
Expeditious Asymmetric Synthesis of Polypropionates Relying on Sulfur Dioxide-Induced C–C Bond Forming Reactions

1
Basic Sciences Faculty, Swiss Institute of Technology in Lausanne (EPFL), 1015 Lausanne, Switzerland
2
Departamento de Química Física y Analítica, Facutad de Química, Universidad de Oviedo, 33006 Oviedo, Spain
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(11), 1267; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111267
Submission received: 1 October 2021
/
Revised: 17 October 2021
/
Accepted: 19 October 2021
/
Published: 21 October 2021
(This article belongs to the Special Issue Catalysts in Carbon-Carbon Coupling Reactions)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:For a long time, the organic chemistry of sulfur dioxide (SO2) consisted of sulfinates that react with carbon electrophiles to generate sulfones. With alkenes and other unsaturated compounds, SO2 generates polymeric materials such as polysulfones. More recently, H-ene, sila-ene and hetero-Diels–Alder reactions of SO2 have been realized under conditions that avoid polymer formation. Sultines resulting from the hetero-Diels–Alder reactions of conjugated dienes and SO2 are formed more rapidly than the corresponding more stable sulfolenes resulting from the cheletropic additions. In the presence of a protic or Lewis acid catalyst, the sultines derived from 1-alkoxydienes are ionized into zwitterionic intermediates bearing 1-alkoxyallylic cation moieties which react with electro-rich alkenes such as enol silyl ethers and allylsilanes with high stereoselectivity. (C–C-bond formation through Umpolung induced by SO2). This produces silyl sulfinates that react with carbon electrophiles to give sulfones (one-pot four component asymmetric synthesis of sulfones), or with Cl2, generating the corresponding sulfonamides that can be reacted in situ with primary and secondary amines (one-pot four component asymmetric synthesis of sulfonamides). Alternatively, Pd-catalyzed desulfinylation generates enantiomerically pure polypropionate stereotriads in one-pot operations. The chirons so obtained are flanked by an ethyl ketone moiety on one side and by a prop-1-en-1-yl carboxylate group on the other. They are ready for two-directional chain elongations, realizing expeditious synthesis of long-chain polypropionates and polyketides. The stereotriads have also been converted into simpler polypropionates such as the cyclohexanone moiety of baconipyrone A and B, Kishi’s stereoheptad unit of rifamycin S, Nicolaou’s C1–C11-fragment and Koert’s C16–CI fragment of apoptolidin A. This has also permitted the first total synthesis of (-)-dolabriferol.
1. Introduction
Natural polypropionates are a large subgroup of polyketides (1,3-polyols) constructed by C3-units. They are found in marine organisms including mollusks, sponges, fungi and actinomycetes [1,2,3,4,5], while some of them are also isolated from terrestrial resources [6]. Polypropionates interspace methyl groups in the polyketide chain which arise biochemically directly from the propionate unit or from the acetate–methionine motif [7,8,9]. They are characterized by abundant structural diversities and are important building blocks in the biosynthesis of several kinds of antibiotics such as macrolides, polyethers and cyclic peptides [10]. Most of them exhibit various kinds of bioactivities, especially antitumor and antimicrobial effects [11]. Examples of natural products containing polypropionate fragments are collected in Figure 1.
The stereoselective construction of polypropionates is challenging, mainly because of stereochemical issues. Aldol reactions, crotylations, allenylation and propargylation of aldehydes R1CHO have been used extensively to construct the four possible stereoisomeric R1CH(OH)-CH(Me)-R2 units [12,13,14,15,16,17,18,19]. More recently, the same fragments have been prepared through the transition metal catalyzed enantioselective hydro(hydroxycarbation) of terminal alkene R2CH=CH2 with alcohols R1CH2OH or aldehydes R1CHO + H2 [20]. The latter methods developed by Krische and co-workers have permitted significant shortening of the total syntheses of many natural polyketides and polypropionates [21,22,23,24,25,26,27,28]. The stereoselective construction of all possible stereoisomers of stereotriads R3CH(Me)-CH(OH)-CH(Me)-R4 (6, Figure 2) has been achieved by many methods [29] including those based on aldol or crotylation reactions of an aldehyde or its metallic enolate analogue bearing an α-methyl substituent [30,31,32,33,34,35,36].
Non-aldol formation of stereotriads has been proposed, such as the Sharpless asymmetric epoxidation of allyl alcohol followed by Pd-catalyzed hydrogenolysis of alkenyl oxirane with HCOOH [37,38] and the cuprate addition to epoxides [39,40,41]. A method applying an intramolecular Rh-catalyzed silylformylation/crotylsilylation/“aprotic” Tamao oxidation sequence has been developed by Leighton and co-workers [42,43,44]. Stereotriads have been prepared through double oxidative hydroboration of allenyl alcohols [45]. Carreira and co-workers used enantiomerically pure chiral nitrile oxides and allylic alcohols to generate enantiomerically pure isoxazolines [46,47]. This permitted the preparation of all four possible dipropionates diastereoisomers, in a protected form, starting with the same set of reagents. Erythronolide A has been obtained by this method [48]. The Diels–Alder additions of alkenes to 1,3-dienes produce cyclohexenes containing up to four stereocenters. Danishefsky and co-workers have generated polypropionates via the Diels–Alder reaction. Cycloadditions of aldehydes to 1-methoxy-2-methyl-3-silyloxypenta-1,3-diene produce pyrans, a molecular fragment in many polypropionates [49,50]. The Diels–Alder reaction between 2,4-dimethylfuran and enantiomerically pure 1-cyanovinyl carboxylates produces enantiomerically pure 2-cyano-1,5-dimethyl-7-oxabicyclo[2.2.1]hept-5-en-2-yl esters. The alkaline hydrolysis of the latter liberates the chiral auxiliary (a carboxylic acid) which can be recycled. The bicyclic compounds so obtained permitted Vogel and co-workers to prepare several stereotetrads [51,52]. Alternatively, Plumet and Arjona used a Diels–Alder reaction between furan and acrylic acid which provided, after several transformations, a complete library of all possible stereotetrads [53,54]. Hunt and Grieco obtained polypropionates starting with the opening at the bridgehead of oxabicyclo[3.2.1]octenes employing silyl ketene acetals [55]. On their side, Toste and co-workers applied the enantioselective amine-catalyzed Kornblum–DeLaMare rearrangement, a reaction first described by Hagenbuch and Vogel in 1980 [56], on a 3-benzyloxy-2,4,8-trimethyl-6,7-dioxabicyclo[3.2.2]non-8-ene derivative to generate the two polypropionate fragments of dolabriferol (Figure 1) [57].
2. One-Pot Synthesis of Polypropionate Stereotriads Ready for Bidirectional Chain Elongations
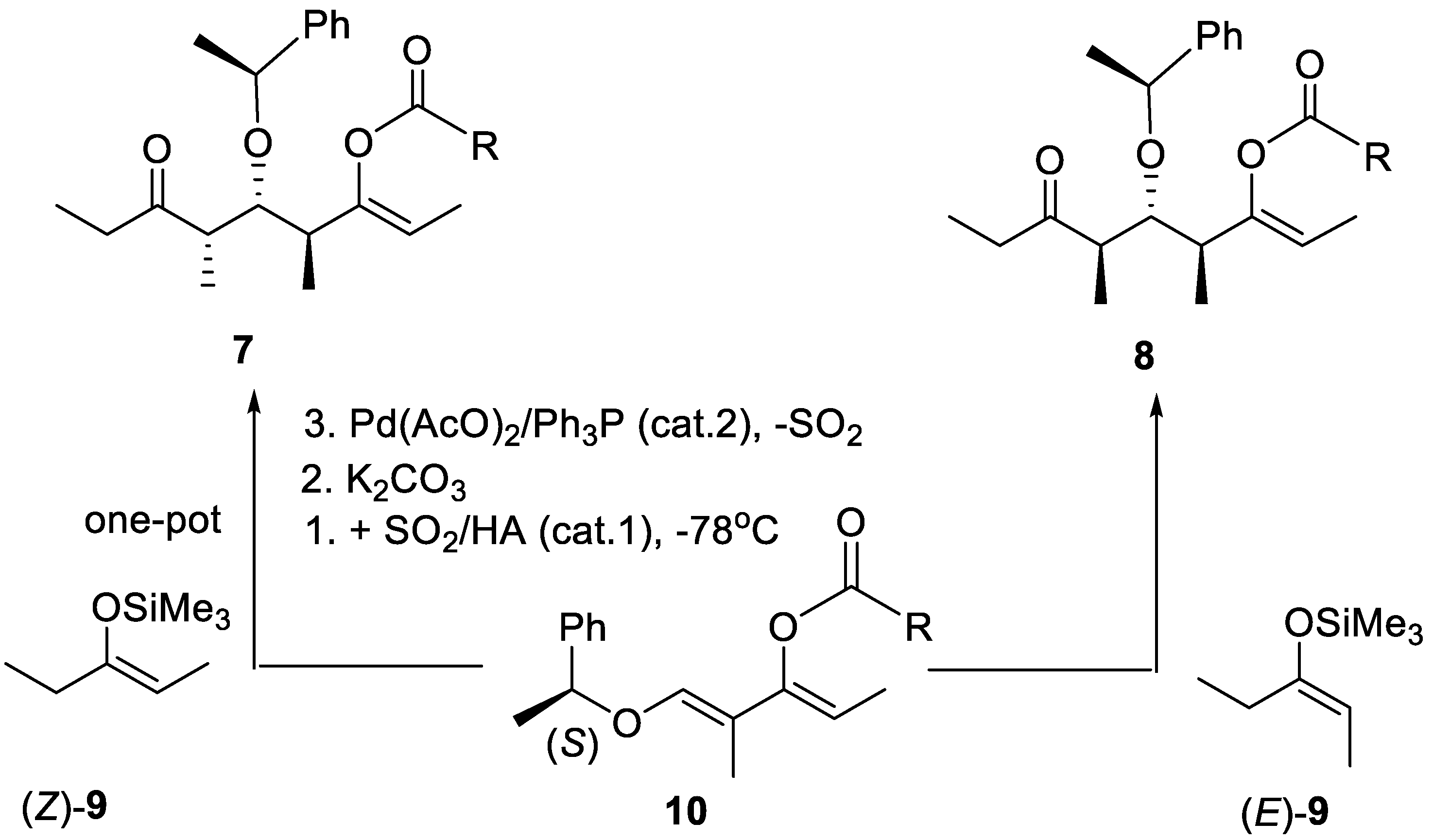
For the stereotriads 6 and their enantiomeric forms (Figure 2) to become useful chirons (enantiomericallly pure synthetic intermediates) in the constructions of complicated polypropionates and analogues, their alcoholic moiety must be protected adequately and their R3 et R4 terminus should be functions that can enter directly in stereoselective C–C bond forming reactions, typically metal aldol reactions. Chirons of type 7 and 8 (and their enantiomers) have been obtained in one-pot operations (Scheme 1) [16]. Their group R3 is an ethyl ketone which can be elongated into a system containing up to two further stereogenic centers via a direct aldol reaction, while the R4 terminus is a protected ethyl ketone under the form of (Z)-prop-1-en-1-yl carboxylate (ethyl ketone enol carboxylate) that stays intact and can be used in the second chain elongation reaction, also producing up to two further stereogenic centers (see below Section 4).
The enantiomers of stereotriads 7 and 8 are obtained in the same way using (R)-1-phenylethanol instead of (S)-1-phenylethanol to generate dienes 10. Other 1-arylethanols can be used to generate more enantiomerically pure dienes (see below). The SO2 is recovered at the end of the process. With the acid catalyst (e.g., HA = (CF3SO2)2NH) it induces the Umpolung of the electron-rich diene into an alkoxyallylic cation intermediate (see below Section 3). R can be an alkyl (e.g., Me2CH) or an aryl group (e.g., Ph).
3. Sulfur Dioxide as Umpolung Agent to Promote Carbon–Carbon Bond Forming Reactions between Alkenes and Dienes
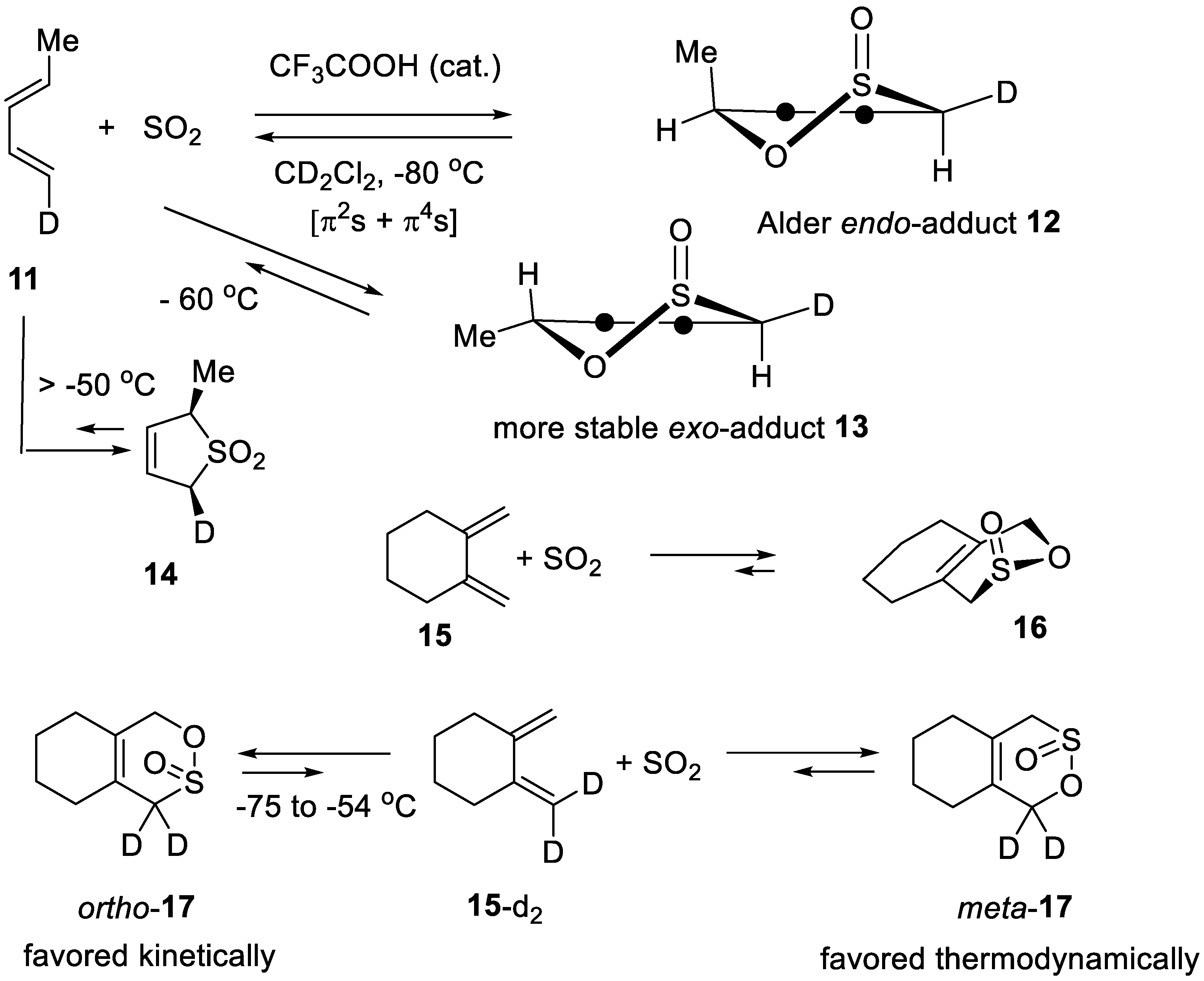
In the presence of SO2, alkenes, alkynes and polyenes produce polymeric materials, including polysulfones (copolymer with sulfur dioxide) [58,59,60]. Competing with these reactions, SO2-catalyzed alkene isomerization can be observed. We have demonstrated that the latter reaction may not imply a hetero-ene reaction of the alkene with SO2, followed by a fast [1,3]-sigmatropic shift of the intermediate β,γ-sulfinic acid and retro-ene reaction [61,62]. Instead, an allylic hydrogen atom is abstracted form the alkene by an alkanesulfonyl radical intermediate equilibrating with the polysulfone [63,64,65]. Since 1914 [66] it has been well-known that 1,3-dienes (that can adopt a s-cis-conformation) and sulfur dioxide equilibrate with their sulfolenes (cheletropic additions). On heating (>100 °C) the sulfolenes undergo cheletropic eliminations giving back the 1,3-dienes and SO2. The first examples of hetero-Diels–Alder additions of SO2 involved highly reactive dienes such as 1,4,5,6-tetramethyl-2,3-dimethylidenetriclo[2.1.1.05,6]hexane [67] and orthoquinodimethane [68]. Below −60 °C simple 1,3-dienes such as isoprene and piperylene react with SO2 equilibrating with their sultines resulting from hetero-Diels–Alder reactions that are much faster than the corresponding cheletropic additions. Deguin and Vogel showed in 1992 that (E,E)-deuteriopiperylene (11) equilibrates with sultine 12 at −80 °C in the presence of an acid catalyst such as CF3COOH. At −60 °C 12 is isomerized into the more stable isomeric sultine 13, by cycloreversion into the initial cycloaddents and re-addition in a second hetero-Diels–Alder reaction (Scheme 2). Both the [4+2]-cycloadditions 11 + SO2 → 12 and 11 + SO2 → 13 are highly regio- and stereoselective [69]. As for many Diels–Alder reactions, the acid-catalyzed hetero-Diels–Alder reactions of SO2 adheres to the Alder-endo rule and to the Woodward–Hoffmann rule of suprafaciality for the diene [69]. Sulfur dioxide itself catalyzes its cycloadditions [70,71,72,73]. In the absence of acid, the secondary deuterium kinetic isotopic effects of the SO2 reaction with the dideuterodiene 15-d2 induced a regioselectivity opposite to the equilibrium isotopic regioselectivity. Sultine ortho-17 formed faster than regioisomer meta-17. On staying at −55 °C, ortho-17 was slowly isomerized into meta-17 (Scheme 2). This demonstrates that the hetero-Diels–Alder reaction of SO2 follows a mechanism with asynchronous formation of the C–S and C–O bonds in the transition state: the C–S bond is formed to a greater extent than the C–O bond [74].
The sulfolenes arising from the cheletropic additions of SO2 to alkyl substituted 1,3-dienes are about 10 kcal mol−1 more stable than their isomeric sultines. In contrast, fluorosultines that result from the hetero-Diels–Alder reactions of SO2 with 1-fluoro-1,3-dienes are more stable than their isomeric sulfolenes. This is assigned to an enthalpic anomeric effect of the F–C–O(S=O) moiety in the sultines [75].
With 1-alkoxy- 3-acyloxy-1,3-dienes 10 (prepared in four steps from butan-3-one and (S)-1-phenylethanol [76], the corresponding sultines 19 are not seen at −100 °C (large excess of SO2, CH2Cl2 or toluene as co-solvent) as these dienes generate the corresponding sulfolenes at this temperature already. Nevertheless, sultines 19 are believed to be formed as intermediates before the isomeric sulfolenes (Scheme 3). In the presence of an acid catalyst (protic acid or Lewis acid), they equilibrate with zwitterionic intermediates 20 that can be reacted with electron-rich alkenes such as enoxysilane (Z)-9. This generates the silyl sulfinates 24. The role of SO2 is to convert the electron-rich dienes into 1-alkoxyallyl cation intermediates, realizing an inversion of polarity (Umpolung) that make possible the C–C coupling reaction between the two nucleophilic partners 10 and (Z)-9 [77].
After removal of SO2 in excess and solvent under vacuum, enough K2CO3 in CH3CN was added to neutralize the acid catalyst. Then a 1:1 mixture of Pd(AcO)2/Ph3P and isopropanol was added, and heating to 80 °C produced the final stereotriads. The stereoselectivity of the reaction cascade is explained in the following way (Scheme 3). The hetero-Diels–Alder reaction of SO2 is face selective because the chiral auxiliary (1-alkoxy substituent of the diene) favors transition structure 18, the least encumbered face of the diene reacting preferentially. The acid catalyzed ionization of the resulting sultines 19 generate ion-pairs 20 in which the sulfinate anion remains closed to the 1-alkoxyallyl cation moieties. This forces the nucleophile (e.g., enoxysilane (Z)-12) to attack 20 on the face opposite to that occupied by the sulfinate anion (transition structure 21). The resulting adducts 22 undergo intramolecular silyl group transfers via conformations 22′. Alternatively, two molecules of 22 could undergo a double intramolecular silyl group transfer giving the silyl sulfinates 23. Alcoholysis of 23 gives sulfinic acids 24 which undergo H-retro-ene reactions generating 7, the stereoselectivity of which is controlled by steric factors making transition structures 24 preferred to 24′. For the reaction cascade using enoxysilane (Z)-9 and diene 10 with R = i-Pr, R* = (S)-1-phenylethyl) (1:1 SO2/toluene, catalyst AH = (CF3SO2)2NH) the corresponding stereotriads 7 and 8 were isolated in 67 and 13 % yield, respectively, after column chromatography. Using Greene’s chiral auxiliaries ((S)-1-[2,4,6- tris(isopropyl)phenyl]ethanol) [78]) instead of inexpensive (S)-1-phenylethanol the diastereoselectivity syn,anti vs. anti,anti-stereotriad was better than 95:5 [79].
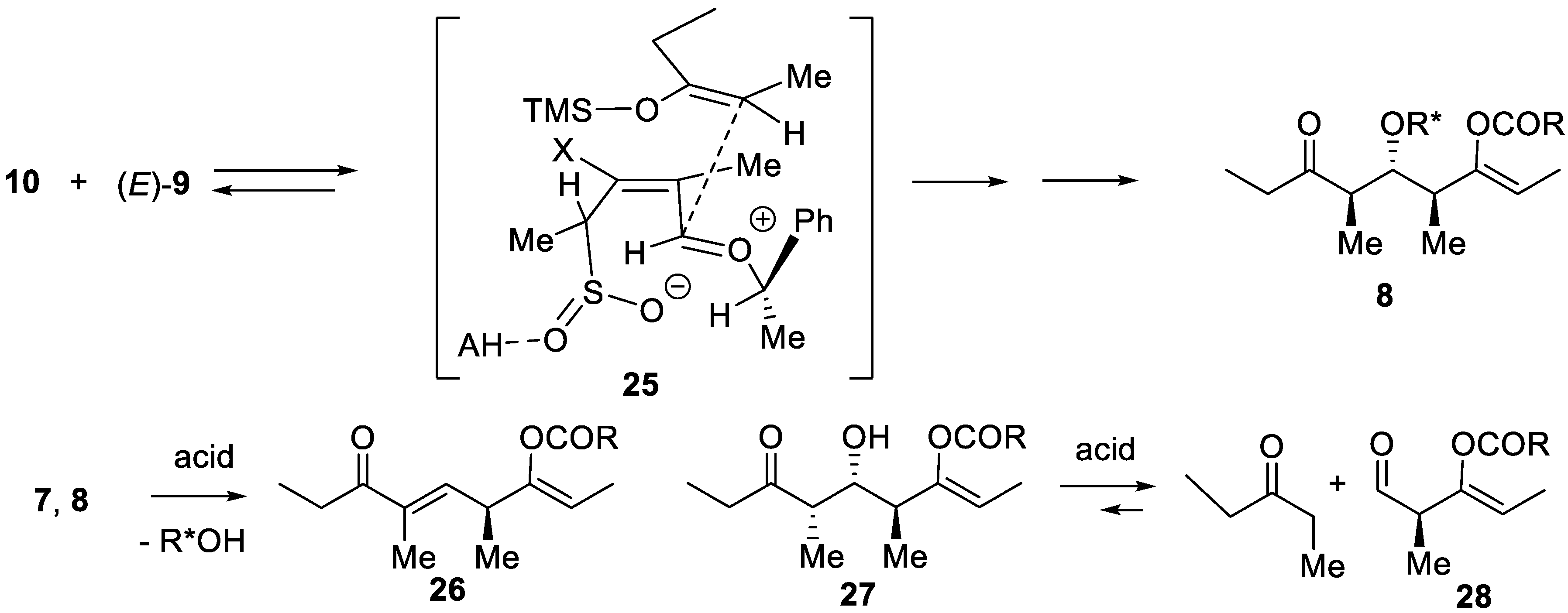
The silyl sulfinates 23 can be isolated, or converted in situ into sulfinate salts that are quenched by all kinds of electrophiles to give sulfones (four-component synthesis of polyfunctional sulfones [80]), or converted in situ (with Cl2 or N-chlorosuccinimide) into sulfonyl chlorides that react with amines to produce sulfonamides (four-component synthesis of polyfunctional sulfonamides [81,82]). Acidic treatment of 23 also leads to desulfinylation producing the stereotriads in a lower yield, due to elimination of 1-phenylethanol-generating dienes 26 and aldols 27, the latter undergoing retro-aldol decomposition into penta-3-one and aldehydes 28 (Scheme 4). This is avoided when the silyl sulfinates are treated under neutral or slightly basic conditions (K2CO3) in isopropanol in the presence of a catalytic amount of Pd(AcO)2 and Ph3P. Without Ph3P the reaction does not occur (formation of Pd(0) species as catalyst). One can envisage a Pd(0) complex intermediate which adds oxidatively (retention of configuration) into the allylic C–SO2SiMe3 bond of 23. Subsequent desulfinylation and protolysis of the Pd–SiMe3 bond gives i-PrOSiMe3 (driving force) and an (allyl)palladium hydride that undergoes regioselective and stereoselective β-insertion of hydride into another (allyl)Pd intermediate. An alternative mechanism (Scheme 3) is to invoke that the Pd(0) role is just to promote the Si-sulfinate bond cleavage under non-acidic conditions generating the corresponding β,γ-unsaturated sulfinic acids that, in turn, undergo H-retro-ene elimination of SO2 [83].
4. Long-Chain Polypropionates through Bidirectional Chain Elongation
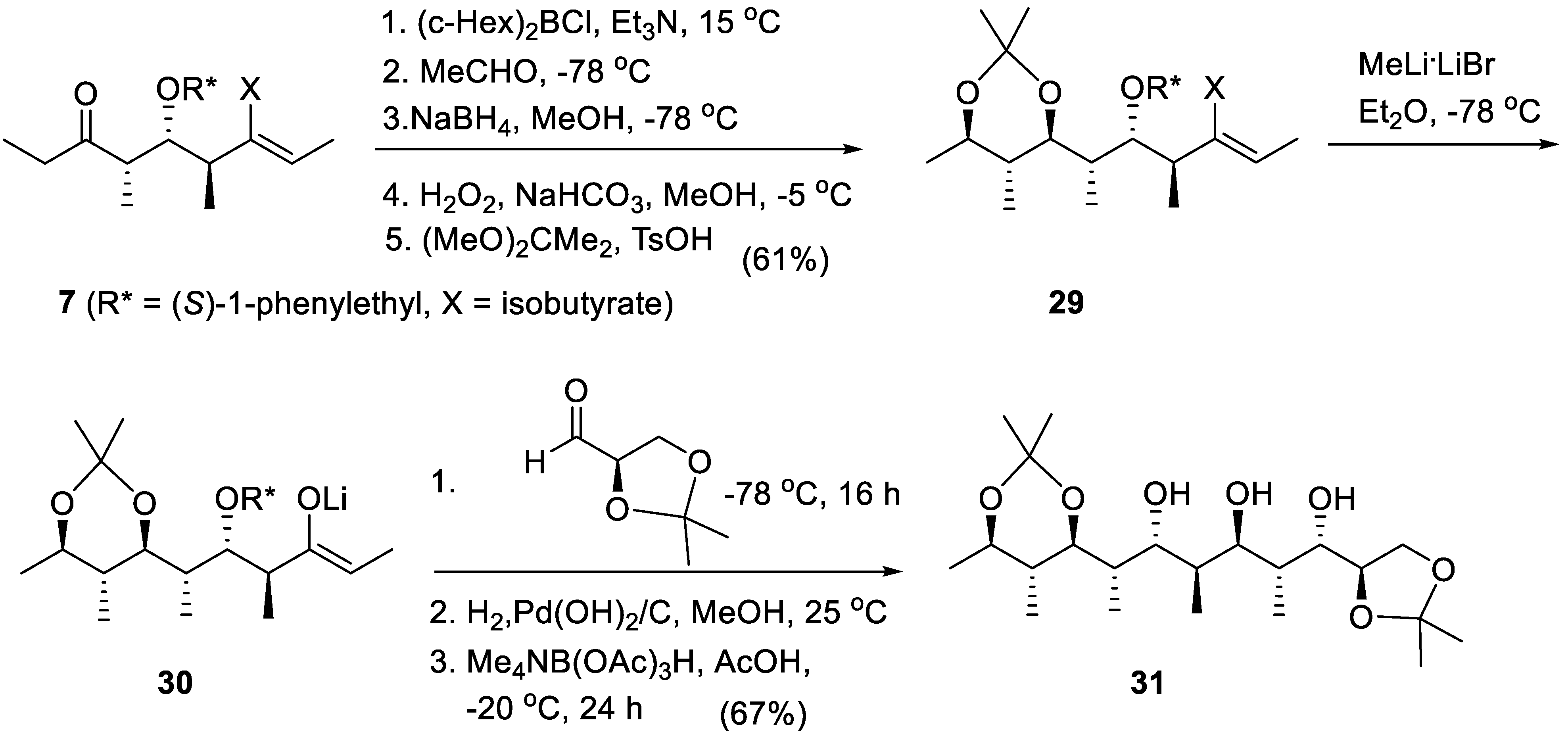
The polypropionate 31 (a stereodecad) containing 10 contiguous stereogenic centers has been obtained by two successive metal-aldol reactions of stereotriad 7 (Scheme 5) [84]. Applying Paterson’s method for direct formation of enoxyboranes [85,86,87] the dicyclohexyl(enoxy)borane derived from 7 reacted with acetaldehyde giving an boron anti-2-methylaldolate that was reduced directly with NaBH4 generating a anti,anti-2-methyl-1,3-diol moiety, which was protected as the acetonide 29 (a stereohexad in one pot operation) in 61% overall yield. The (Z)-enol isobutyrate group of 29 was converted with retention into the (Z) lithium enolate 30 by reaction with MeLi.LiBr in ether. The latter added to the acetonide of D-glyceraldehyde giving a major lithium aldolate. Its phenylethyl ether was hydrogenolyzed under standard conditions and the aldol was reduced with Me4NB(AcO)3H [88,89] selectively into the corresponding anti-1,3-diol 31 (67% overall yield).
5. First Total Asymmetric Synthesis of the Cyclohexanol Subunit of Baconipyrones A and B
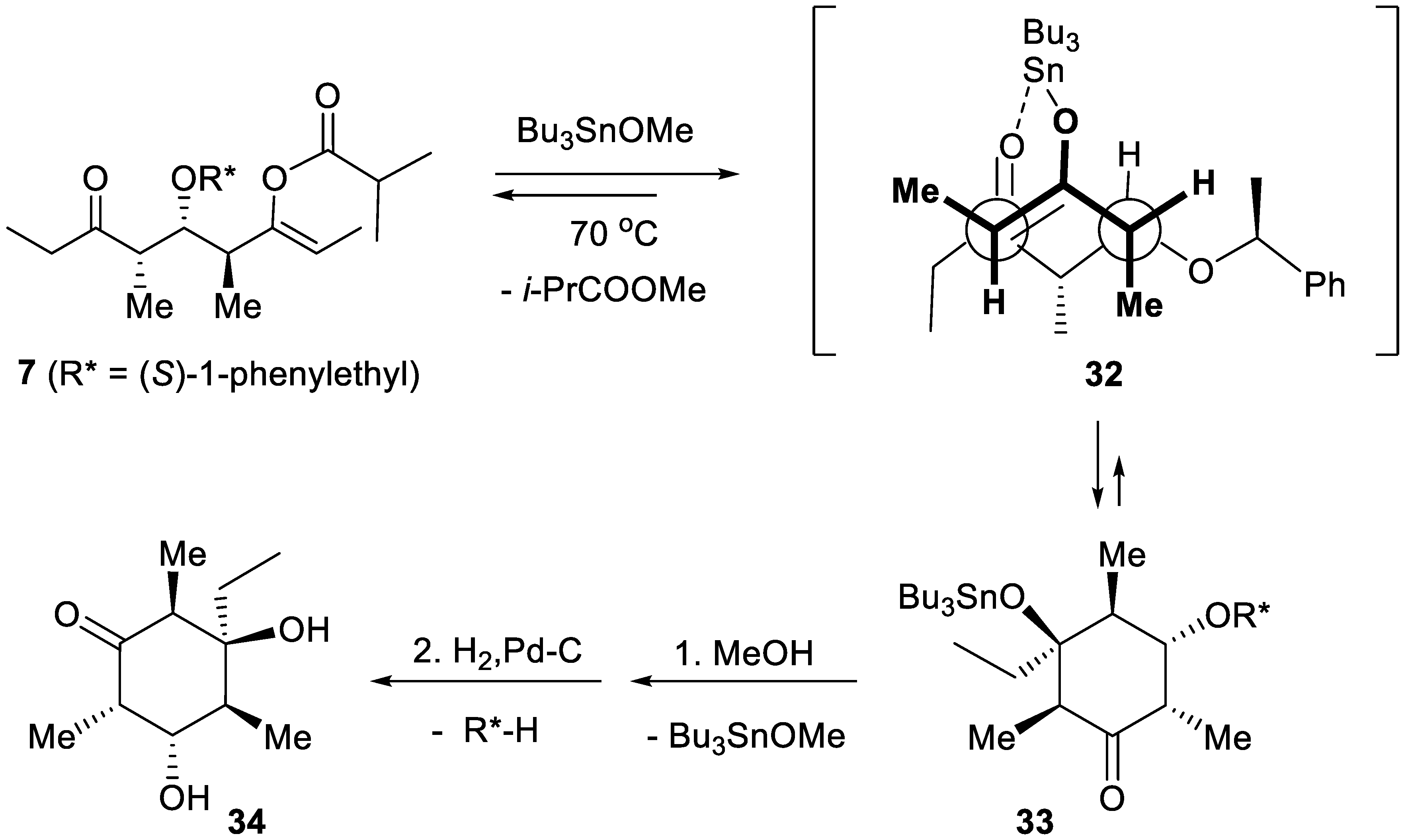
Baconipyrones A-D were isolated in 1989 by Faulkner and co-workers from Siphonaria baconi [90]. The stereotriad 7 (R = i-Pr, R* = (S)-1-phenylethyl) has been converted in two steps into cyclohexanone 34 (overall yield: 86%), subunit of baconipyrones A and B (Figure 1) [91]. Transesterification of enol isobutyrate 7 (Scheme 6) with Bu3SnOMe [92,93] induced the desired stereoselective intramolecular aldol reaction, giving 33. Transition structure 32 was proposed to explain the stereoselectivity hydrogenolysis of 33 and provided 34 quantitatively.
6. First Total Asymmetric Synthesis of (-)-Dolabriferol
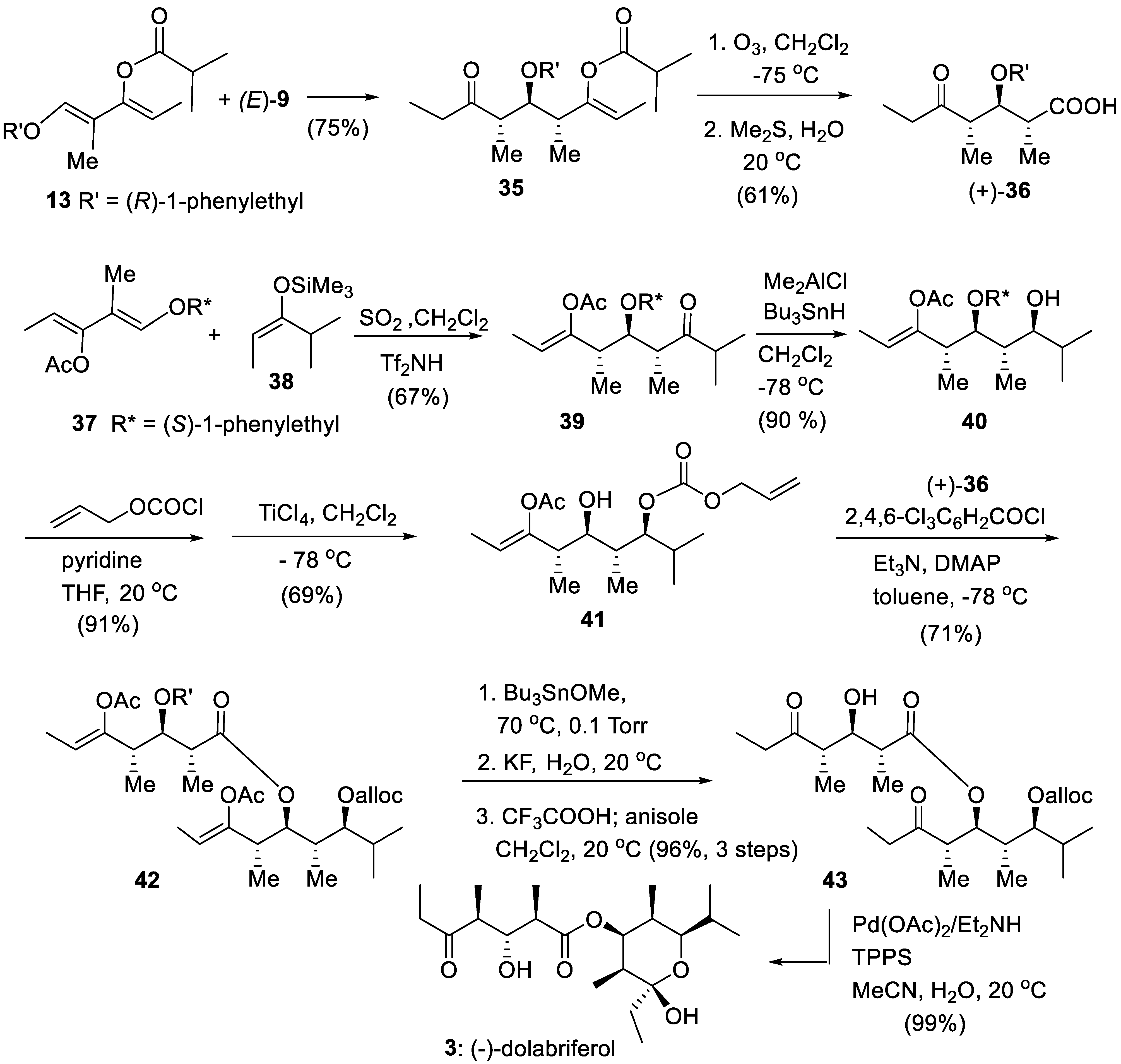
(-)-Dolabriferol was isolated from Dolabrifera dolabrifera, a shell-less mollusk. (-)- Dolabriferol is assumed to protect the mollusk from predators [94]. This natural product is made of two polypropionates subunits linked by an ester function, a structural motif which is also found in baconipyrones (Figure 1). Ozonolysis of pure 35 arising from the reaction of diene 13 (R’ = (R)-1-phenylethyl) and enoxysilane (E)-9 provided the carboxylic subunit (+)-36 of (-)-dolabriferol (two steps from diene 13). Similarly, reaction of diene 37 with enoxysilane 38 resulted in a stereotriad 39 (67%, single diastereomer, diasteroselectivity better than 95:5), that was reduced with stereoselectivity into alcohol 40 (a stereotetrad in two steps from diene 37). Protection as an allyl carbonate (alloc), followed by TiCl4-induced E1 cleavage of the 1-phenylethyl ether moiety furnished alcohol 41. The esterification of 41 with carboxylic acid (+)-36 using Paterson’s protocol [95] produced 42 (DMAP = 4-dimethylaminopyridine). Selective removal of the acetyl group was realized by treatment in pure Bu3SnOMe at 70 °C, followed by KF/H2O work-up. Subsequent treatment with CF3COOH removed the phenylethyl ether, giving 43. Final deprotection of the alloc group (TPPS = 3,3′,3″-phosphinidynetris(benzenesulfonic acid) trisodium salt) and formation of the cyclic hemiacetal gave (-)-dolabriferol (Scheme 7).[96] Since this first total synthesis which established the absolute configuration of (-)-dolabriferol, several other synthetic approaches have been proposed, sometimes requiring more steps [57,97,98,99,100].
7. Expeditious Asymmetric Synthesis of the Stereoheptad C19–C27 of Rifamycins: Formal Total Synthesis of Rifamycin S
Rifamycins [101,102,103] are antibiotics belonging to the group of naphthalenic ansamycins [104] characterized by an aliphatic bridge (polypropionate chain) linking two non-adjacent centers of an aromatic moiety. They are produced from Streptomyces mediterranei [105] and are active against a large variety of organisms, including bacteria, eukaryotes and viruses [106]. Rifamycins have shown also antitumour activity [107] and anti-inflammatory activity [108]. At present, rifamycins and analogues are applied in the treatment of tuberculosis. They inhibit bacterial DNA-dependent RNA polymerase [109,110,111]. Rifamycin S (4) and several analogues showing promising activities have been prepared [112,113,114,115].
The first total synthesis of rifamycin S (4) was reported by Kishi and co-workers in 1980 [116,117,118,119]. The stereoheptad (-)-48 is a key intermediate for the construction of the ansa chain. It was obtained in 26 steps and 5.2% overall yield from (2S)-3-benzyloxy-2-methylpropanal. Since then, several total asymmetric syntheses of the polypropionate fragment have been proposed [120,121,122,123]. The construction of the C19–C27 fragment ((-)-48 and analogues) of this antibiotic has become a challenging target for the testing of asymmetric synthetic methods and strategies [50,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141].
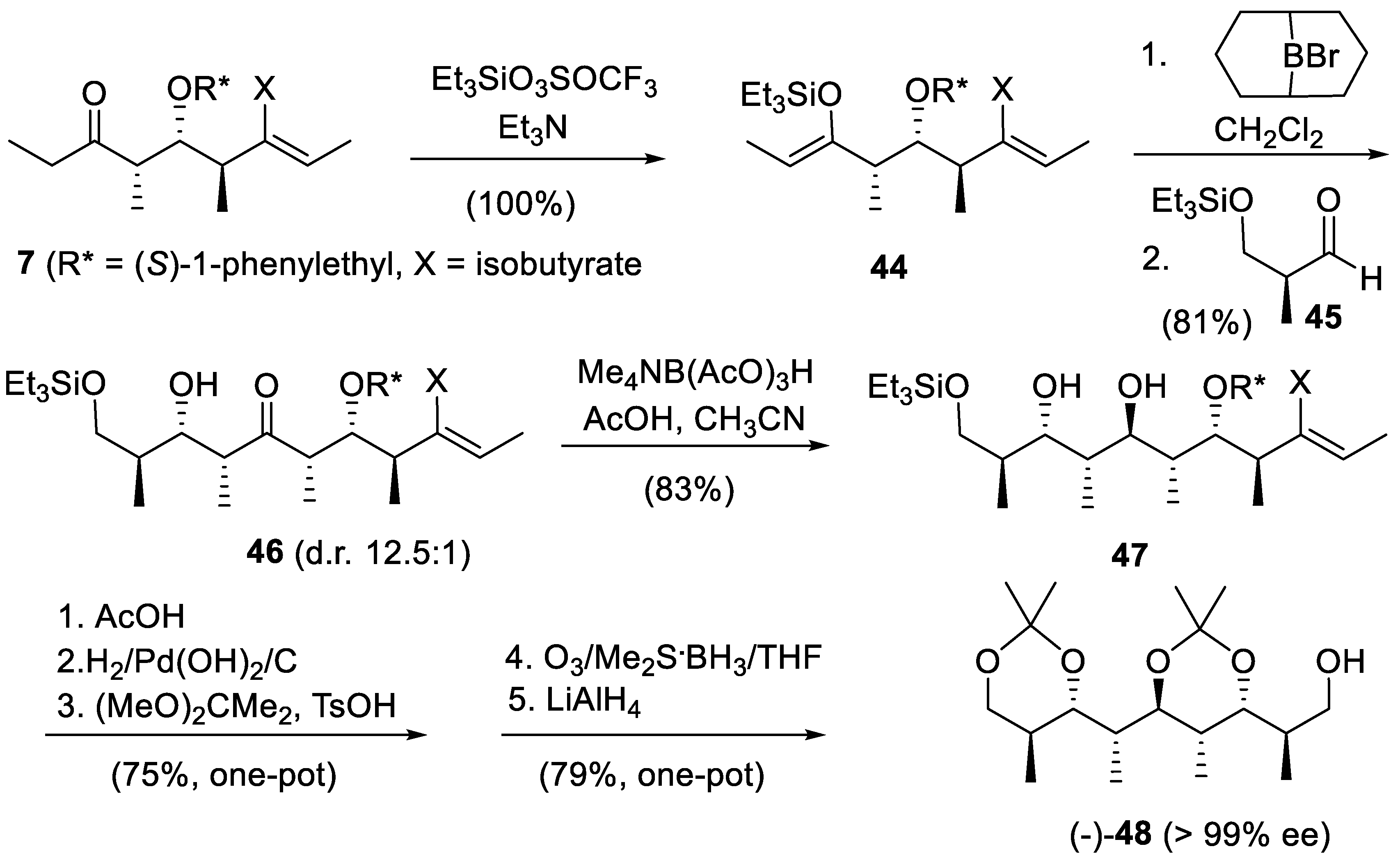
Starting from the readily available diene 10 (R* = (S)-1-phenylethyl, X = isobutyrate, Scheme 1) Kishi’s intermediate (-)-48 was obtained in 25% yield requiring the isolation of only four synthetic intermediates (Scheme 8). The (Z)-enol ether 44 resulting from the silylation of ethyl ketone 7 (derived from diene 10) reacted with 9-bromo-9-borabicyclo[3.3.1]nonane (BrBBN) in CH2Cl2 (silyl/boron exchange) and then with aldehyde 45 to produce a 12.5:1 mixture of 46 and 9-epimer in 81% yield. Pure 46 was reduced under Evans’ conditions [88,89] to give diol 47. The next five operations were carried out in the same pot without isolating any intermediate. Treatment of 47 with AcOH cleaved the silyl ether. Then hydrogenolysis removed the phenylethyl group. The crude tetrol so obtained was converted into the corresponding diacetonide. Ozonolysis of the enol isobutyrate moiety gave a mixed anhydride that was reduced with LiAlH4 into (-)-48 [142].
8. Generalization of the SO2-Induced Umpolung. Short Synthesis of the C16–C28 Fragment of Apoptolidinone: Formal Total Synthesis of Apoptolidin A
Apoptolidin A (isolated from Nocardiopsis sp.) [143,144] and the natural analogues B, C, D, E and F [145,146,147] are leads for the chemotherapy of cancers [148,149,150,151]. They selectively induce apoptosis in cancer cells. The groups of Nicolaou [152,153] and Koert [154,155] have presented the first syntheses of apoptolidin A. The groups of Sulikowsky [156,157] and Crimmins [158] have reported syntheses of the aglycon apoptolidinone A. Fragments of this aglycon has been prepared by other groups [159].
Applying our SO2-induced Umpolung reaction, rapid access (nine steps) to Koert’s C16–C28 polyketide fragment 57 (Scheme 9) of apoptolidinones A has been realized [160]. This work illustrates that other dienes and enoxysilanes than those presented in Scheme 1 can be used in our reaction cascade. Fragment 57 is adequately protected for the glycosidation steps necessary in the construction of apoptolidin A.
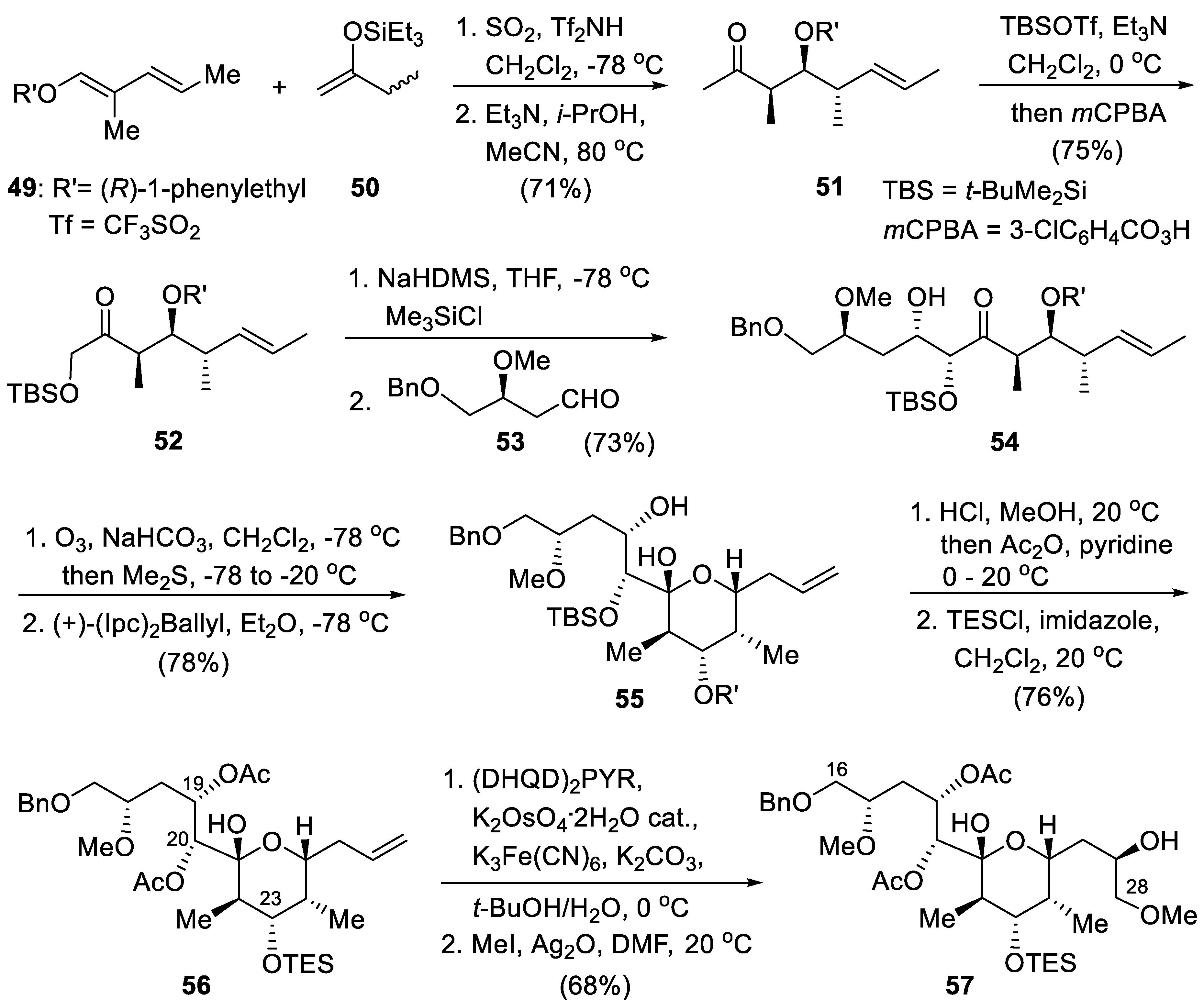
Diene 49 (derived from inexpensive (R)-1-phenylethanol) and silyl ethers 50 (1:1 E/Z mixture) were reacted with a catalytic amount of (CF3SO2)2NH in SO2/CH2Cl2 (5:1) cooled to −78 °C. After neutralization of the acid catalyst with Et3N and solvent evaporation, alcoholysis with i-PrOH (80 °C) gave a 4:1 mixture of stereotriad 51 and its α,β,γ--anti,anti stereomer. This mixture was converted into their kinetic silyl enol ethers and oxidized with mCPBA (Rubottom oxidation [161]) giving 52 that underwent Mukaiyama aldol coupling with aldehyde 53 [162,163], producing alkene 54. Ozonolysis of 54 followed by treatment with Me2S gave an aldehyde that was allylated under Brown’s conditions [164]. The resulting homoallylic alcohol was equilibrated with the hemiacetal 55 that underwent desilylation, debenzylation and Fischer glycosidation on treatment with HCl/MeOH at 50 °C. The resulting triol was then acetylated selectively into a diacetate (at C19 and C20); the most sterically hindered alcohol at C23 was then silylated giving 56. Sharpless asymmetric dihydroxylation [165] of 56 furnished a 4.5:1 mixture of the corresponding diol that was selectively monomethylated with MeI/Ag2O giving Koert’s intermediate 57.
9. The One-Pot Four-Component Synthesis of Polyfunctional Sulfones: Application to a Short Synthesis of the C1–C11 Fragment of Apoptolidin A
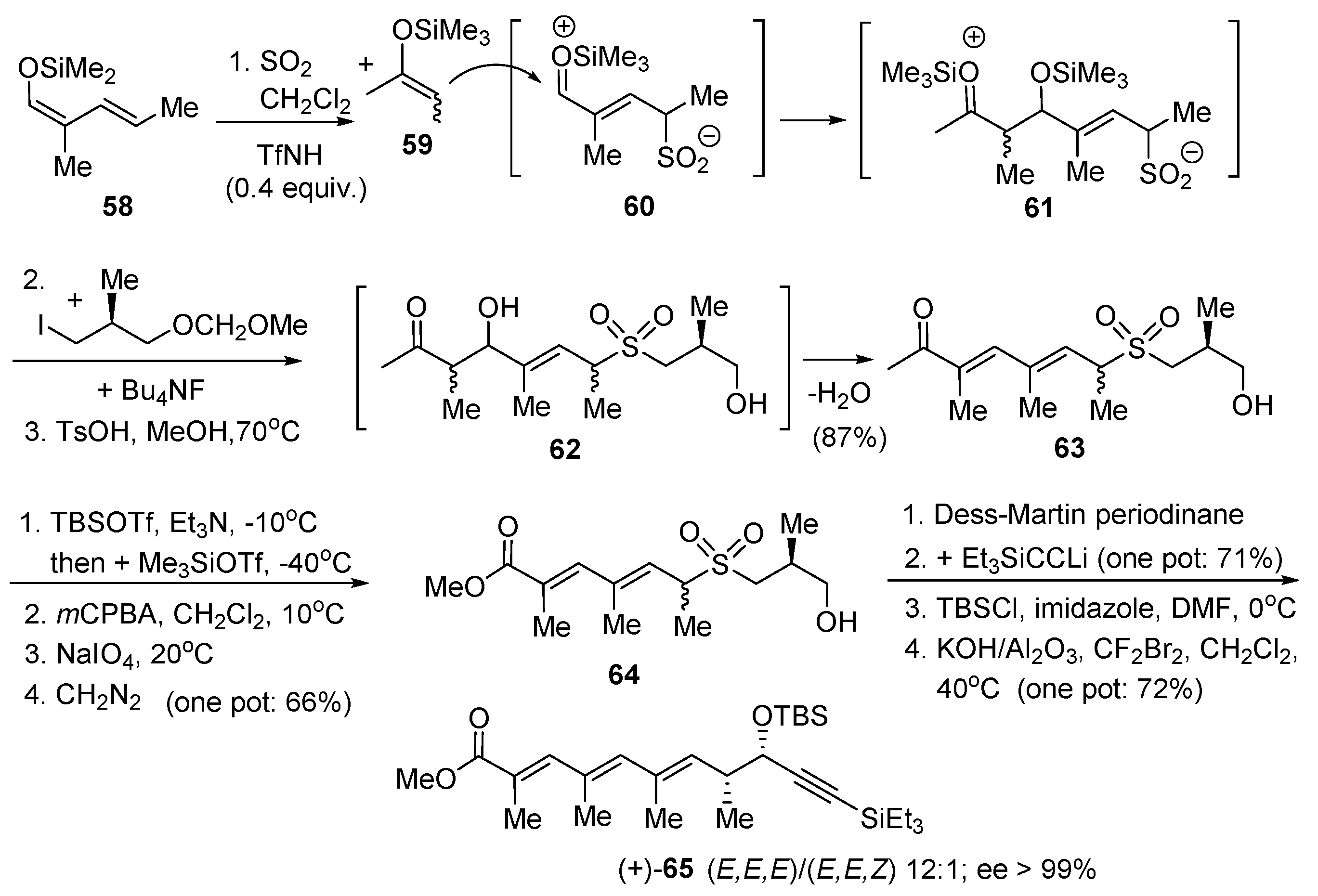
Another key intermediate in the total synthesis of apoptolidin A is the Nicolaou’s C1–C11-fragment (+)-65 the preparation of which requires 11 steps [152]. Applying our SO2-induced Umpolung reaction, an expeditious synthesis of this intermediate was realized with an overall yield of 29% starting with simple diene 58, enoxysilane 59 and the known enantiomerically pure (S)-3-methoxymethoxy-2-methylprop-1-yl iodide. The method required the isolation of only three synthetic intermediates (Scheme 10) [166]. In the presence of 0.4 equivalent of a strong acid such as (CF3SO2)2NH sulfur dioxide adds to the s-trans form of diene 58 equilibrating with a zwitterionic intermediate 60 that was quenched by the enoxysilane 59. One assumes that another zwitterionic intermediate 61 was formed, which after treatment with tetrabutylammonium fluoride generated the dihydroxyketone 62, an aldol that loses one equivalent of water under acidic conditions (p-TsOH, MeOH, 70 °C) to give the (E,E)-dienone 63 (one pot: 87% yield). Silyl ether and enol silyl ether formation was followed by oxidation with mCPBA. This generated an α-hydroxyketone which was not isolated but directly submitted to the Malaprade oxidation, giving a carboxylic acid that was esterified in situ with diazomethane-producing ester 64. Dess–Martin oxidation of the primary alcohol of 64 gave an aldehyde that was reacted, without purification, with Et3SiCC-Li to give a 5:1 mixture of diastereomeric propargylic alcohols. They were silylated and the sulfone moiety underwent a Ramberg–Bäcklund rearrangement [167,168,169] providing a 12:1 mixture of (E,E,E)-/(E,E,Z)-triene-ester (+)-65.
10. Allylsilanes as Nucleophiles: Development of Two-Directional Polypropionate–Polyketide Synthesis
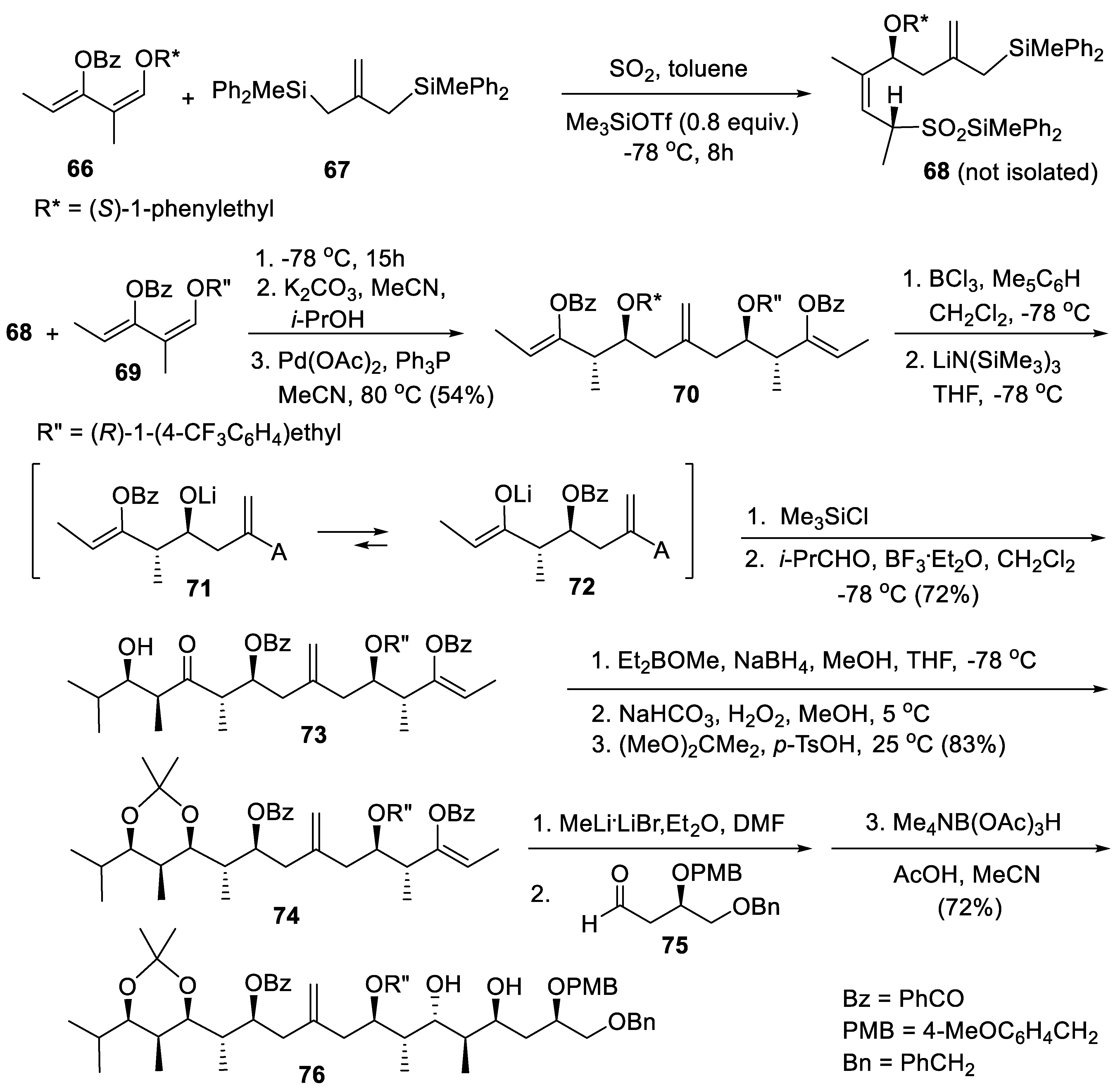
Like enol silyl ethers, allylsilanes are nucleophiles that can be used in our SO2-induced CC bond-forming reaction. Using allyldisilane 67 and two different dienes, 66 and 69, the stereotetrads 70 can be prepared in one-pot operations (Scheme 11). Diene 66 possesses a 1-(1-phenyl)ethoxy group whereas diene bears a 1-[1-(4-fluorophenyl]ethoxy substituent. The SN1 and E1 cleavage of the benzylic C–O ether bond of the 1-phenylethoxy group are faster than the SN1 and E1 C–O benzylic bond cleavage of the 1-(4-fluorophenyl)ethoxy group; the pseudo-symmetrical stereotetrad is suitable for two successive aldol reactions on both their chain terminus. This permits the expeditious syntheses of long-chain polyketides and polypropionates.
Reaction of diene 66 with 67 and SO2 promoted by Me3SiOTf generated the product of mono-alkoxyallylation 68. The reaction 66 + 67 → 68 is faster than the reaction of monoallysilane 68 with diene 67 + SO2 because bisallylsilane 66 enjoys twice the β-silicon effect. In the presence of one equivalent of diene 66 the product of double oxyallytion is not formed; only sulfinate 68 is formed. It can be reacted without purification with SO2 and diene 69, providing a bis(silyl sulfinate) which is not isolated but submitted directly to the double Pd(0)-catalyzed desilylation and desulfitation reactions, furnishing the stereotetrad 70, isolated in 54% yield (one-pot). Selective debenzylation of 70 with BCl3/pentamethylbenzene eliminated the phenylethyl group, giving a homoallylic alcohol that was not isolated but treated directly with (Me3Si)2NLi to engender the corresponding lithium alcoholate 71 (one pot). The latter underwent rapid acyl group migration from the neighboring enol benzoate forming lithium enolate 72. Without isolation, the latter enolate reacted with Me3SiCl giving a (Z)-enoxysilane that was reacted in situ with isobutyraldehyde and BF3 etherate, providing aldol 73 which was a single stereomer isolated in 72% yield. Reduction of aldol 72 under Narasaka’s conditions [170] gave the corresponding syn-1,3-diol that was converted in situ into its acetonide 74 (83%, overall). The treatment of enol benzoate 74 with MeLi.LiBr furnished the corresponding lithium (Z)-enolate. It was quenched by the chiral aldehyde 75 producing a major aldol that was not isolated but directly reduced under Evans’ conditions [88,89]. This furnished 76, a polyketides containing 11 stereogenic centers. As the configuration of the 1-oxydienes 66 and 69 can be either (R) or (S) and since the two successive aldol reactions on the intermediate stereotetrad can used a wide variety of aldehydes under well-chosen conditions, a very large library of polyketides can be prepared applying our method illustrated in Scheme 11 [171,172].
11. Conclusions
At low temperature, and in the presence of a protic or Lewis acid catalyst, 1-alkoxy-1,3-dienes undergo fast hetero-Diels–Alder reactions with SO2, forming unstable sultines that are converted rapidly into zwitterionic intermediates containing 1-oxyallyl cation moieties. The latter are quenched in situ by electron-rich alkenes such as silyl enol ethers generating β,γ-unsaturated silyl sulfinates. Sufur dioxide induces a stereoselective C–C bond forming reactions between electron-rich dienes and alkenes (Umpolung through SO2). The silyl sulfinates so obtained can be converted in situ into stereotriads that are flanked by an ethyl ketone group at one side and by an enol ester of an ethyl ketone on the other side. In a few synthetic steps the synthesis of the cyclohexanone unit of baconipyrones and of the two fragments of (-)-dolabriferol have been realized. With the first total synthesis of (-)-dolabriferol we could establish its absolute configuration. Aldol condensation of the ethyl ketone group of one of our stereotriads has opened a very short route to Kishi’s stereoheptad, which he used to construct rifamycin-S. A similar strategy has permitted us to obtain the Koert’s C16–C28 polyketide fragment of apoptolidin A. Under strongly acidic conditions, s-trans-1-alkoxydienes and enoxysilanes react with SO2 forming sulfinates that are quenched in situ with electrophiles to generate polyfunctional sulfones with a conjugated (E,E)-dienone moiety. The method has permitted an efficient synthesis of Nicolaou’s C1–C11 fragment of apoptolidin A. The stereotriads undergo two successive metal aldol reactions that produce complicated polyketides and polypropionates in a few steps. Allylsilanes can be used instead of enoxysilanes in our SO2-induced Umpolung reaction. With 2-[(methyldiphenyl)methyl]allylmethyl(diphenyl)silane), two successive alkoxyallylations with two different 1-alkoxydienes can be run in the same pot, thus generating stereotetrads ready for two successive aldol reactions (two-directional chain elongations). The strategy permits us to construct, in a few steps, complicated polyketides and polypropionates in a combinatorial fashion.
Funding
The research was financed by the University of Lausanne, the Ecole Polytechnique Fédérale de Lausanne (EPFL), the Swiss National Science Foundation and the University of Oviedo.
Acknowledgments
The authors wish to gratefully thank all their former co-workers for their intellectual input and their fantastic experimental studies. Their names appear in the citations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davies-Coleman, M.T.; Garson, M.J. Marine polypropionates. Nat. Prod. Rep. 1998, 15, 477–493. [Google Scholar] [CrossRef]
- Müller, W.E.G. Marine Molecular Biotechnology; Springer: Berlin, Germany, 2006; Chapter 1.2; Volume 71, pp. 570–575. [Google Scholar]
- Liu, Z.; Liu, H.; Zhang, W. Natural Polypropionates in 1999–2020: An Overview of Chemical and Biological Diversity. Mar. Drugs 2020, 18, 569. [Google Scholar] [CrossRef]
- Wu, Q.; Li, S.-W.; Xu, H.; Wang, H.; Hu, P.; Zhang, H.; Luo, C.; Chen, K.-X.; Nay, B.; Guo, Y.-W.; et al. Complex polypropionates from a South China Sea photosynthetic mollusk: Isolation and biomimetic synthesis highlighting novel rearrangements. Angew. Chem. Int. Ed. 2020, 59, 12105–12112. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, D.; Yu, M.; Liu, Y.; Liu, P.; Zhang, X. A Review on Metabolites from Onchidium Genus: Chemistry and Bioactivity. Chem. Biodivers. 2020, 18, e2000580. [Google Scholar] [CrossRef]
- Lin, S.; Wu, Y.-Z.; Chen, K.-Y.; Ye, J.; Yang, X.-W.; Zhang, W.-D. Polyketides from the fungus Penicillium decumbens. J. Asian Nat. Prod. Res. 2018, 20, 445–450. [Google Scholar] [CrossRef]
- Koskinen, A.M.P.; Karisalmi, K. Polyketide stereotetrads in natural products. Chem. Soc. Rev. 2005, 34, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Hertweck, C. The Biosynthetic Logic of Polyketide Diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J.; Leadlay, P. Combinatorial biosynthesis of reduced polyketides. Nat. Rev. Genet. 2005, 3, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. Nature Builds Macrocycles and Heterocycles into Its Antimicrobial Frameworks: Deciphering Biosynthetic Strategy. ACS Infect. Dis. 2018, 4, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Panek, J.S.; Jain, N.F. Total Synthesis of Rutamycin B and Oligomycin C. J. Org. Chem. 2001, 66, 2747–2756. [Google Scholar] [CrossRef]
- Crossman, J.S.; Perkins, M.V. Total Synthesis and Structural Elucidation of (−)-Maurenone. J. Org. Chem. 2005, 71, 117–124. [Google Scholar] [CrossRef]
- Ward, D.E. The thiopyran route to polypropionates. Chem. Commun. 2011, 47, 11375–11393. [Google Scholar] [CrossRef]
- Ward, D.E.; Kazemeini, A. Aldol Reactions with Kinetic Resolution: Scope and Limitations of Ketal- and Dithioketal-Protected β-Ketoaldehydes. J. Org. Chem. 2012, 77, 10789–10803. [Google Scholar] [CrossRef]
- Turks, M.; Laclef, S.; Vogel, P. Construction of Polypropionate Fragments in Natural Product Synthesis. Stereoselective Synthesis of Drugs and Natural Products; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 1, pp. 271–318. [Google Scholar]
- Alagiri, K.; Lin, S.; Kumagai, N.; Shibasaki, M. Iterative Direct Aldol Strategy for Polypropionates: Enantioselective Total Synthesis of (−)-Membrenone A and B. Org. Lett. 2014, 16, 5301–5303. [Google Scholar] [CrossRef] [PubMed]
- Miles, W.H.; Madison, C.M.; Mastria, M.L.; Tang, P.-I. Synthesis of the C3–C7 fragment of tylonolide by the γ-hydroxybutenolide approach. Tetrahedron Lett. 2016, 57, 3929–3932. [Google Scholar] [CrossRef]
- Hosokawa, S. Remote Asymmetric Induction Reactions using a E,E-Vinylketene Silyl N,O-Acetal and the Wide Range Stereocontrol Strategy for the Synthesis of Polypropionates. Acc. Chem. Res. 2018, 51, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Santana, C.G.; Krische, M.J. From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present, and Future. ACS Catal. 2021, 11, 5572–5585. [Google Scholar] [CrossRef] [PubMed]
- Dechert-Schmitt, A.-M.R.; Schmitt, D.C.; Gao, X.; Itoh, T.; Krische, M.J. Polyketide construction via hydrohydroxyalkylation and related alcohol C–H functionalizations: Reinventing the chemistry of carbonyl addition. Nat. Prod. Rep. 2014, 31, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Kasun, Z.A.; Krische, M.J. Enantioselective Alcohol C–H Functionalization for Polyketide Construction: Unlocking Redox-Economy and Site-Selectivity for Ideal Chemical Synthesis. J. Am. Chem. Soc. 2016, 138, 5467–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.D.; Park, B.Y.; Luong, T.; Sato, H.; Garza, V.J.; Krische, M.J. Metal-catalyzed reductive coupling of olefin-derived nucleophiles: Reinventing carbonyl addition. Science 2016, 354, aah5133. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Zhang, W.; Krische, M.J. Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res. 2017, 50, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.; Schwartz, L.A.; Krische, M.J. Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lin, S.; Jacobsen, K.M.; Poulsen, T.B. Chemical Syntheses and Chemical Biology of Carboxyl Polyether Ionophores: Recent Highlights. Angew. Chem. Int. Ed. 2019, 58, 13630–13642. [Google Scholar] [CrossRef] [PubMed]
- Doerksen, R.S.; Meyer, C.C.; Krische, M.J. Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed. 2019, 58, 14055–14064. [Google Scholar] [CrossRef] [PubMed]
- Siu, Y.-M.; Roane, J.; Krische, M.J. Total Synthesis of Leiodermatolide A via Transfer Hydrogenative Allylation, Crotylation, and Propargylation: Polyketide Construction beyond Discrete Allyl- or Allenylmetal Reagents. J. Am. Chem. Soc. 2021, 143, 10590–10595. [Google Scholar] [CrossRef]
- Li, J.; Menche, D. Direct methods for stereoselective polypropionate synthesis: A survey. Synthesis 2009, 14, 2293–2315. [Google Scholar]
- Solsona, J.G.; Romea, A.P.; Urpí, F. Studies Directed toward the Construction of the Polypropionate Fragment of Superstolide A. Org. Lett. 2003, 5, 4681–4684. [Google Scholar] [CrossRef]
- Yadav, J.S.; Srinivas, R.; Sathaiah, K. Total synthesis of natural (+)-membrenone C and its 7-epimer. Tetrahedron Lett. 2006, 47, 1603–1606. [Google Scholar] [CrossRef]
- Chandra, B.; Fu, D.; Nelson, S.G. Catalytic Asymmetric Synthesis of Complex Polypropionates: Lewis Base Catalyzed Aldol Equivalents in the Synthesis of Erythronolide B. Angew. Chem. Int. Ed. 2010, 49, 2591–2594. [Google Scholar] [CrossRef]
- Mochirian, P.; Godin, F.; Katsoulis, I.; Fontaine, I.; Brazeau, J.-F.; Guindon, Y. A Bidirectional Approach to the Synthesis of Polypropionates: Synthesis of C1–C13 Fragment of Zincophorin and Related Isomers. J. Org. Chem. 2011, 76, 7654–7676. [Google Scholar] [CrossRef]
- Brady, P.B.; Yamamoto, H. Rapid and Stereochemically Flexible Synthesis of Polypropionates: Super-Silyl-Governed Aldol Cascades. Angew. Chem. Int. Ed. 2012, 51, 1942–1946. [Google Scholar] [CrossRef] [Green Version]
- Becerril-Jiménez, F.; Ward, D.E. On the Origin of Siphonariid Polypropionates: Total Synthesis of Caloundrin B and Its Isomerization to Siphonarin B. Org. Lett. 2012, 14, 1648–1651. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.E. Polypropionate synthesis via substrate-controlled stereoselective aldol couplings of chiral fragments. In Modern Methods in Stereoselective Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 377–429. [Google Scholar]
- Oshima, M.; Yamazaki, H.; Shimizu, I.; Nisar, M.; Tsuji, J. Palladium-catalyzed selective hydrogenolysis of alkenyloxiranes with formic acid. Stereoselectivity and synthetic utility. J. Am. Chem. Soc. 1989, 111, 6280–6287. [Google Scholar] [CrossRef]
- Nagasawa, K.; Shimizu, I.; Nakata, T. ChemInform Abstract: Total Synthesis of Preswinholide A. Part 1. Stereoselective Synthesis of the C11-C23 Segment. ChemInform 2010, 28, 6881–6884. [Google Scholar] [CrossRef]
- Jung, M.E.; Chaumontet, M.; Salehi-Rad, R. Total Synthesis of Auripyrone B Using a Non-Aldol Aldol−Cuprate Opening Process. Org. Lett. 2010, 12, 2872–2875. [Google Scholar] [CrossRef] [Green Version]
- Bandaru, A.; Kaliappan, K.P. Synthesis of the C1-C10 fragment of muamvatin. Chem. Asian J. 2020, 15, 2208–2211. [Google Scholar] [CrossRef]
- Si, D.; Kaliappan, K.P. Synthesis of C9-C13 and C15-C21 subunits of discodermolide. Asian J. Org. Chem. 2020, 9, 1205–1212. [Google Scholar] [CrossRef]
- Zacuto, M.J.; O’Malley, S.J.; Leighton, J.L. Tandem silylformylation–allyl(crotyl)silylation: A new approach to polyketide synthesis. Tetrahedron 2003, 59, 8889–8900. [Google Scholar] [CrossRef]
- Foley, C.N.; Leighton, J.L. Beyond the Roche Ester: A New Approach to Polypropionate Stereotriad Synthesis. Org. Lett. 2014, 16, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.N.; Leighton, J.L. A Highly stereoselective, efficient, and scalable synthesis of the C(1)–C(9) fragment of the epothilones. Org. Lett. 2015, 17, 5858–5861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Kong, L.; Gu, Q.; Shao, S.; Lin, G.-Q.; Hong, R. Stereoselective Access to Polypropionates Expedited by the Double Hydroboration of Allenes: Total Synthesis of Antitumor (−)-Pironetin. CCS Chem. 2021, 3, 769–779. [Google Scholar] [CrossRef]
- Bode, J.W.; Fraefel, N.; Muri, D.; Carreira, E.M. A General solution to the modular synthesis of polyketide building blocks by Kanemasa hydroxy-directed nitrile oxide cycloadditions. Angew. Chem. Int. Ed. 2001, 40, 2082–2085. [Google Scholar] [CrossRef]
- Fader, L.D.; Carreira, E.M. Stereochemically Rich Pentaketides from Bis(isoxazolines): A General Strategy for Efficient Polyketide Synthesis. Org. Lett. 2004, 6, 2485–2488. [Google Scholar] [CrossRef]
- Muri, D.; Carreira, E. Stereoselective Synthesis of Erythronolide A via Nitrile Oxide Cycloadditions and Related Studies. J. Org. Chem. 2009, 74, 8695–8712. [Google Scholar] [CrossRef]
- Danishefsky, S.; Harvey, D.F. A new approach to polypropionates—Routes to subunits of nonensin and tirandamycin. J. Am. Chem. Soc. 1985, 107, 6647–6652. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Myles, D.C.; Harvey, D.F. Expeditious synthesis of the polypropionate sector of rifamycin S by reiterative Diene-Aldehyde cyclocondensation reactions. J. Am. Chem. Soc. 1987, 109, 862–867. [Google Scholar] [CrossRef]
- Vogel, P.; Sevin, A.-F.; Kernen, P.; Bialecki, M. ’Naked sugars of the second generation’: Asymmetric synthesis of long-chain polypropionates and analogues starting with acetone. Pure Appl. Chem. 1996, 68, 719–722. [Google Scholar] [CrossRef]
- Vogel, P.; Cossy, J.; Plumet, J.; Arjona, O. Derivatives of 7-oxabicyclo[2.2.1]heptane in nature and as useful synthetic intermediates. Tetrahedron 1999, 55, 13521–13642. [Google Scholar] [CrossRef]
- Arjona, O.; Menchaca, R.; Plumet, J. Building a small polypropionate library. Synthesis of all possible stereotetrads (building blocks for polyketide synthesis) from furan. J. Org. Chem. 2001, 66, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Arjona, O.; Plumet, J. The Ring Opening of Oxabicyclic Compounds Controlled by a Phenylsulfonyl Group: Synthetic Applications. ChemInform 2003, 34, 571–595. [Google Scholar] [CrossRef]
- Hunt, K.W.; Grieco, P.A. Oxabicyclo[3.2.1]octenes in organic synthesis—Direct ring opening of oxabicyclo[3.2.1] systems employing silyl ketene acetals in concentrated solutions of lithium perchlorate-diethyl ether: Application to the synthesis of the C(19)-C(27) fragment of rifamycin S. Org. Lett. 2001, 3, 481–484. [Google Scholar] [PubMed]
- Hagenbuch, J.-P.; Vogel, P. Asymmetric induction in the rearrangement of monocyclic endoperoxides into gamma-hydroxy-alpha,beta-unsaturated aldehydes. J. Chem. Soc. Chem. Commun. 1980, 22, 1062–1063. [Google Scholar] [CrossRef]
- Gesinski, M.R.; Brenzovich, W.E.; Staben, S.T.; Srinilta, D.J.; Toste, F.D. A divergent/convergent approach to dolabriferol: The Kornblum-DeLaMare enantiomeric resolution. Tetraedron Lett. 2015, 56, 3643–3646. [Google Scholar] [CrossRef] [Green Version]
- Tokura, N. Olefin-sulfur dioxide copolymers. Encycl. Polym. Sci. Technol. 1968, 9, 460–485. [Google Scholar]
- Gray, D.N. The status of olefin-sulfur dioxide copolymers as biomaterials. Polym. Sci. Technol. 1981, 14, 21–27. [Google Scholar]
- Fawcett, A.H. Olefin-sulfur dioxide copolymers. Encycl. Polym. Sci. Eng. 1987, 10, 408–432. [Google Scholar]
- Masilamani, D.; Reuman, M.E.; Rogic, M.M. Ene-type reaction through the intermediacy of the 1,4-dipolar ion in the reaction of tetracyanoethylene with nucleophilic double-bonds in liquid sulfur dioxide. J. Org. Chem. 1980, 45, 4602–4605. [Google Scholar] [CrossRef]
- Masilamani, D.; Rogic, M.M. Organic reactions of sulfur dioxide. 4. Facile regiospecific hydrogen-deuterium exchange in olefins—Consequence of intermediacy of allylic sulfinic acids in ene reaction of sulfur dioxide with double bonds. J. Am. Chem. Soc. 1978, 100, 4634–4635. [Google Scholar] [CrossRef]
- Marković, D.; Vogel, P. Polysulfones: Catalysts for Alkene Isomerization. Angew. Chem. Int. Ed. 2004, 43, 2928–2930. [Google Scholar] [CrossRef]
- Markovic, D.; Vogel, P. Allyl, Methallyl, Prenyl, and Methylprenyl Ethers as Protected Alcohols: Their Selective Cleavage with Diphenyldisulfone under Neutral Conditions. Org. Lett. 2004, 6, 2693–2696. [Google Scholar] [CrossRef]
- Marković, D.; Varela-Álvarez, A.; Sordo, J.A.; Vogel, P. Mechanism of the Diphenyldisulfone-Catalyzed Isomerization of Alkenes. Origin of the Chemoselectivity: Experimental and Quantum Chemistry Studies. J. Am. Chem. Soc. 2006, 128, 7782–7795. [Google Scholar] [CrossRef]
- Backer, H.J.; Strating, J. Cyclical sulphones, derivatives and butadienes. Recl. Trav. Chim. Pays-Bas 1934, 53, 525–543. [Google Scholar] [CrossRef]
- Heldeweg, R.F.; Hogeveen, H. (2+4) (π+π,π) and (2+4)(n+π-π) modes of addition in reaction between SO2 and a diene—Kinetic vs thermodynamic control. J. Am. Chem. Soc. 1976, 98, 2341–2342. [Google Scholar] [CrossRef]
- Durst, T.; Tetreault-Ryan, L. Reaction of ortho-quinodimethane with sulfur dioxide. Competition between π + π-π and n + π-π cycloadditions. Tetrahedron Lett. 1978, 26, 2353–2354. [Google Scholar] [CrossRef]
- Deguin, B.; Vogel, P. Hetero-Diels-Alder addition of sulfur dioxide to 1,3-dienes. Suprafaciality, regioselectivity, and stereoselectivity. J. Am. Chem. Soc. 1992, 114, 9210–9211. [Google Scholar] [CrossRef]
- Fernandez, T.; Sordo, J.A.; Monnat, F.; Deguin, B.; Vogel, P. Sulfur dioxide promotes its hetero-Diels-Alder and cheletropic additions to 1,2-dimethylenecyclohexane. J. Am. Chem. Soc. 1998, 120, 13276–13277. [Google Scholar] [CrossRef]
- Monnat, F.; Vogel, P.; Rayón, V.M.; Sordo, J.A. Ab Initio and Experimental Studies on the Hetero-Diels−Alder and Cheletropic Additions of Sulfur Dioxide to (E)-1-Methoxybutadiene: A Mechanism Involving Three Molecules of SO2. J. Org. Chem. 2002, 67, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Sordo, J. Hetero-Diels-Alder and Cheletropic Additions of Sulfur Dioxide to Conjugated Dienes. Experimental Facts and Theoretical Analysis. Curr. Org. Chem. 2006, 10, 2007–2036. [Google Scholar] [CrossRef]
- Markovic, D.; Roversi, E.; Scoppelliti, R.; Vogel, P.; Meana, R.; Sordo, J.A. The hetero-Diels-Alder addition of sulfur dioxide: The pseudo-chair conformation of a 4,5-dialkylsultine. Chem. Eur. J. 2003, 9, 4911–4915. [Google Scholar] [CrossRef] [PubMed]
- Monnat, F.; Vogel, P.; Meana, R.; Sordo, J.A. Equilibrium and Kinetic Deuterium Isotope Effects on the Hetero-Diels–Alder Addition of Sulfur Dioxide. Angew. Chem. Int. Ed. 2003, 42, 3924–3927. [Google Scholar] [CrossRef] [PubMed]
- Roversi, E.; Scopelliti, R.; Solari, E.; Estoppey, R.; Vogel, P.; Braña, P.; Menéndez, B.; Sordo, J.A. The Hetero-Diels-Alder Addition of Sulfur Dioxide to 1-Fluorobuta-1,3-dienes: The Sofa Conformations Preferred by 6-Fluorosultines (6-Fluoro-3,6-dihydro-1,2-oxathiin-2-oxides) Enjoy Enthalpic and Conformational Anomeric Effects. Chem. A Eur. J. 2002, 8, 1336–1355. [Google Scholar] [CrossRef]
- Laclef, S.; Exner, C.J.; Turks, M.; Videtta, V.; Vogel, P. Synthesis of (E,Z)-1-Alkoxy-3-acyloxy-2-methylpenta-1,3-dienes via Danishefsky-Type Dienes or O-Acylation of Enones. J. Org. Chem. 2009, 74, 8882–8885. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Turks, M.; Exner, C.; Hamel, C. Umpolung with Sulfur Dioxide: Carbon-Carbon Cross-Coupling of Electron-Rich 1,3-Dienes and Alkenes; Application to the Enantioselective Synthesis of Long-Chain Polyketide Fragments. Synthesis 2009, 2009, 1065–1074. [Google Scholar] [CrossRef]
- Kanazawa, A.; Delair, P.; Pourashraf, M.; Greene, A.E. Convergent, enantioselective synthesis of the novel furanoditerpene (+)-taonianone through facially selective chiral olefin–ketene [2+2] cycloaddition. J. Chem. Soc. Perkin Trans. 1997, 13, 1911–1912. [Google Scholar] [CrossRef]
- Narkevitch, V.; Megevand, S.; Schenk, K.; Vogel, P. Development of a New Carbon−Carbon Bond Forming Reaction. New Organic Chemistry of Sulfur Dioxide. Asymmetric Four-Component Synthesis of Polyfunctional Sulfones. J. Org. Chem. 2001, 66, 5080–5093. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Huang, X. One-Pot, Four-Component Synthesis of Polyfunctional Sulfones. Synthesis 2002, 2002, 0232–0236. [Google Scholar] [CrossRef]
- Bouchez, L.C.; Dubbaka, S.R.; Turks, M.; Vogel, P. Sulfur Dioxide Mediated One-Pot, Three- and Four-Component Syntheses of Polyfunctional Sulfonamides and Sulfonic Esters: Study of the Stereoselectivity of the Ene Reaction of Sulfur Dioxide. J. Org. Chem. 2004, 69, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, L.C.; Turks, M.; Dubbaka, S.R.; Fonquerne, F.; Craita, C.; Laclef, S.; Vogel, P. Sulfur dioxide mediated one-pot, four-component synthesis of polyfunctional sulfones and sulfonamides, including medium-ring cyclic derivatives. Tetrahedron 2005, 61, 11473–11487. [Google Scholar] [CrossRef]
- Vogel, P.; Turks, M.; Bouchez, L.; Craita, C.; Murcia, M.C.; Fonquerne, F.; Didier, C.; Huang, X.; Flowers, C. Use of sultines in the asymmetric synthesis of polypropionate antibiotics. Pure Appl. Chem. 2008, 80, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Turks, M.; Fairweather, K.A.; Scopelliti, R.; Vogel, P. Efficient Asymmetric Synthesis of Long-Chain Polyketides Containing up to Ten Contiguous Stereogenic Centres by Double Chain Elongation. Eur. J. Org. Chem. 2011, 2011, 3317–3328. [Google Scholar] [CrossRef]
- Paterson, I.; Florence, G.J.; Gerlach, K.; Scott, J.; Sereinig, N. Total synthesis of the immunosuppressive agent (-)-discodermolide. J. Am. Chem. Soc. 2001, 123, 9535–9544. [Google Scholar] [CrossRef]
- Paterson, I.; Perkins, M.V. Total Synthesis of (−)-Denticulatins A and B Using Efficient Methods of Acyclic Stereocontrol. Tetrahedron 1996, 52, 1811–1834. [Google Scholar] [CrossRef]
- Stockdale, T.P.; Lam, N.Y.S.; Anketell, M.J.; Paterson, I. The Stereocontrolled Total Synthesis of Polyketide Natural Products: A Thirty-Year Journey. Bull. Chem. Soc. Jpn. 2021, 94, 713–731. [Google Scholar] [CrossRef]
- Evans, D.A.; Chapman, K.T. The directed reduction of β-hydroxy ketones employing Me4NB(OAc)3H. Tetrahedron Lett. 1986, 27, 5939–5942. [Google Scholar] [CrossRef]
- Evans, D.A.; Chapman, K.T.; Carreira, E.M. Directed reduction of beta.-hydroxy ketones employing tetramethylammonium triacetoxyborohydride. J. Am. Chem. Soc. 1988, 110, 3560–3578. [Google Scholar] [CrossRef]
- Manker, D.C.; Faulkner, D.J.; Stout, T.J.; Clardy, J. The baconipyrones. Novel polypropionates from the pulmonate Siphonaria baconi. J. Org. Chem. 1989, 54, 5371–5374. [Google Scholar] [CrossRef]
- Turks, M.; Murcia, M.C.; Scopelliti, R.; Vogel, P. First Asymmetric Synthesis of the Cyclohexanone Subunit of Baconipyrones A and B. Revision of Its Structure. Org. Lett. 2004, 6, 3031–3034. [Google Scholar] [CrossRef]
- Pereyre, M.; Bellegarde, B.; Mendelsohn, J.; Valade, J. Action of triorganotin alkaloids on enol esters—Problem of obtaining C-or O-stannylated compounds. J. Organomet. Chem. 1968, 11, 97–110. [Google Scholar] [CrossRef]
- Labadie, S.S.; Stille, J.K. Stereoselective aldol condensations of organotin reagents with aldehydes. Tetrahedron 1984, 40, 2329–2336. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Gavagnin, M.; Puliti, R.; Cimino, G.; Martinez, E.; Ortea, J.; Mattia, C.A. Dolabriferol: A new polypropionate from the skin of the anaspidean mollusc Dolabrifera dolabrifera. Tetrahedron 1996, 52, 12831–12838. [Google Scholar] [CrossRef]
- Paterson, I.; Chen, D.Y.K.; Acena, J.L.; Franklin, A.S. Studies in marine polypropionate synthesis: Total synthesis of (−)-baconipyrone C. Org. Lett. 2000, 2, 1513–1516. [Google Scholar] [CrossRef]
- Laclef, S.; Turks, M.; Vogel, P. Total Synthesis and Determination of the Absolute Configuration of (−)-Dolabriferol. Angew. Chem. Int. Ed. 2010, 49, 8525–8527. [Google Scholar] [CrossRef]
- Currie, R.H.; Goodman, J.M. In Silico Inspired Total Synthesis of (−)-Dolabriferol. Angew. Chem. Int. Ed. 2012, 51, 4695–4697. [Google Scholar] [CrossRef]
- Karagiannis, A.; Diddi, N.; Ward, D.E. On the Origin of Dolabriferol: Total Synthesis via Its Putative Contiguous Precursor. Org. Lett. 2016, 18, 3794–3797. [Google Scholar] [CrossRef] [PubMed]
- Gantasala, N.; Borra, S.; Pabbaraja, S.; Srihari, P. Stereoselective Total Synthesis of the Non-Contiguous Polyketide Natural Product (-)-Dolabriferol. Eur. J. Org. Chem. 2018, 2018, 1230–1240. [Google Scholar] [CrossRef]
- Bandaru, A.; Si, D.; Kaliappan, K.P. Synthesis of C1-C9 and C10-C21 fragments of (-)-dolabriferol. Asian J. Org. Chem. 2020, 9, 1045–1052. [Google Scholar] [CrossRef]
- Rinehart, K.L. Antibiotics with ansa rings. Acc. Chem. Res. 1972, 5, 57–64. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Shield, S. Chemistry of the ansamycin antibiotics. Fortsch. Chem. Org. Naturst. 1976, 33, 231–307. [Google Scholar]
- Wehrli, W. Ansamycins Chemistry, biosynthesis and biological activity. Top. Curr. Chem. 1977, 72, 21–49. [Google Scholar]
- Prelog, V. Conformation and reactivity of medium-sized ring compounds. Pure Appl. Chem. 1963, 6, 545–560. [Google Scholar] [CrossRef] [Green Version]
- Sensi, P.; Furesz, S.; Maggi, G.; Maffi, G. Chemical modifications and biological properties of rifamycins. Antimicrob. Agents Chemother. 1966, 6, 699–714. [Google Scholar]
- Arora, S.K. Correlation of structure and activity in ansamycins—Structure, conformation, and interactions of antibiotic rifamycin-S. J. Med. Chem. 1985, 28, 1099–1102. [Google Scholar] [CrossRef]
- Joss, U.R.; Hughes, A.M.; Calvin, M. Effect of dimethylbenzyldesmethyl-rifamycin (Dmb) on chemically induced mammary-tumors in rats. Nat.-New Biol. 1973, 242, 88–90. [Google Scholar] [CrossRef]
- Spisani, S.; Traniello, S.; Martuccio, C.; Rizzuti, O.; Cellai, L. Rifamycins inhibit human neutrophil functions: New derivatives with potential antiinflammatory activity. Inflammation 1997, 21, 391–400. [Google Scholar] [CrossRef]
- Hartmann, G.; Honikel, K.O.; Knüsel, F.; Nüesch, J. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1967, 145, 843–844. [Google Scholar] [CrossRef]
- Bacchi, A.; Pelizzi, G.; Nebuloni, M.; Ferrari, P. Comprehensive Study on Structure−Activity Relationships of Rifamycins: Discussion of Molecular and Crystal Structure and Spectroscopic and Thermochemical Properties of Rifamycin O. J. Med. Chem. 1998, 41, 2319–2332. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.-W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Lyle, M.A.; Snider, J.D.E. Rifabutin (Ansamycin LM 427): A New Rifamycin-S Derivative for the Treatment of Mycobacterial Diseases. Clin. Infect. Dis. 1987, 9, 519–530. [Google Scholar] [CrossRef]
- Brogden, R.N.; Fitton, A. Rifabutin—A review of its antimicrobial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1994, 47, 983–1009. [Google Scholar] [CrossRef]
- Barluenga, J.; Aznar, F.; García, A.B.; Cabal, M.P.; Palacios, J.J.; Menéndez, M.A. New rifabutin analogs: Synthesis and biological activity against mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2006, 16, 5717–5722. [Google Scholar] [CrossRef]
- Selva, E.; Lancini, G. Rifamycins, Antibacterial Antibiotics and Their New Applications; Fischer, J., Ganellin, C.R., Eds.; Analogue-Based Drug Discovery II; Wiley-VCH Verlag GmbH & CO. KGaA: Weinheim, Germany, 2010; pp. 173–187. [Google Scholar]
- Nagaoka, H.; Rutsch, W.; Schmid, G.; Iio, H.; Johnson, M.R.; Kishi, Y. Total synthesis of rifamycins. 1. Stereocontrolled synthesis of the aliphatic building block. J. Am. Chem. Soc. 1980, 102, 7962–7965. [Google Scholar] [CrossRef]
- Iio, H.; Nagaoka, H.; Kishi, Y. Total synthesis of rifamycins. 2. Total synthesis of racemic rifamycin-S. J. Am. Chem. Soc. 1980, 102, 7965–7967. [Google Scholar] [CrossRef]
- Kishi, Y. Total synthesis of rifamycin S. Pure Appl. Chem. 1981, 53, 1163–1180. [Google Scholar] [CrossRef]
- Nagaoka, H.; Kishi, Y. Further synthetic studies on rifamycin s. Tetrahedron 1981, 37, 3873–3888. [Google Scholar] [CrossRef]
- Hanessian, S.; Pougny, J.R.; Boessenkool, I.K. Total synthesis of the C19-C29 aliphatic segment of (+)-rifamycin-S. J. Am. Chem. Soc. 1982, 104, 6164–6166. [Google Scholar] [CrossRef]
- Katsuki, T.; Hanamoto, T.; Yamaguchi, M. Synthesis of C19–C27Fragment of Ansa Chain Part of Rifamycin S. Chem. Lett. 1989, 18, 117–118. [Google Scholar] [CrossRef]
- Paterson, I.; McClure, C.K.; Schumann, R.C. A short asymmetric synthesis of a C19–C27 segment of rifamycin S. Kinetic resolution in the aldol reactions of ethylketones using chiral boron reagents. Tetrahedron Lett. 1989, 30, 1293–1296. [Google Scholar] [CrossRef]
- Chênevert, R.; Rose, Y.S. Enzymatic Desymmetrization of a Meso Polyol Corresponding to the C(19)−C(27) Segment of Rifamycin S. J. Org. Chem. 2000, 65, 1707–1709. [Google Scholar] [CrossRef]
- Corey, E.J.; Hase, T. Studies on the total synthesis of rifamycin—Highly stereoselective synthesis of intermediates for construction of the C(15) to C(29) chain. Tetrahedron Lett. 1979, 4, 335–338. [Google Scholar] [CrossRef]
- Masamune, S.; Imperiali, B.; Garvey, D.S. Synthesis of ansamycins: The ansa chain of rifamycin S. J. Am. Chem. Soc. 1982, 104, 5528–5531. [Google Scholar] [CrossRef]
- Paterson, I.; Mansuri, M.M. Recent developments in the total synthesis of macrolide antibiotics. Chem. Informationsdienst 1985, 41, 3569–3624. [Google Scholar] [CrossRef]
- Rao, A.V.R.; Yadav, J.S.; Vidyasagar, V. Stereoselective synthesis of the C-21 to C-27 segment of rifamycin-S. J. Chem. Soc. Chem. Commun. 1985, 55–56. [Google Scholar] [CrossRef]
- Rao, A.V.R.; Yadav, J.S.; Vidyasagar, V. Stereoselective synthesis of the C-21 to C-27 segment of rifamycin-S. Tetrahedron Lett. 1986, 27, 3297–3298. [Google Scholar] [CrossRef]
- Roush, W.R.; Palkowitz, A.D. Applications of tartrate ester modified allylic boronates in organic synthesis—An efficient, highly stereoselective synthesis of the C(19)-C(29) segment of rifamycin-S. J. Am. Chem. Soc. 1987, 109, 953–955. [Google Scholar] [CrossRef]
- Ziegler, F.E.; Kneisley, A. 3-Methyl-gamma-butyrolactone as a source of 2-methyl-3-hydroxyketones and 2-methyl-1,3-diols—A synthesis of the C19-C27 fragment of rifamycin-S by linear iteration. Tetrahedron Lett. 1987, 28, 1725–1728. [Google Scholar] [CrossRef]
- Ziegler, F.E.; Cain, W.T.; Kneisley, A.; Stirchak, E.P.; Wester, R.T. Applications of the 3-methyl-gamma-butyrolactone strategy to the synthesis of polypropionates—the Prelog-Djerassi lactonic ester, ent-invictolide, and the C19-C27 fragment of rifamycin S. J. Am. Chem. Soc. 1988, 110, 5442–5452. [Google Scholar] [CrossRef]
- Tarara, G.; Hoppe, D. Total synthesis of protected D-altro-3,6-dideoxy-3-C-methylhexose and D-galacto-3,6-dideoxy-3-C-methylhexose—Key intermediates of a rifamycin S synthesis. Synthesis 1989, 2, 89–92. [Google Scholar] [CrossRef]
- Born, M.; Tamm, C. Stereoselective Synthesis of the C(19)-to-C(27) Segment of Rifamycin S. Helvetica Chim. Acta 1990, 73, 2242–2250. [Google Scholar] [CrossRef]
- Roush, W.R.; Palkowitz, A.D.; Ando, K. Acyclic diastereoselective synthesis using tartrate ester-modified crotylboronates. Double asymmetric reactions with alpha.-methyl chiral aldehydes and synthesis of the C(19)-C(29) segment of rifamycin S. J. Am. Chem. Soc. 1990, 112, 6348–6359. [Google Scholar] [CrossRef]
- Harada, T.; Kagamihara, Y.; Tanaka, S.; Sakamoto, K.; Oku, A. A highly convergent asymmetric synthesis of the C(19)-C(27) segment of rifamycin S: An application of enantiodifferentiating acetalization with menthone. J. Org. Chem. 1992, 57, 1637–1639. [Google Scholar] [CrossRef]
- Lautens, M.; Belter, R.K. The effect of remote oxygens on the ring-opening reactions of oxabicyclic compounds with organolithium reagents. Synthesis of the C21–C27 segment of rifamycin S. Tetrahedron Lett. 1992, 33, 2617–2620. [Google Scholar] [CrossRef]
- Miyashita, M.; Yoshihara, K.; Kawamine, K.; Hoshino, M.; Irie, H. Synthetic studies on polypropionate antibiotics based on the stereospecific methylation of gamma,delta-epoxy acrylates by trimethylaluminum—A highly stereoselective construction of the 8 contiguous chiral centers of ansa-chains of rifamycins. Tetrahedron Lett. 1993, 34, 6285–6288. [Google Scholar] [CrossRef]
- Harada, T.; Oku, A. Enantiodifferentiating transformation of prochiral polyols by using menthone as chiral template. Synlett 1994, 2, 95–104. [Google Scholar] [CrossRef]
- Yadav, J.S.; Rao, C.S.; Chandrasekhar, S.; Rao, A.V.R. Asymmetric synthesis of C-19 to C-27 fragment of rifamycin-S. Tetrahedron Lett. 1995, 36, 7717–7720. [Google Scholar] [CrossRef]
- Hanessian, S.; Wang, W.; Gai, Y.; Olivier, E. A General and Stereocontrolled Strategy for the Iterative Assembly of Enantiopure Polypropionate Subunits: Synthesis of the C19−C28 Segment of Rifamycin S from a Single Chiron. J. Am. Chem. Soc. 1997, 119, 10034–10041. [Google Scholar] [CrossRef]
- Marshall, J.A.; Palovich, M.R. Synthesis of Stereopentad Subunits of Zincophorin and Rifamycin-S through Use of Chiral Allenyltin Reagents. J. Org. Chem. 1998, 63, 3701–3705. [Google Scholar] [CrossRef]
- Turks, M.; Huang, X.; Vogel, P. Expeditious Asymmetric Synthesis of a Stereoheptad Corresponding to the C(19)-C(27)-Ansa Chain of Rifamycins: Formal Total Synthesis of Rifamycin S. Chem. A Eur. J. 2004, 11, 465–476. [Google Scholar] [CrossRef]
- Kim, J.W.; Adachi, H.; Shin, Y.K.; Hayakawa, Y.; Seto, H. Apoptolidin, a new apoptosis inducer in transformed cells from Nocardiopsis sp. J. Antibiot. 1997, 50, 628–630. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, Y.; Kim, J.W.; Adachi, H.; Shin-Ya, K.; Fujita, A.K.-I.; Seto, H. Structure of Apoptolidin, a Specific Apoptosis Inducer in Transformed Cells. J. Am. Chem. Soc. 1998, 120, 3524–3525. [Google Scholar] [CrossRef]
- Wender, P.A.; Sukopp, M.; Longcore, K. Apoptolidins B and C: Isolation, Structure Determination, and Biological Activity. Org. Lett. 2005, 7, 3025–3028. [Google Scholar] [CrossRef]
- Wender, P.A.; Longcore, K.E. Isolation, Structure Determination, and Anti-Cancer Activity of Apoptolidin D. Org. Lett. 2007, 9, 691–694. [Google Scholar] [CrossRef]
- Wender, P.A.; Longcore, K.E. Apoptolidins E and F, New Glycosylated Macrolactones Isolated from Nocardiopsis sp. Org. Lett. 2009, 11, 5474–5477. [Google Scholar] [CrossRef] [Green Version]
- Salomon, A.R.; Voehringer, D.W.; Herzenberg, L.A.; Khosla, C. Understanding and exploiting the mechanistic basis for selectivity of polyketide inhibitors of F0F1-ATPase. Proc. Natl. Acad. Sci. USA 2000, 97, 14766–14771. [Google Scholar] [CrossRef] [Green Version]
- Benitez-Bribiesca, L. Assessment of apoptosis in tumor growth: Importance in clinical oncology and cancer therapy. In When Cells Die; Lockshin, R.A., Zakeri, Z., Tilly, J.L., Eds.; Wiley-Liss: New York, NK, USA, 1998; pp. 453–492. [Google Scholar]
- Salomon, A.R.; Vöhringer, D.W.; Herzenberg, L.A.; Khosla, C. Apoptolidin, a selective cytotoxic agent, is an inhibitor of F0F1-ATPase. Chem. Biol. 2001, 8, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Salomon, A.R.; Zhang, Y.; Seto, H.; Khosla, C. Structure−Activity Relationships within a Family of Selectively Cytotoxic Macrolide Natural Products. Org. Lett. 2000, 3, 57–59. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, Y.; Fylaktakidou, K.C.; Mitchell, H.J.; Sugita, K. Total Synthesis of Apoptolidin: Part 2. Coupling of Key Building Blocks and Completion of the Synthesis. Angew. Chem. Int. Ed. 2001, 40, 3854–3857. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Fylaktakidou, K.C.; Monenschein, H.; Li, Y.; Weyershausen, B.; Mitchell, H.J.; Wei, H.-X.; Guntupalli, P.; Hepworth, D.; Sugita, K. Total Synthesis of Apoptolidin: Construction of Enantiomerically Pure Fragments. J. Am. Chem. Soc. 2003, 125, 15433–15442. [Google Scholar] [CrossRef]
- Wehlan, H.; Dauber, M.; Fernaud, M.-T.M.; Schuppan, J.; Mahrwald, R.; Ziemer, B.; Garcia, M.-E.J.; Koert, U. Total Synthesis of Apoptolidin. Angew. Chem. Int. Ed. 2004, 43, 4597–4601. [Google Scholar] [CrossRef]
- Wehlan, H.; Dauber, M.; Fernaud, M.T.M.; Schuppan, J.; Keiper, S.; Mahrwald, R.; Garcia, M.-E.J.; Koert, U. Apoptolidin A: Total Synthesis and Partially Glycosylated Analogues. Chem.—A Eur. J. 2006, 12, 7378–7397. [Google Scholar] [CrossRef]
- Wu, B.; Liu, Q.; Sulikowski, G.A. Total Synthesis of Apoptolidinone. Angew. Chem. Int. Ed. 2004, 43, 6673–6675. [Google Scholar] [CrossRef]
- Ghidu, V.P.; Wang, J.Q.; Wu, B.; Liu, Q.; Jacobs, A.; Marnett, L.J.; Sulikowski, G.A. Synthesis and Evaluation of the Cytotoxicity of Apoptolidinones A and D. J. Org. Chem. 2008, 73, 4949–4955. [Google Scholar] [CrossRef] [Green Version]
- Crimmins, M.T.; Christie, H.S.; Chaudhary, A.K.; Long, A. Enantioselective Synthesis of Apoptolidinone: Exploiting the Versatility of Thiazolidinethione Chiral Auxiliaries. J. Am. Chem. Soc. 2005, 127, 13810–13812. [Google Scholar] [CrossRef]
- Chau, S.T.; Sulikowski, G.A.; Wu, B. Studies on the synthesis of the apoptolidins. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Elsevier Science Publ. Co. Inc.: San Diego, CA, USA, 2012; Volume 8, pp. 375–394. [Google Scholar]
- Craita, C.; Didier, C.; Vogel, P. Short synthesis of the C16–C28polyketide fragment of apoptolidin A aglycone. Chem. Commun. 2007, 23, 2411–2413. [Google Scholar] [CrossRef] [PubMed]
- Rubottom, G.M.; Vazquez, M.A.; Pelegrina, D.R. Peracid oxidation of trimethylsilyl enol ethers: A facile α-hydroxylation procedure. Tetrahedron Lett. 1974, 15, 4319–4322. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; Pullez, M.; Solladie, G.; Colobert, F. Convergent Highly Stereoselective Preparation of the C12−C24 Fragment of Macrolactin A. J. Org. Chem. 2004, 69, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Hartung, I.V.; Niess, B.; Haustedt, L.O.; Hoffmann, H.M.R. Toward the Total Synthesis of Disorazole A1 and C1: Asymmetric Synthesis of a Masked Southern Segment. Org. Lett. 2002, 4, 3239–3242. [Google Scholar] [CrossRef]
- Brown, H.C.; Bhat, K.S.; Randad, R.S. Chiral synthesis via organoboranes. 21. Allylboration and crotylboration of alpha-chiral aldehydes with diisopinocampheylboron as the chiral auxiliary. J. Org. Chem. 1989, 54, 1570–1576. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.; Sharpless, K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Bouchez, L.C.; Vogel, P. Synthesis of the C(1)-C(11) Polyene Fragment of Apoptolidin with a New Sulfur Dioxide-Based Organic Chemistry. Chem. A Eur. J. 2005, 11, 4609–4620. [Google Scholar] [CrossRef]
- Ramberg, L.; Bäcklung, B. The reactions of some monohalogen derivatives of diethyl sulfone. Ark. Kemi. Mineral. Geol. 1940, 13A, 1–50. [Google Scholar]
- Chan, T.L.; Fong, S.; Li, Y.; Man, T.O.; Poon, C.D. A new one-flask Ramberg-Bäcklund reaction. J. Chem. Soc. 1994, 15, 1771–1772. [Google Scholar] [CrossRef]
- Cao, X.P. Stereoselective synthesis of substituted all-trans-1,3,5,7-octatetraenes by a modified Ramberg-Bäcklund reaction. Tetrahedron 2002, 58, 1301–1307. [Google Scholar] [CrossRef]
- Narasaka, K.; Pai, H.C. ChemInform Abstract: STEREOSELECTIVE SYNTHESIS OF MESO (OR ERYTHRO) 1,3-DIOLS FROM β-HYDROXYKETONES. Chem. Inf. 1981, 12. [Google Scholar] [CrossRef]
- Exner, C.J.; Laclef, S.; Poli, F.; Turks, M.; Vogel, P. Total asymmetric syntheses of β-hydroxy-δ-lactones via Umpolung with sulfur dioxide. J. Org. Chem. 2011, 76, 840–845. [Google Scholar] [CrossRef]
- Exner, C.J.; Turks, M.; Fonquerne, F.; Vogel, P. Concise synthesis of complicated polypropionates through one-pot disymmetrical two-directional chain elongation. Chem. Eur. J. 2011, 17, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
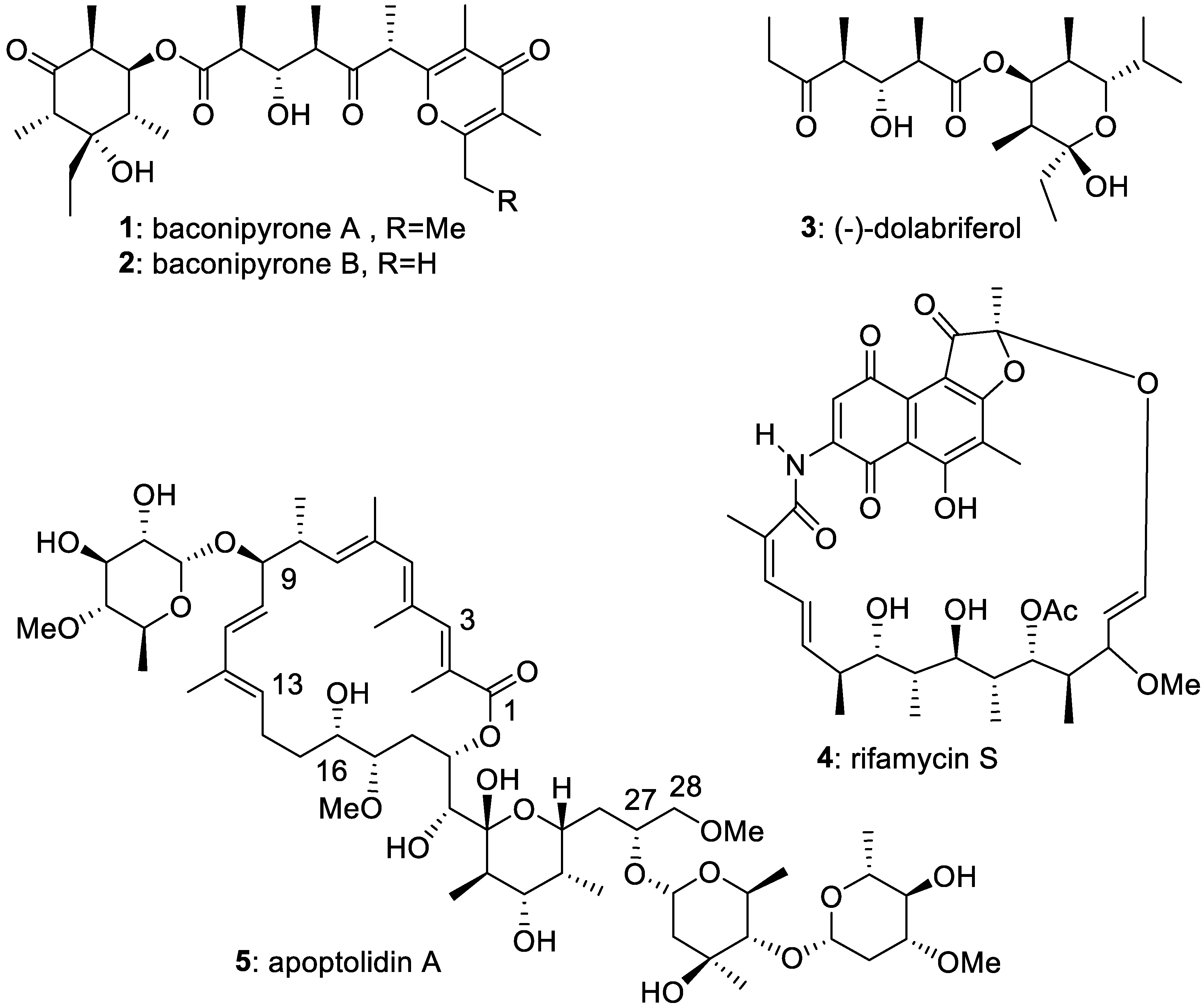
Figure 1.
Examples of natural products containing polypropionate fragments, their synthesis being presented in this report.
Figure 1.
Examples of natural products containing polypropionate fragments, their synthesis being presented in this report.

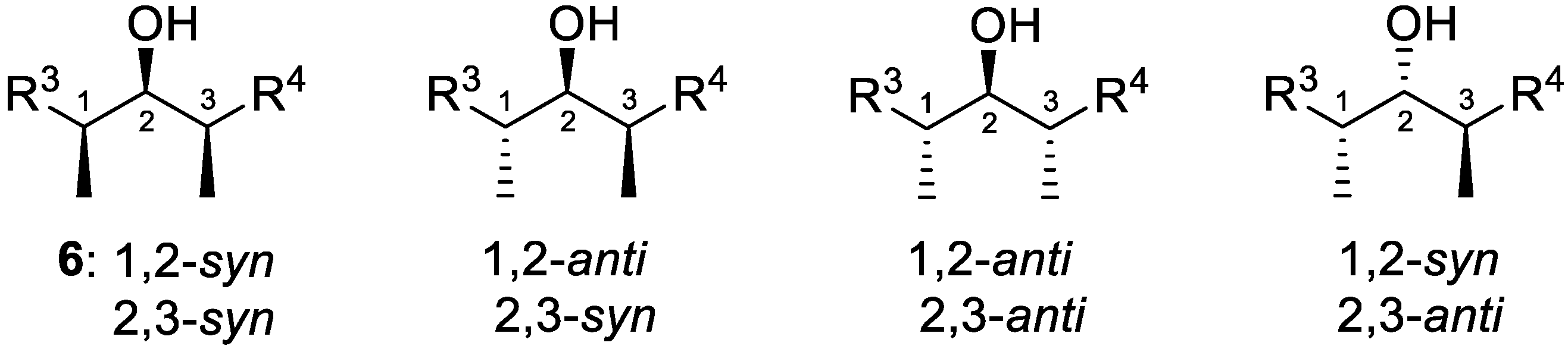
Figure 2.
Four possible diastereomeric polypropionate stereotriads of type R3-CH(Me)-CH(OH)-CH(Me)-R4. Their enantiomers are not shown. Other stereotriads could be of the type R3-CH(OH)-CH(Me)-CH(OH)-R4.
Figure 2.
Four possible diastereomeric polypropionate stereotriads of type R3-CH(Me)-CH(OH)-CH(Me)-R4. Their enantiomers are not shown. Other stereotriads could be of the type R3-CH(OH)-CH(Me)-CH(OH)-R4.

Scheme 1.
One-pot synthesis of syn,anti and anti,anti stereotriads 7 and 8 through C–C bond formation between an 1-((1S)-phenylethyloxy)-2-methylpenta-1,3-dien-3-yl carboxylic esters 10 and the (E)- or (Z)-enoxysilane derived from butan-3-one ((E)-9, (Z)-9). HA is either a protic (e.g., (CF3SO2)2NH) or Lewis acid (e.g,: Me3SiOSO2CF3) catalyst.
Scheme 1.
One-pot synthesis of syn,anti and anti,anti stereotriads 7 and 8 through C–C bond formation between an 1-((1S)-phenylethyloxy)-2-methylpenta-1,3-dien-3-yl carboxylic esters 10 and the (E)- or (Z)-enoxysilane derived from butan-3-one ((E)-9, (Z)-9). HA is either a protic (e.g., (CF3SO2)2NH) or Lewis acid (e.g,: Me3SiOSO2CF3) catalyst.

Scheme 2.
Simple dienes that can adopt a s-cis conformation undergo SO2 and acid-catalyzed hetero-Diels–Alder reactions with SO2 at low temperature, giving sultines that are about 10 kcal mol−1 less stable than the corresponding sulfolenes.
Scheme 2.
Simple dienes that can adopt a s-cis conformation undergo SO2 and acid-catalyzed hetero-Diels–Alder reactions with SO2 at low temperature, giving sultines that are about 10 kcal mol−1 less stable than the corresponding sulfolenes.

Scheme 3.
Proposed mechanism for the reaction cascade producing the syn,anti-stereotriad 7 as major product: (1) face-selective acid-catalyzed hetero-Diels–Alder reaction of SO2, (2) immediate ionization of the sultine so-obtained into a zwitterion, (3) face-selective quenching of the alkoxyallyl cation intermediate by the enoxysilane, (4) intramolecular or intermolecular silyl transfer forming a silyl sulfinate (can be isolated), (5) its Pd-catalyzed alcoholysis with isopropanol forming the corresponding, β,γ-unsaturated sulfinic acid which (6) undergoes a face-selective H-retro-ene reaction.
Scheme 3.
Proposed mechanism for the reaction cascade producing the syn,anti-stereotriad 7 as major product: (1) face-selective acid-catalyzed hetero-Diels–Alder reaction of SO2, (2) immediate ionization of the sultine so-obtained into a zwitterion, (3) face-selective quenching of the alkoxyallyl cation intermediate by the enoxysilane, (4) intramolecular or intermolecular silyl transfer forming a silyl sulfinate (can be isolated), (5) its Pd-catalyzed alcoholysis with isopropanol forming the corresponding, β,γ-unsaturated sulfinic acid which (6) undergoes a face-selective H-retro-ene reaction.

Scheme 4.
Mechanism for the preparation of the anti,anti-stereotriad 8 and formation of co-products under acid-induced desulfinylation.
Scheme 4.
Mechanism for the preparation of the anti,anti-stereotriad 8 and formation of co-products under acid-induced desulfinylation.

Scheme 5.
An example of bidirectional chain elongation of stereotriad 7 by two successive metal-aldol reactions; synthesis of a stereodecad from a stereotriad requiring the isolation of one synthetic intermediate.
Scheme 5.
An example of bidirectional chain elongation of stereotriad 7 by two successive metal-aldol reactions; synthesis of a stereodecad from a stereotriad requiring the isolation of one synthetic intermediate.

Scheme 6.
Synthesis of the cyclohexanone subunit of baconipyrones A and B.

Scheme 7.
Total asymmetric synthesis of (-)-dolabriferol.

Scheme 8.
Expeditious asymmetric synthesis of Kishi’s stereoheptad: formal total synthesis of rifamycin S.
Scheme 8.
Expeditious asymmetric synthesis of Kishi’s stereoheptad: formal total synthesis of rifamycin S.

Scheme 9.
Synthesis of Koert’s C16–C28 polyketide fragment of apoptolidin A.

Scheme 10.
An expeditious synthesis of the Nicolaou’s C1–C11 fragment of apoptolidin A.

Scheme 11.
Convergent synthesis of complicated polypropionate–polyketide fragments through dissymmetrical two-directional chain elongation.
Scheme 11.
Convergent synthesis of complicated polypropionate–polyketide fragments through dissymmetrical two-directional chain elongation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vogel, P.; Sordo Gonzalo, J.A. Expeditious Asymmetric Synthesis of Polypropionates Relying on Sulfur Dioxide-Induced C–C Bond Forming Reactions. Catalysts 2021, 11, 1267. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111267
AMA Style
Vogel P, Sordo Gonzalo JA. Expeditious Asymmetric Synthesis of Polypropionates Relying on Sulfur Dioxide-Induced C–C Bond Forming Reactions. Catalysts. 2021; 11(11):1267. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111267
Chicago/Turabian StyleVogel, Pierre, and José Angel Sordo Gonzalo. 2021. "Expeditious Asymmetric Synthesis of Polypropionates Relying on Sulfur Dioxide-Induced C–C Bond Forming Reactions" Catalysts 11, no. 11: 1267. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111267
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.