by Val decreased activity mainly towards pAP glycosides (substrates with natural aglycone). The replacement of Ser504 by Trp (bulky side chain and lacking hydroxyl groups) resulted in rapid decrease of activity towards diglycosides (less than 2%), in the case of monoglycosides only to 20–30%. Replacement by Ala resulted in an increase of activity to 113% toward furcatin but slightly decreased toward other substrates. Analysis of kinetic parameters determined with pNP β-primeveroside revealed that replacement of Ala419 decreased Km values so enhanced affinity of mutants, but decreased catalytic rates (kcat), thus decrease overall activity (kcat/Km). Replacement of Ser504 by Trp decreased both affinity and catalytic rates, so indicated that bulky side chain prevented binding of pNP β-primeveroside by steric hindrance. The replacement by Ala resulted in a comparable catalytic rate, but Km was slightly increased, which suggests that the substrate was bound to mutate at an accurate position in the active site [38].
In silico analysis of the sequence suggested that FH has subcellular localization in chloroplasts (putative signal peptide for chloroplast transport at N-terminus). This corresponds to results obtained after expression of GFP (Green fluorescent protein) fused protein in Arabidopsis leaf cells and onion epidermal cells [18].
Phylogenetic analysis of β-glycosidase from Dalbergia nigrescens with acuminosidase activity showed that both enzymes are most closely related to legume isoflavonoid β-glucosidases and cyanogenic β-glucosidases with distant relationship to furcatin hydrolase and β-primeverosidase. Moreover, the analysis suggested that isoflavonoid β-glucosidases, which hydrolyze also glycosides with 6-O-modified glucose are derived from the same branch of GH1 as the defense-related glucosidases and are more closely related to them than to non-isoflavonoid diglycosidases. Full length cDNA sequence (GenBank Accession No. AF163097) consisted of 1964 nucleotides (ORF 1593) encoding 531 amino acid precursor protein (including predicted 23 AA signal sequence). The calculated molecular weight of mature protein (508 AA) was 60,509 Da. The predicted N-terminal sequence, however, did not correspond to the previous chemical sequencing of isolated protein, so possible proteolysis occurred during purification or in the plant. The results also indicated that at least three of the five putative glycosylation sites were glycosylated [35].
Catalytic nucleophile (Glu396) was, again, as in case of FH, found in the (Y)ITENG motif and the catalytic acid/base (Glu182) within (TI)NEP sequence motif both conserved within GH1 family. Ten AA residues were analyzed to determine aglycone specificity. Homology modelling based on the most related cyanogenic β-glucosidase (PDB: 1CBG, the same as used for FH) identified residues most likely involved in the binding site. Further comparison with D. cochinchinensis β-glucosidase revealed that residues 454 and 455 differ between both enzymes and therefore may contribute to significantly different catalytic efficiency toward mono and diglycosides. These residues were therefore mutated in D. cochinchinensis β-glucosidase and kinetic parameters of the mutants were compared. Double mutation of A454S/E455G had little effect on hydrolysis of pNP glycosides, but improved hydrolysis of natural acuminosides by D. nigrescens glycosidase in such a manner which could not be explained by combination of effects of individual mutations, which has no ability to hydrolyze these substrates, indicated certain synergism or cooperation between two residues could explain this effect. The improved catalytic efficiency was anyway very low (2–3%). The combination of Ser454 and Gly455 apparently helped the enzyme to bind the isoflavonoid ring in the proper position for hydrolysis of 7-O-glycosyl more than Ala and Glu in D. cochinchinensis β-glucosidase. Further analysis suggested that apiosyl residue would be expected to sit in the entry cleft across from or even against the aglycone, where it might interact with putative aglycone binding residues and O6 of glucose sits 9-10 Å away from Ser454, so apiosyl binding may be indirect unlike FH and primeverosidase, which can better explain the hydrolysis of isoflavone glucosides than diglycosides [35].
4. Potential Applications of Apiose-Relevant Glycosidases
A pioneering and very extensive study of the role of apiosidases and acuminosidases in aroma development during the process of wine production was done by the research team around Ziya Günata [19,20,23,24,26,29]. The sensory perception of wine is very complex, being formed by a variety of taste and odour-active compounds. Among them, monoterpenols play a very significant role. In some types of wines, up to 90% of the monoterpenols are glycosylated [39], and therefore do not affect the grape odor. These glycosides, mainly diglycosides and β-glucosides [40], can be hydrolyzed during the process of grape processing and wine maturation and thus significantly contribute to the final sensory properties of wine. Among the grape glycosides, acuminosides can be hydrolyzed either directly by acuminosidases or sequentially by cooperation of added apiosidase and (added or naturally occurring) β-glucosidase. As mentioned in the Section 3.1.3., fungal apiosidase hydrolyzed the extracted or synthetic grape aroma precursors linalyl, geranyl, neryl and linalyloxide acuminosides [23,26]. The authors, in their patent [41], claim use of pectinase Klerzyme 200 (comprising apiosidase and β-glycosidase activities) in the complete hydrolysis of neryl and geranyl acuminosides in Muscat wine accompanied by the increase of the free terpenols nerol and geraniol. It is, however, a fact that β-apiosidases are presently used in the winemaking more-less non-intentionally and empirically, since the enzymes are not marketed as such, but are present as a side-activity of used industrial glycanases. A substantial part of the commercial enzymes comprising β-apiosidase summarized in Table 1 are glycanases dedicated for winemaking, but only a few of them are claimed by their producers to contain apiosidase. A similar situation occurs in the role of acuminosidase in winemaking. Although there is no industrial acuminosidase available, grape berry skin comprises rather high levels of endoglycosidase able to hydrolyze various monoterpenyl acuminosides [20]. The enzymatic hydrolysis of these precursors thus depends on the particular technology enabling manifestation of this activity in berry skin.
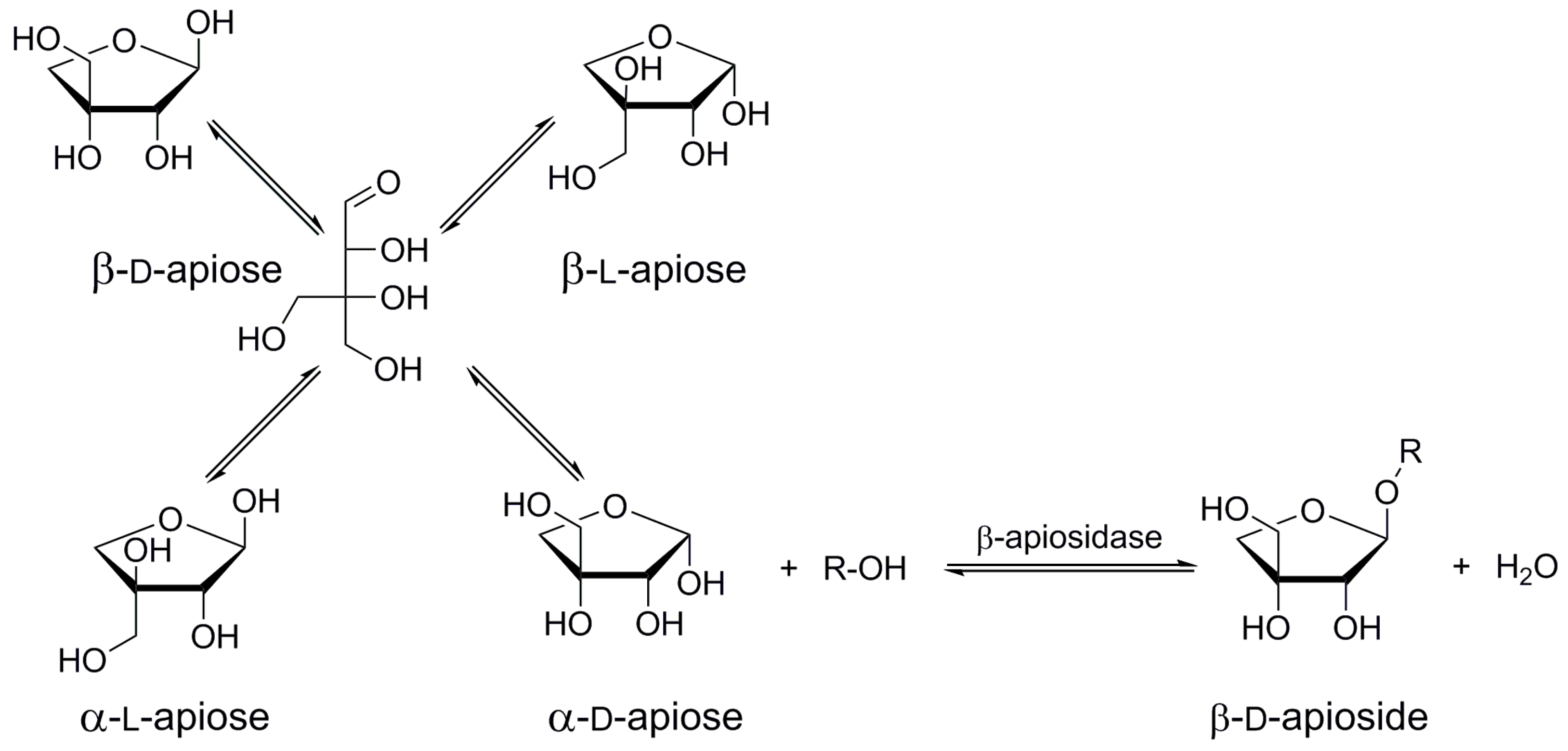
Due to a growing knowledge of natural complex glycosides responsible for biological activities of medical plants, there is an interest in the synthesis of such valuable substances. The use of glycosidases in their preparation is a logical alternative to processes of organic synthesis. Direct glycosidase-driven apiosylation of free aglycones or simple glycosides is however hampered by the practical unavailability of retaining apiosidases. A very recent study of β-apiosidase from Aspergillus aculeatus revealed its inverting reaction mechanism [31]. The inverting glycosidases are unable to catalyze transglycosylations and can form a glycosidic bond only by reverse hydrolysis starting from free monosaccharide. Free apiose, however, does not occur naturally and its synthesis may in fact be more complicated than the preparation of apiosides. Moreover, as a branched sugar, free apiose exists in four anomeric forms in solution (Figure 8), thus lowering the probability of reverse hydrolysis, since only α-d-anomer can take part in the reaction catalyzed by the inverting β-d-apiosidase. This anomer stands for ca. 30% of all anomeric forms present in the equilibrated solution of free apiose [31]. Most of the identified β-apiosidases are produced by the genus Aspergillus [22,26,27,28]; it is therefore probable that the enzymes are working under the same inverting mechanism. The only exception is the endoapiosidase from Bacteroides thetaiotaomicron acting by retention mechanism of glycoside hydrolysis [30]. Although this enzyme is already marketed, its price and availability of natural substrates (fragments of rhamnogalacturonan II) do not make it a practical biocatalyst in the synthesis of apiosides and apiosylated glycosides at a reasonable scale. The search for new retaining β-apiosidases accepting simple synthetic apiosides as substrates for transapiosylations therefore remains a permanent challenge in the field of synthesis of apiosylated bioactive molecules. In the present situation, when there are inexpensive inverting apiosidases widely available on the market, it seems, however, reasonable to focus also on the development of a simple multi-gram method of synthesis of free apiose, enabling biocatalytic preparation of relevant β-apiosides by reverse hydrolysis. It should, however, be stressed that to date, enzymatic synthesis catalyzed by apiosidases (neither by reverse β-apiosylation nor by transapiosylation) has not been reported.
Several diglycosidases were proven to act by the retaining mechanism or positively tested in the ability to catalyze transglycosylations [32,42,43,44,45,46]. This fact makes them good candidates for catalysis of one-step synthesis of new diglycosidic structures. Their high specificity toward the aglycone (especially in case of plant diglycosidases like rutinosidases [47]) may ultimately be a limiting factor due to a narrow pool of suitable substrates like natural glycosides. Contrary to rutinosides, the natural acuminosides are available on the market in amounts and prices prohibitive for their synthetic use. The unavailability of appropriate substrates is probably the reason why there is no reported screening of microbial acuminosidases. Synthetic acuminosides serving as the enzyme probes as well as substrates for synthetic reactions (transacuminosylations) are therefore highly demanded. Actually, the only so far realized transacuminosylation was reported as early as in 1961 by Imaseki and Yamamoto [17]. The authors used furcatin to acuminosylate various aliphatic alcohols with furcatin hydrolase and had observed that the products could also serve as FH substrates. The later study on Ahn et al. [18] confirmed the retention mechanism of FH. Acceptation of simple alkyl acuminosides together with methods of expression of the enzyme [18] makes this enzyme a promising catalyst for the production of targeted acuminosides. Another interesting enzyme, the grape endoglycosidase deserves also attention of scientists since, although not yet cloned, it is obtainable from the crop produced worldwide and has an interestingly wide substrate specificity, making it a universal diglycosidase. The search for new sources of acuminosidases or endo-glycosidases accepting acuminosides remains a task for researchers in the field of aroma development in fermented foods and beverages, as well as in biocatalysis.
5. Conclusions
In conclusion, we presented here the biochemical characteristics and potential industrial and biocatalytic applications of glycosidases specific for D-apiose and acuminose. Although these enzymes may find use in the food and beverage industries as well as in the synthesis of pharmacologically active substances, their new sources are highly demanded along with available substrates serving either as detection probes or glycosyl donors in synthetic applications.
Author Contributions
Conceptualization, V.M. and M.M.; writing—original draft preparation, E.K.P.; writing—review and editing, V.M. and E.K.P.; supervision, V.M.; project administration, M.M.; funding acquisition, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovak Research and Development Agency, grant number APVV-18-0188 and by the Slovak Grant Agency for Science VEGA, grant number 2/0126/19.
Acknowledgments
The contribution of COST Action CA18103 “INNOGLY-Innovation with Glycans: new frontiers from synthesis to new biological targets” supported by COST (European Cooperation in Science and Technology), in promoting interaction, exchange of knowledge and collaborations in the field of glycosciences is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pičmanová, M.; Møller, B.L. Apiose: One of nature’s witty games. Glycobiology 2016, 26, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Darvill, A.G.; McNeil, M.; Albersheim, P. Structure of plant cell walls: VIII. A new pectic polysaccharide. Plant Physiol. 1978, 62, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Nepogodiev, S.A.; Fais, M.; Hughes, D.L.; Field, R.A. Synthesis of apiose-containing oligosaccharide fragments of the plant cell wall: Fragments of rhamnogalacturonan-II side chains A and B, and apiogalacturonan. Org. Biomol. Chem. 2011, 9, 6670–6684. [Google Scholar] [CrossRef] [PubMed]
- Duff, R. The occurrence of apiose in Lemna (duckweed) and other angiosperms. Biochem. J. 1965, 94, 768–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Toumy, S.A.; Omara, E.A.; Nada, S.A.; Bermejo, J. Flavone C-glycosides from Montanoa bipinnatifida stems and evaluation of hepatoprotective activity of extract. J. Med. Plants Res. 2011, 5, 1291–1296. [Google Scholar] [CrossRef]
- Gupta, S.R.; Seshadri, T.R. A study of apiin from the parsley seeds and plant. Proc. Indian Acad. Sci.-Sect. A 1952, 35, 242. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Xu, L.; Hao, S.Y.; Li, Y.; Zhang, Z.Q. HPLC method determination of isoliquiritin apioside and isoliquiritin in rat plasma for application in pharmacokinetic study after an oral administration of Zhigancao extract. J. Anal. Methods Chem. 2012, 1, 364013. [Google Scholar] [CrossRef]
- Kernan, M.R.; Amarquaye, A.; Chen, J.L.; Chan, J.; Sesin, D.F.; Parkinson, N.; Ye, Z.; Barrett, M.; Bales, C.; Stoddart, C.A.; et al. Antiviral phenylpropanoid glycosides from the medicinal plant Markhamia lutea. J. Nat. Prod. 1998, 61, 564–570. [Google Scholar] [CrossRef]
- Koike, K.; Li, W.; Liu, L.; Hata, E.; Nikaido, T. New phenolic glycosides from the seeds of Cucurbita moschata. Chem. Pharm. Bull. 2005, 53, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Koike, K.; Tatsuzaki, M.; Koide, A.; Nikaido, T. Cucurbitosides F-M, acylated phenolic glycosides from the seeds of Cucurbita pepo. J. Nat. Prod. 2005, 68, 1754–1757. [Google Scholar] [CrossRef]
- Voirin, S.; Baumes, R.; Bayonove, C.; M’Bairaroua, O.; Tapiero, C. Synthesis and NMR spectral properties of grape monoterpenyl glycosides. Carbohydr. Res. 1990, 207, 39–56. [Google Scholar] [CrossRef]
- Wu, B.; Takahashi, T.; Kashiwagi, T.; Tebayashi, S.; Kim, C.S. New flavonoid glycosides from the leaves of Solidago altissima. Chem. Pharm. Bull. 2007, 55, 815–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuankhayan, P.; Hua, Y.; Svasti, J.; Sakdarat, S.; Sullivan, P.A.; Ketudat Cairns, J.R. Purification of an isoflavonoid 7-O-beta-apiosyl-glucoside beta-glycosidase and its substrates from Dalbergia nigrescens Kurz. Phytochemistry 2005, 66, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sung, S.H.; Kim, Y.C. Two new hepatoprotective stilbene glycosides from Acer mono leaves. J. Nat. Prod. 2005, 68, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Jia, X. Two new coumarin biosides from Angelica dahurica. Chem. Nat. Compd. 2008, 44, 692–695. [Google Scholar] [CrossRef]
- Lee-Juian, L.; Long, Z.L.; Ruangrungsi, N.; Cordell, G.A. 3-Hydroxycoumarin glycosides from Alyxia reinwardti var. Lucida. Phytochemistry 1993, 34, 825–830. [Google Scholar] [CrossRef]
- Imaseki, H.; Yamamoto, T. A furcatin hydrolyzing glycosidase of Viburnum furcatum Blume. Arch. Biochem. Biophys. 1961, 92, 467–474. [Google Scholar] [CrossRef]
- Ahn, Y.O.; Mizutani, M.; Saino, H.; Sakata, K. Furcatin hydrolase from Viburnum furcatum Blume is a novel disaccharide-specific acuminosidase in glycosyl hydrolase family 1. J. Biol Chem. 2004, 279, 23405–23414. [Google Scholar] [CrossRef] [Green Version]
- Günata, Z.; Bitteur, S.; Brillouet, J.-M.; Bayonove, C.; Cordonnier, R. Sequential enzymic hydrolysis of potentially aromatic glycosides from grape. Carbohydr. Res. 1988, 184, 139–149. [Google Scholar] [CrossRef]
- Günata, Z.; Blondeel, C.; Vallier, M.J.; Lepoutre, J.P.; Sapis, J.C.; Watanabe, N. An endoglycosidase from grape berry skin of cv. M. Alexandria hydrolyzing potentially aromatic disaccharide glycosides. J. Agric. Food Chem. 1998, 46, 2748–2753. [Google Scholar] [CrossRef]
- Ogawa, K.; Ijima, Y.; Guo, W.; Watanabe, N.; Usui, T.; Dong, S.; Tong, Q.; Sakata, K. Purification of a β-primeverosidase concerned with alcoholic aroma formation in tea leaves (cv. Shuixian) to be processed to Oolong tea. J. Agric. Food Chem. 1997, 45, 877–882. [Google Scholar] [CrossRef]
- Günata, Z.; Dugelay, I.; Vallier, M.J.; Sapis, J.C.; Bayonove, C. Multiple forms of glycosidases in an enzyme preparation from Aspergillus niger: Partial characterization of a β-apiosidase. Enzym. Microb. Technol. 1997, 21, 39–44. [Google Scholar] [CrossRef]
- Guo, W.; Salmon, J.M.; Baumes, R.; Tapiero, C.; Günata, Z. Purification and some properties of an Aspergillus niger β-apiosidase from an enzyme preparation hydrolyzing aroma precursors. J. Agric. Food Chem. 1999, 47, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Sarry, J.-E.; Günata, Z. Plant and microbial glycoside hydrolases: Volatile release from glycosidic aroma precursors. Food Chem. 2004, 87, 509–521. [Google Scholar] [CrossRef]
- Bojarová-Fialová, P.; Křen, V. Enzymatic approaches to O-glycoside introduction: Glycosidases. In Comprehensive Glycoscience; Elsevier: Amsterdam, The Netherlands, 2007; pp. 453–487. [Google Scholar] [CrossRef]
- Dupin, I.; Gunata, Z.; Sapis, J.C.; Bayonove, C.; Mbairaroua, O.; Tapiero, C. Production of beta-apiosidase by Aspergillus-niger–partial purification, properties, and effect on terpenyl apiosylglucosides from grape. J. Agric. Food Chem. 1992, 40, 1886–1891. [Google Scholar] [CrossRef]
- Kis, P.; Potocká, E.; Mastihuba, V.; Mastihubová, M. Efficient chemoenzymatic synthesis of 4-nitrophenyl β-d-apiofuranoside and its use in screening of β-d-apiofuranosidases. Carbohydr. Res. 2016, 430, 48–53. [Google Scholar] [CrossRef]
- Karkeszová, K.; Illeová, V.; Kis, P.; Mastihuba, V.; Polakovič, M. Apiin-induction of β-apiosidase production by Aspergillus sp. strains. Acta Chim. Slov. 2020, 13, 72–76. [Google Scholar] [CrossRef]
- Sarry, J.E.; Grimplet, J.; Sommerer, N.; Vallier, M.J.; Pradal, M.; Mondolot, L.; Andary, C.; Günata, Z.; Romieu, C. Combined mass mapping and biochemical characterization of grape β-glycosidase-enriched extract. Protein J. 2008, 27, 258–266. [Google Scholar] [CrossRef]
- Ndeh, D.; Rogowski, A.; Cartmell, A.; Luis, A.S.; Baslé, A.; Gray, J.; Venditto, I.; Briggs, J.; Zhang, X.; Labourel, A.; et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 2017, 544, 65–70. [Google Scholar] [CrossRef]
- Mastihuba, V.; Karnisova Potocka, E.; Uhliarikova, I.; Kis, P.; Kozmon, S.; Mastihubova, M. Reaction mechanism of beta-apiosidase from Aspergillus aculeatus. Food Chem. 2019, 274, 543–546. [Google Scholar] [CrossRef]
- Mizutani, M.; Nakanishi, H.; Ema, J.; Ma, S.J.; Noguchi, E.; Inohara-Ochiai, M.; Fukuchi-Mizutani, M.; Nakao, M.; Sakata, K. Cloning of beta-primeverosidase from tea leaves, a key enzyme in tea aroma formation. Plant Physiol. 2002, 130, 2164–2176. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, L.S.; Breccia, J.D. Functional and biotechnological insights into diglycosidases. Biocatal. Biotransformation 2011, 29, 103–112. [Google Scholar] [CrossRef]
- Hosel, W.; Barz, W. Beta-glucosidases from Cicer arietinum L. Purification and properties of isoflavone-7-O-glucoside-specific beta-glucosidases. Eur. J. Biochem. 1975, 57, 607–616. [Google Scholar] [CrossRef]
- Chuankhayan, P.; Rimlumduan, T.; Tantanuch, W.; Mothong, N.; Kongsaeree, P.T.; Metheenukul, P.; Svasti, J.; Jensen, O.N.; Ketudat Cairns, J.R. Functional and structural differences between isoflavonoid β-glycosidases from Dalbergia sp. Arch. Biochem. Biophys. 2007, 468, 205–216. [Google Scholar] [CrossRef]
- Baumgertel, A.; Grimm, R.; Eisenbeiss, W.; Kreis, W. Purification and characterization of a flavonol 3-O-beta-heterodisaccharidase from the dried herb of Fagopyrum esculentum Moench. Phytochemistry 2003, 64, 411–418. [Google Scholar] [CrossRef]
- Mazzaferro, L.S.; Breccia, J.D. Quantification of hesperidin in citrus-based foods using a fungal diglycosidase. Food Chem. 2012, 134, 2338–2344. [Google Scholar] [CrossRef]
- Daiyasu, H.; Saino, H.; Tomoto, H.; Mizutani, M.; Sakata, K.; Toh, H. Computational and experimental analyses of furcatin hydrolase for substrate specificity studies of disaccharide-specific glycosidases. J. Biochem. 2008, 144, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Morrison, J.C.; Adams, D.O.; Noble, A.C. Distribution of free and glycosidically bound monoterpenes in the skin and mesocarp of Muscat of Alexandria grapes during development. J. Agric. Food Chem. 1991, 39, 514–518. [Google Scholar] [CrossRef]
- Mateo, J.J.; Jiménez, M. Monoterpenes in grape juice and wines. J. Chromatogr. A. 2000, 881, 557–567. [Google Scholar] [CrossRef]
- Gunata, Z.; Bitteur, S.; Baumes, R.; Brillouet, J.-M.; Tapiero, C.; Bayonove, C.; Cordonnier, R. Process for Obtaining Aroma Components and Aromas from Their Precursors of a Glycosidic Nature, and Aroma Components and Aromas Thereby Obtained. Patent No. EP0416713B2, 21 September 1990. [Google Scholar]
- Karnišová Potocká, E.; Mastihubová, M.; Mastihuba, V. Transrutinosylation of tyrosol by flower buds of Sophora japonica. Food Chem. 2021, 336, 127674. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Ohno, F.; Yamauchi, Y.; Kato, M.; Makabe, H.; Nakamura, S. Enzymatic synthesis of novel phenol acid rutinosides using rutinase and their antiviral activity in vitro. J. Agric. Food Chem. 2013, 61, 9617–9622. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, L.; Piñuel, L.; Minig, M.; Breccia, J.D. Extracellular monoenzyme deglycosylation system of 7-O-linked flavonoid β-rutinosides and its disaccharide transglycosylation activity from Stilbella fimetaria. Arch. Microbiol. 2010, 192, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, L.S.; Piñuel, L.; Erra-Balsells, R.; Giudicessi, S.L.; Breccia, J.D. Transglycosylation specificity of Acremonium sp. α-rhamnosyl-β-glucosidase and its application to the synthesis of the new fluorogenic substrate 4-methylumbelliferyl-rutinoside. Carbohydr. Res. 2012, 347, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Tsuruhami, K.; Mori, S.; Amarume, S.; Saruwatari, S.; Murata, T.; Hirakake, J.; Sakata, K.; Usui, T. Isolation and characterization of a β-primeverosidase-like enzyme from Penicillium multicolor. Biosci. Biotech. Biochem. 2006, 70, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Koseki, T.; Ishikawa, M.; Kawasaki, M.; Shiono, Y. β-Diglycosidases from microorganisms as industrial biocatalysts: Biochemical characteristics and potential applications. Appl. Microbiol. Biotechnol. 2018, 102, 8717–8723. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
General structure of β-d-apiofuranoside (3-C-(hydroxymethyl)-d-erythrofura-noside).

Figure 2.
Structures of two natural apioglucoses.

Figure 3.
Sequential vs. one-step hydrolysis of β-acuminosides.

Figure 4.
Hydrolysis of synthetic probes and oligosaccharide in apiosidase assays.

Figure 5.
Structures of selected acuminosidase substrates.

Figure 6.
Phylogenetic tree of diglycosidases and related monoglycosidases according to BLAST taken from Daiyasu et al. [37], but updated with the most related furcatin hydrolase-like proteins, and with highlighted enzymes comprising acuminosidase activity. Enzymes listed with GenBank accession number, protein name and source name, analyzed by software MEGA v.10.2.6, aligned by Clustal W and phylogenetic tree constructed using Neighbor-Joining method.
Figure 6.
Phylogenetic tree of diglycosidases and related monoglycosidases according to BLAST taken from Daiyasu et al. [37], but updated with the most related furcatin hydrolase-like proteins, and with highlighted enzymes comprising acuminosidase activity. Enzymes listed with GenBank accession number, protein name and source name, analyzed by software MEGA v.10.2.6, aligned by Clustal W and phylogenetic tree constructed using Neighbor-Joining method.

Figure 7.
2D diagram of interactions with pNP β-primeveroside in model structure of FH constructed according to data from [38].
Figure 7.
2D diagram of interactions with pNP β-primeveroside in model structure of FH constructed according to data from [38].

Figure 8.
Theoretical synthesis of β-d-apiosides by reverse hydrolysis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Commercial enzymes comprising β-apiosidase side-activity.
| Enzyme Preparation | Producer | Microbial Source | Original Designation | References |
|---|---|---|---|---|
| Rapidase AR 2000 (Klerzyme 200) | DSM | Aspergillus niger | Pectinases, polygalacturonases, β-glucanases | [22,24,26,27] |
| Pectinase 263 | Gist Brocades | A. niger | Pectinases, polygalacturonases | [22,24,26] |
| Pectolase 3PA 400 | Grinsted | A. niger | Pectinases, polygalacturonases | [22,24,26] |
| Ultrazym 100 | Ciba-Geigy | A. niger | Pectinases, polygalacturonases β-glucanases | [22,24,26,27] |
| Lallzyme BETA | Lallemand | A. niger | Pectinases, polygalacturonases, β-glucanases | [27] |
| Rapidase Expression | DSM | A. niger | Pectinases, polygalacturonases, β-glucanases | [27] |
| α-galactosidase DS | Amano | A. niger | α-Galactosidase | [27] |
| Lallzyme Cuvée Blanc | Lallemand | A. niger | β-Glucosidase | [27] |
| Lipolyve AN | Lyven | A. niger | Lipases | [27] |
| Enzeco Lipase | Enzyme Development Corporation | A. niger | Lipases | [27] |
| Lipase A | Amano | A. niger | Lipases | [27] |
| Viscozyme L | Novozymes | A. aculeatus | β-Glucanases, cellulases | [27] |
| Novozym 188 | Novozymes | A. niger | β-Glucanases, cellulases | [27] |
| Pectinex UF | Novozymes | A. aculeatus | Pectinases, polygalacturonases | [27] |
| Ultrazym AFP | Novozymes | Aspergillus sp. | Pectinases, polygalacturonases | [27] |
| TP714L | Biocatalysts | Aspergillus sp. | Pectinases, polygalacturonases | [27] |
| Peclyve LVG | Lyven | A. niger | Pectinases, polygalacturonases | [27] |
| Depol 692L | Biocatalysts | Aspergillus sp. & Trichoderma | β-Glucanases, cellulases | [27] |
Table 2.
Commercial enzyme comprising β-apiosidase activity.
| Source | Molecular Mass [kDa] | Km 1 [mM] | Vmax 1 [nkat/mg] | Optimal pH | Optimal Temperature [°C] | pI |
|---|---|---|---|---|---|---|
| A. niger [26] | 38 | 16 | 0.192 | 5.6 | 50 | - |
| Klerzyme 200 [22] | 84 | 3.3 | 216 | 5.0–6.0 | 50–60 | - |
| Klerzyme 200 [23] | 120 | 4.16 | 2460 | 5.0 | 40 | 3.5 |
| B. thetaiotaomicron [30] | - | - | - | 6.5–7.5 | 37 | - |
1 Values are determined with 4-nitrophenyl β-d-apiofuranoside as the substrate.
Table 3.
Summarization of known acuminosidases and their activity towards various substrates.
| Source | Tested Activity | |
|---|---|---|
| Confirmed | Zero or Negligible | |
| Viburnum furcatum [17] | Furcatin n-Propyl β-acuminoside n-Butyl β-acuminoside Ethyleneglycol monomethylether β-acuminoside | Apiin Rutin |
| Viburnum furcatum [18] | Furcatin 4-Allylphenyl β-d-glucopyranoside pNP β-primeveroside 2-Phenylethyl β-primeveroside | Vicianin pNP β-gentiobioside pNP β-d-glucopyranoside Amygdalin Prunasin 2-phenylethyl-β-gentiobioside 2-Phenylethyl β-d-glucoside |
| Cicer arietinum L. [34] | Biochanin A 7-O-β-d-glucoside Biochanin A 7-O-β-apioglucoside Formononetin 7-O-β-d-glucoside 2-Methyl-4’-nitro-isoflavone 7-O-β-d-glucoside Apiigenin 7-O-β-d-glucoside 4Mum β-d-glucopyranoside pNP β-d-glucopyranoside pNP β-d-galactopyranoside oNP β-d-glucopyranoside Salicin | Formononetin 7-O-β-cellobioside Genistein 4’-O-β-d-glucoside Apigenin 7-O-β-apioglucoside Kaempferol 3-O-β-d-glucoside Kaempferol 3-O-β-apioglucoside Quercetin 3-O-β-rutinoside 4Mum β-d-glucuronide 4Mum β-d-N-acetylglucosamide pNP α-d-glucoside Phloridzin Cellobiose |
| Dalbergia nigescens Kurz [13] | Dalpatein β-acuminoside Dalnigrein β-acuminoside Genistin (genistein 7-O-Glcp) Daidzin (daidzein 7-O-Glcp) pNP β-d-fucoside pNP β-d-glucopyranoside | - |
| Vitis vinifera [20] | 2-phenylethyl β-rutinoside pNP β-rutinoside Neryl β-rutinoside Linalyl β-rutinoside Neryl 3-O-α-l-arabinofuranosyl- (1→6)-β-d-glucopyranoside Linaloyl 3-O-α-l-arabinofuranosyl- (1→6)-β-d-glucopyranoside Geranyl β-acuminoside Linalyl β-acuminoside 4Mum β-vicianoside Eugenyl β-primeveroside | Peltatoside 1 |
1 Hydrolysis to monosaccharides.
Table 4.
Summarization of molecular and catalytic properties of purified acuminosidase.
| Source | Molecular Mass [kDa] | Oligomer | Km [mM] | Vmax [nkat/mg] | Optimal pH | Optimal Temperature [°C] |
|---|---|---|---|---|---|---|
| Cicer arietinum L. [34] | 68 (subunit) | Dimer | 2 1 0.2 2 | - | 7.0–7.5 (add. minor 4.5–5.0) | 45 |
| Viburnum furcatum Blume [17] | - | - | - | - | 5.8-6.3 | Below 40 |
| Viburnum furcatum Blume [18] | 56 61 (recombinant) | Monomer | 2.2 3 | - | 5 | 40 |
| Dalbergia nigrescens Kurz [13] | 62–63 (subunit) | Tetramer | 14.7 1 0.5 4 (0.7) 4 | 66.6 1 | 5.0–6.0 | 65 |
| Vitis vinifera endoglycosidase [20] | 58 | - | 1.69 5 | 275 5 | 4–5 | 50 |
1. p-Nitrophenyl β-d-glucoside as the substrate. 2. Biochanin A 7-O-β-acuminoside as the substrate. 3. Furcatin as the substrate. 4. Dalpatein 7-O-β-acuminoside and dalnigrein 7-O-β-acuminoside (in parentheses) as the substrate. 5. p-Nitrophenyl β-rutinoside as the substrate.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karnišová Potocká, E.; Mastihubová, M.; Mastihuba, V. Apiose-Relevant Glycosidases. Catalysts 2021, 11, 1251. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101251
AMA Style
Karnišová Potocká E, Mastihubová M, Mastihuba V. Apiose-Relevant Glycosidases. Catalysts. 2021; 11(10):1251. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101251
Chicago/Turabian StyleKarnišová Potocká, Elena, Mária Mastihubová, and Vladimír Mastihuba. 2021. "Apiose-Relevant Glycosidases" Catalysts 11, no. 10: 1251. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101251
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.