
Activation of Pt Nanoclusters on TiO2 via Tuning the Metallic Sites to Promote Low-Temperature CO Oxidation


1
Key Laboratory of Hunan Province for the Synergetic Control and Resource Reuse of the Multi-Pollutants of Flue Gas, Changsha 410205, China
2
National Sintering and Pelletizing Equipment System Engineering Research Center, Zhongye Changtian International Engineering Co., Ltd., Changsha 410205, China
3
Institute of Zhejiang University-Quzhou, Quzhou 324000, China
4
College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310007, China
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(11), 1280; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111280
Submission received: 26 September 2021
/
Revised: 18 October 2021
/
Accepted: 19 October 2021
/
Published: 23 October 2021
(This article belongs to the Special Issue Catalytic CO Oxidation and Preferential CO Oxidation (PROX))
Abstract
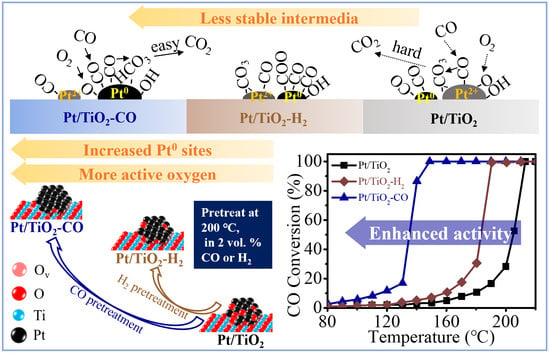
:Metallic Pt sites are imperative in the CO oxidation reaction. Herein, we demonstrate the tuning of Pt sites by treating a Pt catalyst in various reductive atmospheres, influencing the catalyst activities in low-temperature CO oxidation. The H2 pretreatment of Pt clusters at 200 °C decreases the T50 from 208 °C to 183 °C in the 0.1 wt % Pt/TiO2 catalyst. The T50 shows a remarkable improvement using a CO pretreatment, which decreases the T50 further to 135 °C. A comprehensive characterization study reveals the integrated reasons behind this phenomenon: (i) the extent of PtO transition to metallic Pt sites, (ii) the ample surface active oxygen triggered by metallic Pt, (iii) the CO selectively adsorbs on metallic Pt sites which participate in low-temperature CO oxidation, and (iv) the formation of the unstable intermediate such as bicarbonate, contributes together to the enhanced activity of CO pretreated Pt/TiO2.
1. Introduction
Low-temperature CO oxidation is one of the most investigated model reactions in the field of heterogeneous catalysis [1]. The CO oxidation is not only important in practical applications (e.g., automotive emission control [2,3] and remediation of flue gas [4,5]) but also acts as a probe reaction guiding the design of catalysts’ surface properties [6]. The adsorption of CO on active sites of catalyst surface is a crucial step that controls CO oxidation reaction rates, and it is especially true for noble metal catalysts [7]. The linear CO adsorbed on metallic Pt sites that can facilely react with atmospheric O2 is responsible for the high activity in the low-temperature CO oxidation [8]. The ionic Pt reacts with linear CO when the temperature is higher than 100 °C. This required reduction pretreatment of Pt/TiO2 to obtain high activity in the low-temperature CO oxidation. On the other hand, the pretreatment conditions influence the peroxide and superoxide generated by O2 adsorption at the metal-support interface that determines the catalytic performance [9,10]. As a critical step of CO oxidation, pretreatment can modify the chemical properties of catalyst and alter the reaction pathway [11,12]. Maldonado-Hodar’s group [13] studied the influence of treatments in the reductive atmosphere (i.e., H2) and inert atmosphere (i.e., He) on the performance of Pt/TiO2. They found that the reduction pretreatment by H2 favored the formation of oxygen vacancies and facilitated the diffusion of Pt species into the TiO2. The Pt–TiO2 interface and the active surface sites were suppressed by He pretreatment, leading to unsatisfactory activity in the selective citral hydrogenation. Reduction pretreatment in CO also promotes the activity, as evidenced by the CO oxidation over Pt/CeO2-Al2O3 catalyst. The CO pretreatment induces a more reduced Pt with a low Pt-O coordination number than the catalysts pretreated in H2 or C3H6, leading to enhanced activity in diesel oxidation reaction [14]. Moreover, investigations on Au/CeO2 catalyst pretreated in O2, N2, CO, and H2 suggest that the long-term activity is dominated by the reducibility of the support and the particle size [15]. It is generally accepted that the reductive atmosphere has a significant impact on both the chemical status and morphology of noble metal and thus varies the activity of the catalysts. However, the difference of the reductant atmospheres (e.g., CO and H2) on the transition of active sites (e.g., Pt0, Ptδ+ sites) and surface oxygen species (e.g., O2−ads, O−ads) and roles they play in the reaction are still unclear and require detailed investigations.
In this work, the tuning of Pt sites in Pt/TiO2 was carried out by a mild reduction in the atmospheres of H2 and CO, respectively. The influences on the chemical and surface properties of Pt/TiO2 catalyst were investigated by a combination of surface characterizations (e.g., X-ray photoelectron spectroscopy (XPS), H2 temperature-programmed reduction (H2-TPR), O2 temperature-programmed desorption (O2-TPD)). The impact of reduction pretreatment on the structural characters of Pt/TiO2 was studied by the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The transition of Pt active adsorption sites and the reactive intermediates were investigated by the CO adsorption using an in situ diffuse reflectance infrared Fourier transform spectroscopy (In situ DRIFTs). Furthermore, the catalytic performance of Pt/TiO2 catalysts was studied by both in situ and ex situ CO oxidation to identify the influence of pretreatments on catalyst surface species.
2. Results
2.1. Chemical and Structural Modifications
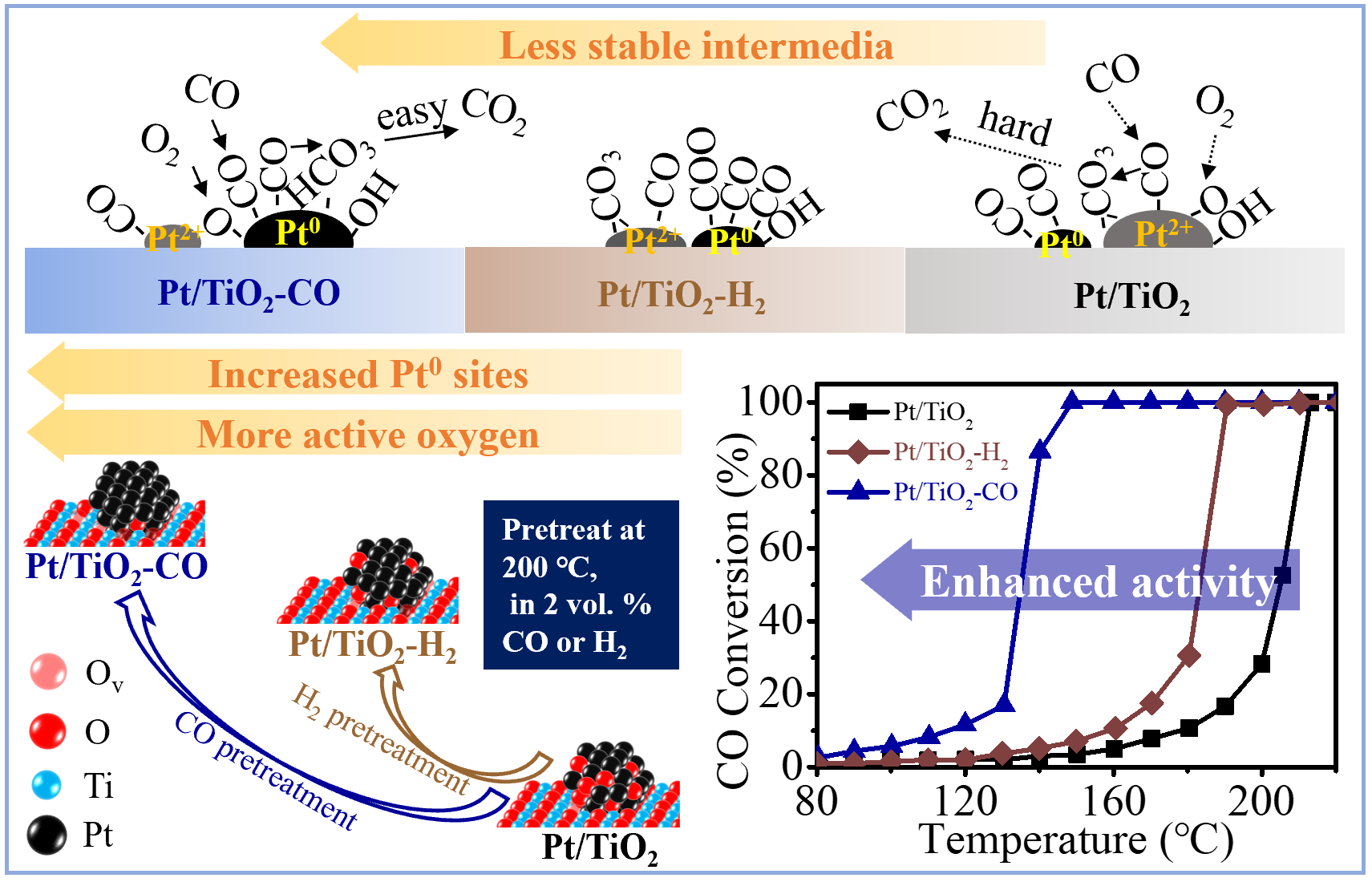
The Pt/TiO2-H2 and Pt/TiO2-CO catalysts have XRD patterns similar to that of Pt/TiO2 (Figure 1a), showing primarily anatase TiO2 (JCPDS No. 21-1272) with crystal planes (101) (004), (200), (105), (211), (204), (116), (220), and (215). The characteristic peak at 27.34° is assigned to TiO2 rutile (110) (JCPDS No. 21-1276). The absence of Pt characteristic peak in the XRD patterns indicates a good dispersion of Pt nanoclusters [16,17]. A slight shift of anatase TiO2 (101) from 25.30° to 25.34° is detected on Pt/TiO2-H2, implying a distortion of TiO2 lattice caused by the interaction of Pt species [18,19]. The crystal sizes of TiO2 for Pt/TiO2 catalysts are 17.1 ± 0.3 nm, as calculated in Table S1 by the Debye–Scherrer equation from the XRD patterns in Figure 1a.
The micro-Raman spectra of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts in Figure 1b are characterized by the vibrational modes of 144 cm−1 (Eg(1)), 197 cm−1 (Eg(2)), 396 cm−1 (B1g), 516 cm−1 (A1g), and 639 cm−1 (Eg(3)) of TiO2 anatase phase, and 453 cm−1 (Eg) of TiO2 rutile phase (Figure 1b) [20]. The blue shifts of Eg(1) modes at 144 cm−1 are observed for Pt/TiO2-H2 and Pt/TiO2-CO that shift to 146 cm−1 and 148 cm−1, respectively. Meanwhile, peak broadening is found for both the Pt/TiO2-H2 and Pt/TiO2-CO catalysts. This is determined by a full width at half maximum (FWHM) of Eg(1) modes of 14 cm−1 and 16 cm−1 for Pt/TiO2-H2 and Pt/TiO2-CO catalysts, respectively, in comparison to an FWHM of 12 cm−1 for Pt/TiO2 (Table S2). The peak shift and broadening indicate the lattice distortion and the defect formation [16] trigger by reduction pretreatments [21]. Additionally, the porous structure of the three Pt/TiO2 catalysts is comparable with a surface area of 64.0 ± 1.8 m2/g and a pore volume of 0.30 ± 0.02 cm3/g (Figure 1c and Table 1).
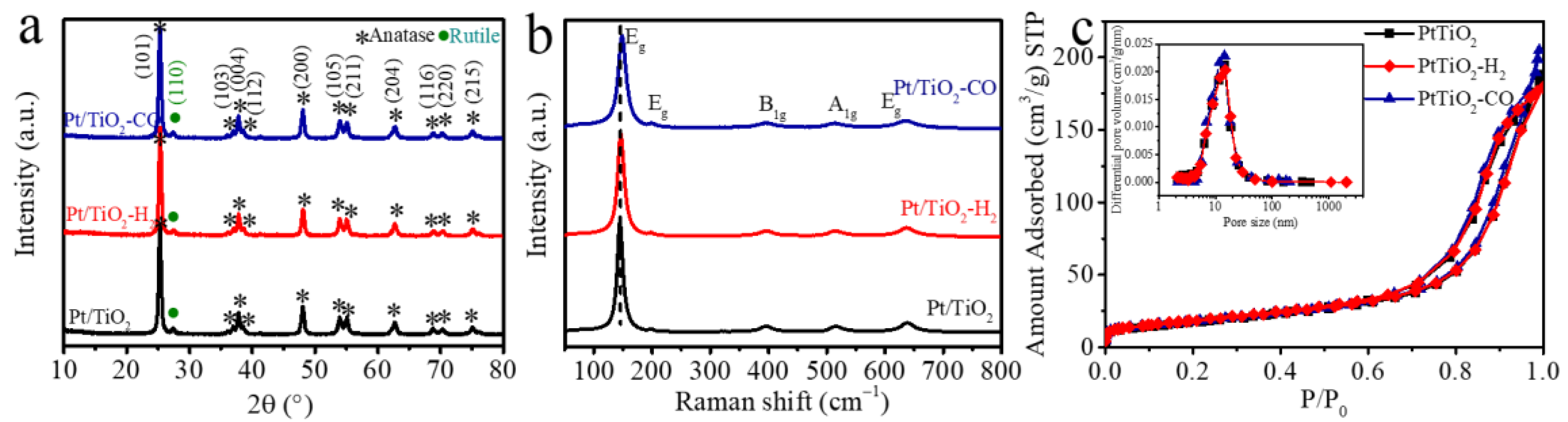
The chemical status of Pt and surface composition of Pt/TiO2 catalysts are characterized by the X-ray photoelectron spectroscopy (XPS) spectra with Pt 4f, O 1s, and Ti 2p narrow scan spectra in Figure 2a–c. The influence of air exposure on a sample before the XPS test is negligible for it happened at room temperature (ca., 25 °C), which is much lower than the reduction pretreatment temperature of 200 °C. In the presence of an overlapping Ti loss peak at around 75 eV [22], the Pt 4f spectra of Pt/TiO2 catalysts exhibits Pt0 at 71.00 eV, 74.35 eV [23] and Pt2+ at 72.40 eV, 75.75 eV [24]. Though the signal-to-noise ratio (S/N) problem occurs for the catalyst with a low loading ratio of Pt on TiO2 (i.e., 0.1 w.t.%) [22], the tendency of Pt valence state change over reduction pretreatments can be recognized. The ratio of metallic Pt0 of Pt/TiO2 catalyst without pretreatment is 16.7% (Table 1), which is lower than the H2-pretreated (i.e., 23.7%) and CO-pretreated catalysts (46.9%). Almost a half of Pt in Pt/TiO2-CO being metallic Pt suggests that CO reduced Pt effectively. Meanwhile, the O 1s spectra in Figure S1 exhibits two peaks belonging to lattice oxygen (OLatt) at 529.78 eV and oxygen species adjacent to oxygen vacancies (OAds) at 531.44 eV [25,26]. The ratio OAds/(OLatt + OAds) of reduced Pt/TiO2 catalysts in Table 1 is close to 17.0%, which is higher than that of un-pretreated Pt/TiO2 (i.e., 12.5%). This indicates that the reduction pretreatment facilitates the adsorption of oxygen on the catalyst surface by producing metallic Pt, as schemed in Figure 2 [27,28].
The structural modifications of Pt/TiO2 catalysts are studied using high-resolution, high-angle annular dark-field (HAADF) scanning transmission electron microscopy. All three catalysts present a spherical morphology with a TiO2 particle size of 20 ± 2 nm that is similar to the P25 TiO2 support. The bright points of 2 nm in the dark-field STEM micrographs and elemental map of Pt in Figure 2d–f indicate the existence of Pt nanoclusters in all the Pt/TiO2 catalysts. A slight aggregation of Pt nanoparticles is observed on the Pt/TiO2 pretreated by H2 in Figure 2e. This might be due to the particle migration and coalescence in H2 during heating treatment [29].
2.2. Surface Reduction Properties
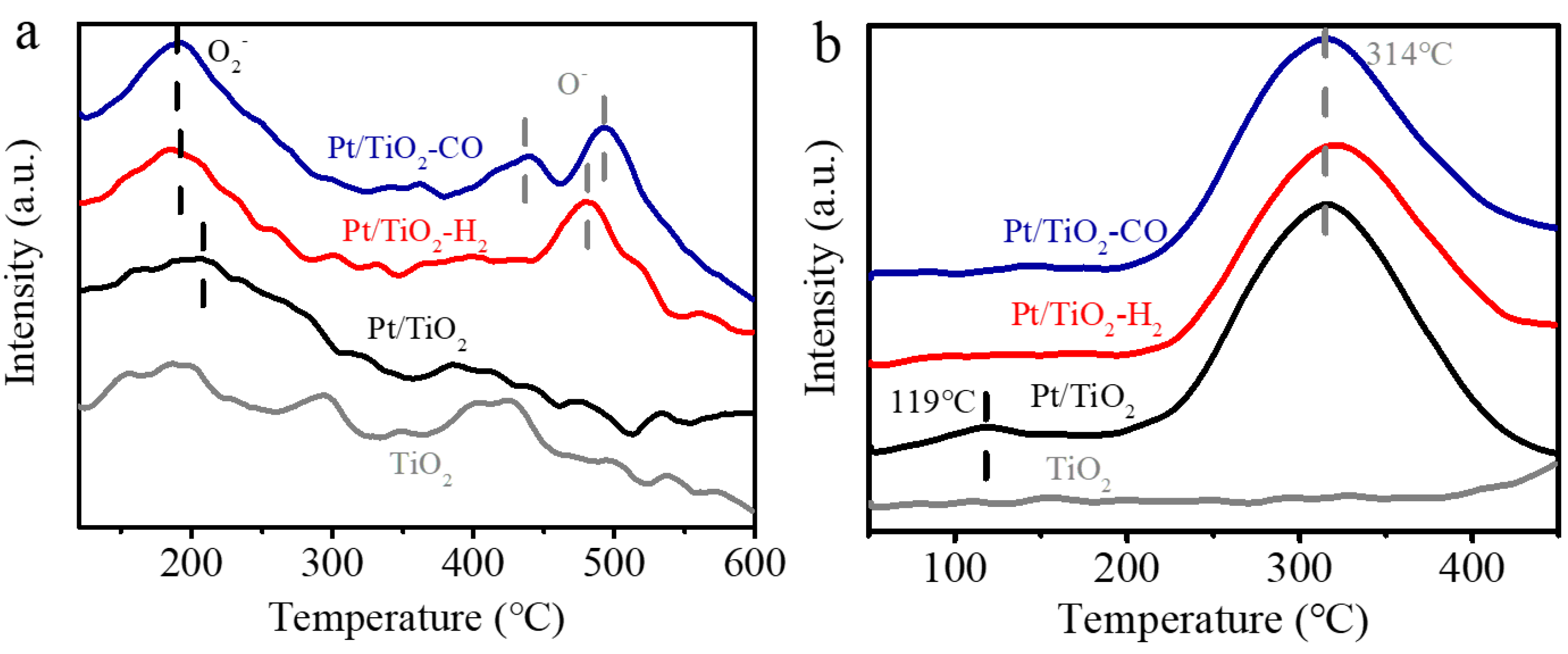
The temperature-programmed studies (i.e., O2-TPD, H2-TPR) are performed to analyze the surface properties of the Pt/TiO2 catalysts prior to and after the reduction pretreatments. The surface adsorbed oxygen is determined by the O2-TPD, and the desorption profiles for the three Pt/TiO2 catalysts are shown in Figure 3a. The desorption peak at 120 °C to 250 °C is attributed to the superoxide O2−ads [30]. The amount of O2−ads is improved after the reduction pretreatment in CO and H2, and the desorption temperatures of O2−ads on Pt/TiO2-CO (i.e., 187 °C) and Pt/TiO2-H2 (i.e., 190 °C) are lower than the one of unpretreated Pt/TiO2 (i.e., 202 °C). The desorption peak from 350 °C to 550 °C is assigned to the dissociatively adsorbed peroxide O−ads [30] on Pt/TiO2 catalysts that participate in low-temperature CO oxidation [31]. The O−ads peak area of the Pt/TiO2-CO at 350 °C to 550 °C is the largest, followed by Pt/TiO2-H2, and Pt/TiO2 shows the smallest O− area, similar to pristine TiO2. The superoxide and peroxide in Pt/TiO2-CO facilitate the oxidation of CO at low temperatures, indicating an elevated activity of the catalyst.
The redox property of the Pt/TiO2, Pt/TiO2-CO, and Pt/TiO2-H2 catalysts is evaluated by H2-TPR profiles shown in Figure 3b, and the H2 consumption summarized in Table S3. The reduction peak at 119 °C that is only observed on the Pt/TiO2 catalyst is ascribed to the reduction of PtOx to metallic Pt [32]. The absence of PtOx reduction on Pt/TiO2-CO, and Pt/TiO2-H2 suggests that the majority of Pt on the catalyst surface are in the form of metallic Pt. The reduction that occurs at 314 °C is assigned to the partial reduction of surface oxygen, as induced by hydrogen spillover from Pt to the TiO2 surface [21,32,33]. The pure TiO2 only presents a reduction of lattice oxygen at a temperature higher than 600 °C [33]. These results show that the reduction peak of surface oxygen only presents in the presence of Pt, which is indicative of a strong Pt-TiO2 interaction that produces oxygen vacancy for the dissociative oxygen adsorption. Furthermore, Pt/TiO2 consumes more H2 (i.e., 0.40 mmol/g) during H2-TPR than the H2 required by Pt/TiO2-CO (i.e., 0.32 mmol/g) and Pt/TiO2-H2 (i.e., 0.29 mmol/g). The suppression of PtOx reduction peak and low H2 consumption for Pt/TiO2-H2 and Pt/TiO2-CO catalysts confirm the transition from ionic Pt to metallic Pt by H2 or CO pretreatment, which is in line with XPS results (Figure 2).
2.3. Catalytic Performance
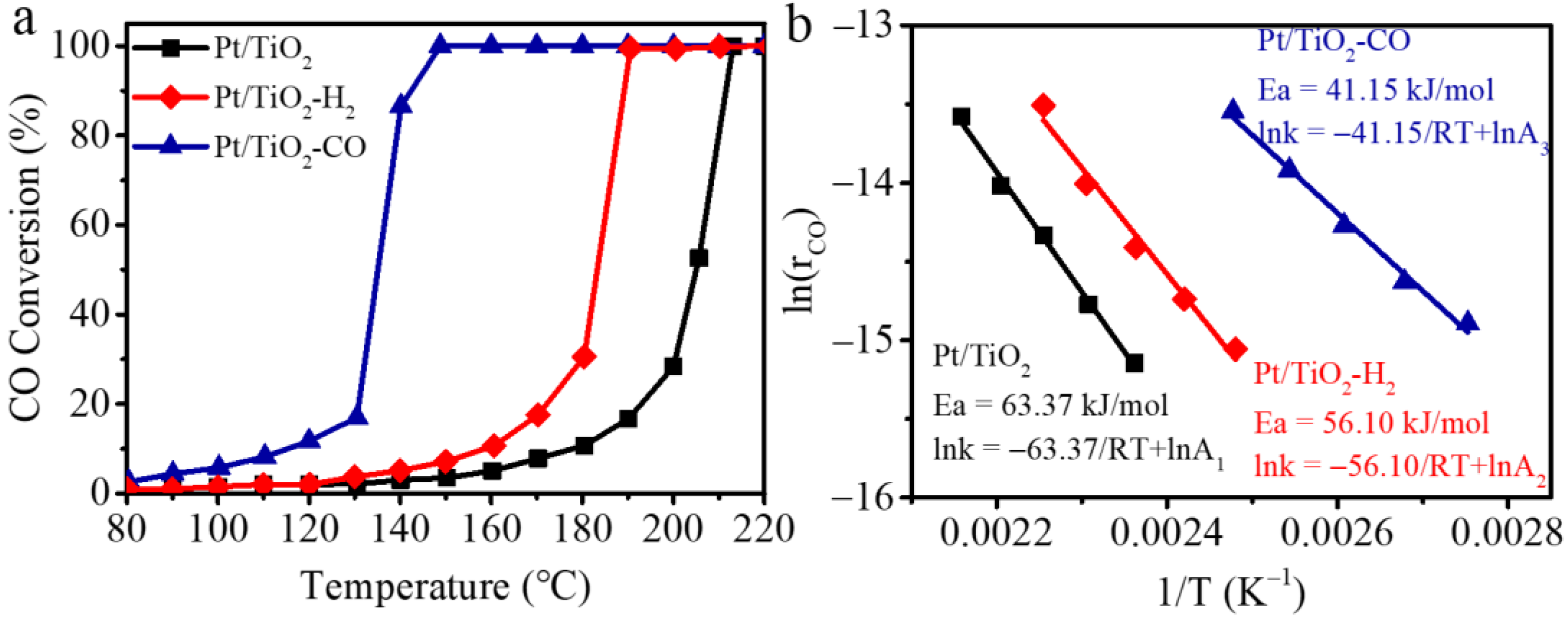
The catalytic performance of un-pretreated Pt/TiO2 and the two pretreated Pt/TiO2 catalysts in H2 (i.e., Pt/TiO2-H2) and CO (i.e., Pt/TiO2-CO) are evaluated by the CO oxidation in Figure 4. It is noteworthy that the Pt/TiO2-CO exhibits a significantly enhanced activity, with a much lower temperature for 50% conversion (i.e., T50) of 135 °C, as compared to 184 °C of Pt/TiO2-H2 and to 203 °C of the catalyst without pretreatment. Complete oxidation of CO is achieved at 150 °C (i.e., T100) by Pt/TiO2-CO, while the Pt/TiO2 and Pt/TiO2-H2 require high temperatures of 210 °C and 190 °C, respectively. Figure 4b presents the Arrhenius plots of the three Pt/TiO2 catalysts. The apparent activation energies calculated from the slopes of the fitting lines are 63.4 kJ/mol, 56.1 kJ/mol, 41.2 kJ/mol for Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO, respectively. It is found that the CO reduction pretreatment produces a high ratio of metallic Pt sites over ionic Pt sites (i.e., 46.9%) as indicated by the XPS spectra in Figure 2, and abundant surface oxygen species (e.g., O2−ads) as suggested by the temperature-programmed studies in Figure 3, which promotes the oxidation at low-temperatures [34]. The related mechanism is discussed in Section 2.4 by in situ DRIFTs that explore the adsorbed intermediates of CO adsorption and the reaction pathway of CO oxidation.
2.4. In Situ DRIFTS Study
To determine the adsorbed intermediates and to investigate the reaction pathway of CO oxidation, two groups of in situ DRIFTs studies are performed by feeding CO and then O2 (i.e., CO + O2), and by feeding the mixture gas of CO and O2 (i.e., CO/O2), respectively.
2.4.1. In Situ CO Adsorption–Desorption–Oxidation
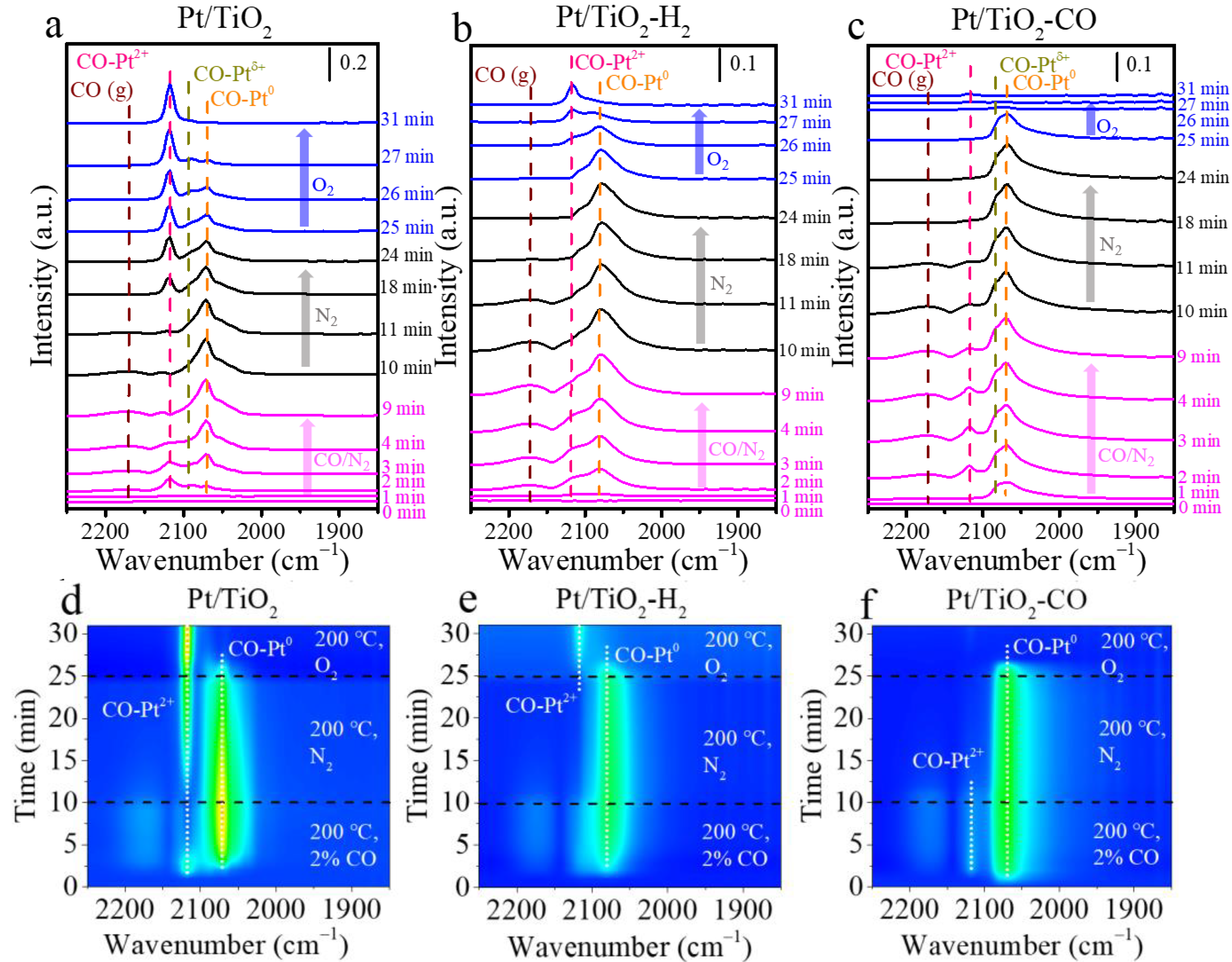
Figure 5 plots in situ DRIFT spectra of CO + O2 condition at 200 °C, where the CO adsorption is carried out in 2 vol.% CO/N2 for 10 min, followed by purging in N2 for 15 min, and the oxidation is carried out by introducing O2 for 6 min. The CO exhibits four types of IR vibrations that centered at 2171 cm−1 for gaseous CO [32,34,35], at 2117 cm−1 for CO adsorbed on ionic Pt sites (Pt2+-CO) [34,35,36], at 2090 cm−1 for CO adsorbed on metallic Pt with Pt(Ox) neighbor (Ptδ+-CO) [32,34,35,37], and at 2078 cm−1 for linear CO adsorbed on metallic Pt sites (Pt0-CO) [34,35,36,38] (Table S1). CO adsorbed on Pt species is barely observed on unpretreated Pt/TiO2 catalyst at 1 min (Figure 5a), implying that CO adsorption on predominating Pt2+ sites takes place slowly at 200 °C. A fast CO adsorption occurred on the pretreated catalysts that have Pt0 sites. Both Pt/TiO2-H2 and Pt/TiO2-CO exhibit the adsorbed CO at 1 min, with Pt0-CO and Pt2+-CO on Pt/TiO2-H2 (Figure 5b), and only Pt0-CO on Pt/TiO2-CO (Figure 5c). The absence of Pt2+-CO on Pt/TiO2-CO at 1 min demonstrates that CO pretreatment converted the surface Pt species to Pt0 more effectively than H2 pretreatment, and Pt0 adsorbed CO faster than Pt2+. When the adsorption is launched for 2 min, the CO is adsorbed on unpretreated Pt/TiO2 primarily in the form of Pt2+-CO, followed by Ptδ+-CO and Pt0-CO (Figure 5a). The CO adsorbed on Pt0 sites accumulates on the surface of Pt/TiO2 catalyst for 2 min, and the intensity of Pt2+-CO declines simultaneously (Figure 5a,d). This indicates that the fed CO in the CO adsorption triggers the reduction of the unpretreated Pt/TiO2 [24,34].
N2 is purged at 200 °C from 10 min to 25 min to remove the weakly adsorbed CO. The CO adsorbed on Pt0 of unpretreated Pt/TiO2 starts to drop at 10 min with the increasing Pt2+-CO in N2 flow (Figure 5a,d), indicating that the Pt0 sites created by the CO adsorption for only 10 min are unstable that facilely transfer to Pt2+. In contrast, a transition from Pt0 to Pt2+ is not observed on the pretreated samples (Figure 5b,c,e,f), instead, the intensity of Pt2+-CO decreases because of the desorption of CO in N2. In the spectra obtained after CO adsorption and N2 purging (Figure S2), the ratio of the CO on metallic Pt peak area of Pt/TiO2-H2 and Pt/TiO2-CO are 99% and 93%, respectively, both higher than that of Pt/TiO2 (i.e., 72%), indicating that H2 and CO reduction triggers the transition of Pt2+ to Ptδ+ and Pt0, which is consistent with the XPS (Figure 2) and H2-TPR (Figure 4b) analysis.
The CO adsorbed on metallic Pt (i.e., Pt0-CO and Ptδ+-CO) for all the three catalysts is completely disappeared after feeding O2 for 6 min, as shown in Figure 5. The CO adsorbed on ionic Pt accumulates on the catalyst surface of unpretreated Pt/TiO2 (Figure 5d) and H2 pretreated Pt/TiO2 (Figure 5e), especially on the unpretreated Pt/TiO2, which is in line with its low activity in Figure 4. This confirms that the metallic Pt sites act as the active sites for the low-temperature CO oxidation [34]. When introducing oxygen at 25 min after the saturation adsorption of CO on the catalysts, a change of CO2 intensity is observed for the three catalysts (Figure S4). The Pt/TiO2-H2 and Pt/TiO2-CO produced more CO2 than Pt/TiO2 as their CO2 intensities increase more than Pt/TiO2. The weaker CO2 peak intensity of Pt/TiO2 at 31 min indicates that not all CO adsorbed on Pt0 converts to CO2, but also transfers to Pt2+ to form Pt2+-CO, where the Pt2+ is originated from the re-oxidation of Pt0 under the oxidative condition. The disappearance of adsorbed CO is monitored on Pt/TiO2-CO when introducing O2 only for 2 min (Figure 5f). The high activity of Pt/TiO2-CO in CO oxidation in Figure 4 is due to the plentiful metallic Pt sites (Figure 2) created by CO pretreatment, which promotes both CO adsorption and CO oxidation at low temperature [34,35]. Furthermore, studies of Pt catalysts pretreated in CO have shown that CO assists the transition of higher coordination number of Pt at larger wavenumber to lower coordination number of metallic Pt (i.e., Pt0-CO) at smaller wavenumber more effectively than H2 [14]. It provides active sites for CO adsorption and low-temperature oxidation [39], considering that the Pt2+ sites are less active in low-temperature CO oxidation and the oxygen-surrounding Ptδ+ partially deactivates the sites [34].
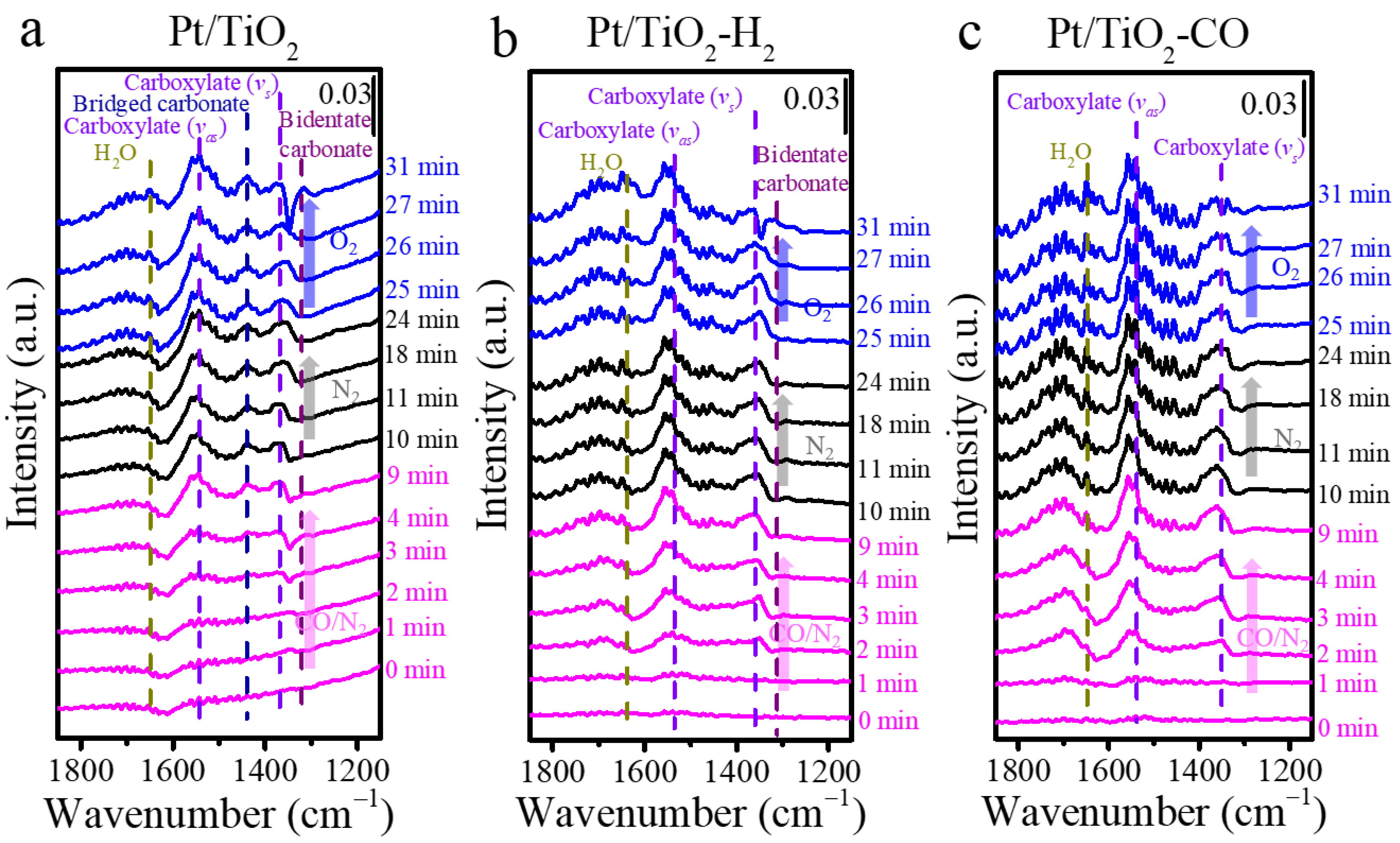
The species that formed during the CO adsorption, desorption and oxidation are presented in Figure 6 for Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts. Carboxylate, which has the structure of -CO2−, with C atom or C and O atoms binding on the support, is found on all the three Pt/TiO2 catalysts with characteristic IR vibrations of asymmetric stretching mode at 1542 cm−1 and symmetric stretching mode at 1361 cm−1 [40]. Carbonate species at 1438 cm−1 [41] is only observed on unpretreated Pt/TiO2 after the CO adsorption (Figure 6a). The carboxylate requires a lower energy barrier to be reacted to CO2 than that of carbonate species [42]. Moreover, the bidentate carbonate at 1315 cm−1 [41] is produced at 31 min on Pt/TiO2 (Figure 6a) and Pt/TiO2-H2 (Figure 6b), along with the partial oxidation of Pt0 to Pt2+ (Figure 5a,b). It implies that adsorbed CO is transformed to bidentate carbonates in O2. No carbonates species are found on in situ DRIFT spectra of Pt/TiO2-CO in Figure 6c for the whole CO adsorption–desorption–oxidation process, which is in line with its high activity in the CO oxidation in Figure 4.
2.4.2. In Situ CO Oxidation
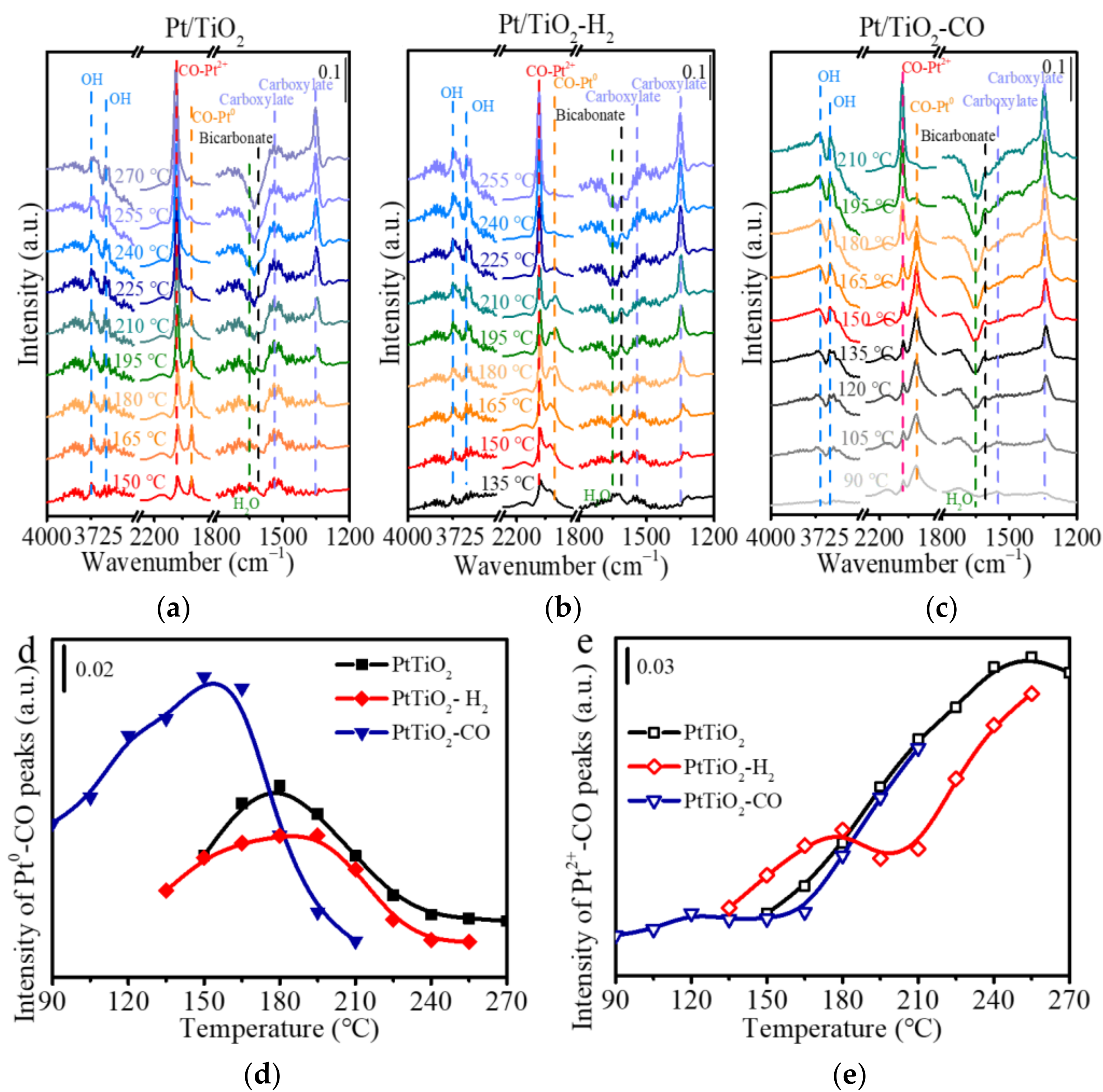
In situ DRIFT spectra of the CO oxidation (i.e., CO/O2 condition) in Figure 7 are obtained in an atmosphere containing 0.7 vol.% CO and 16 vol.% O2. The temperature program with an interval of 15 °C is set up for each catalyst according to the corresponding light-off curve in Figure 5. The unpretreated Pt/TiO2 in Figure 7a displays both Pt2+-CO at 2117 cm−1 and Pt0-CO at 2071 cm−1 at 150 °C, and consumption of the Pt0-CO occurs when the temperature is higher than 180 °C, implying that CO oxidation takes place at the metallic Pt sites with CO adsorbed. The Pt0-CO disappears as it is fully converted to CO2, leaving CO adsorbed on Pt2+ participates in the high-temperature oxidation. Similarly, it is also true for the pretreated Pt/TiO2 catalysts in Figure 7b,c that the ratio of Pt2+-CO to Pt0-CO becomes predominant along with increasing temperatures. Bicarbonates at 1608 cm−1 (υas) and 1627 cm−1 (υs) are reactive intermediates in the CO oxidation for Pt/TiO2 catalysts, implying a reaction pathway that CO converted to bicarbonates before transiting to CO2 (Figure S5) [40].
Figure 7d,e summarizes the IR intensity of Pt0-CO and Pt2+-CO as a function of reaction temperature. The Pt0-CO peak intensity of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO reaches their summit at 180 °C, 180 °C, and 150 °C, respectively, which correspond with their activity tests in Figure 4a (i.e., the T100 of 210 °C, 190 °C, 150 °C, respectively). The Pt0-CO peak intensity of Pt/TiO2-CO at low temperature is higher than those of the Pt/TiO2-H2 and Pt/TiO2-CO, indicating that Pt/TiO2-CO owns high CO adsorption capacity. The Pt2+-CO accumulates up to a temperature of 255 °C in Figure 7e, which is much higher than Pt0-CO, implying that CO is adsorbed more strongly on Pt2+ than Pt0-CO [35,36] upon exposure to the reactant gases. These results are consistent with the literature [35,43] that linear CO adsorbed on metallic Pt is responsible for reacting in the low-temperature CO oxidation, while the CO adsorbed on ionic Pt participates in the CO oxidation along with the increasing temperature. The abundant metallic Pt sites introduced by the reduction pretreatment, especially the CO pretreatment, are responsible for the high activity in the low-temperature CO oxidation.
3. Discussion
The reduction assists the formation of the metallic Pt, which is responsible for the oxygen activation at the metal–support interface. This is related to the electron transfer from platinum to titanium, forming Pt–O–Ti3+ sites, leading to more oxygen vacancy for the dissociative oxygen adsorption (Figure 3) [27]. The abundant adsorbed superoxide and peroxide produced on the catalyst surface participate in the Langmuir–Hinshelwood (L–H) mechanism at low temperatures. The oxygen activation capacity of Pt/TiO2 is elevated by the CO and H2 pretreatment. Moreover, in the reaction gas, the adsorbed oxygen on the catalyst surface assists the direct transition of bicarbonate intermediates to CO2, instead of forming carbonate, which could inhibit CO oxidation [44]. On the basis of the above considerations, the reductive pretreatment of Pt/TiO2 catalyst by H2 and CO enhances the catalytic performance over CO oxidation reaction.
CO adsorption is also influenced by the factors such as dispersion of Pt on TiO2 support and the coordination number of Pt sites to O [34]. The reductive pretreatment at 200 °C in CO can not trigger the particle migration and coalescence that enlarge the particle size [14,29]. The Pt/TiO2 after CO and H2 pretreatment varies from pristine Pt/TiO2 in valence state, presenting more metallic Pt sites. The reduction extent of H2 and CO in this work is consistent with the results reported on Pt/CeO2-Al2O3 catalyst [14]. CO pretreatment generates more metallic Pt with a low Pt-O coordination number than H2 pretreatment. The strength of CO adsorption on metallic Pt is weaker than ionic Pt [36], and thus the CO on metallic Pt reacts with oxygen at lower temperatures [34], leading to a remarkable CO oxidation activity.
4. Materials and Methods
4.1. Materials
The PtCl4 (99%) and TiO2 (P25, 20 nm, 99%) were purchased from Macklin Inc. (Shanghai, China). All chemicals were used as received without further purification. The deionized water was generated from a Master-Q (Hitech, Shanghai, China) water purification system.
4.2. Catalyst Preparation
The 0.1 wt.% Pt/TiO2 catalyst was synthesized by an incipient wetness impregnation method. The P25 TiO2 powder (0.1 mol, 8 g) was dispersed in 5.5 mL deionized water by stirring to obtain a homogenous slurry. The slurry was dried at 120 °C for 6 h, then calcinated in a programmable furnace at 450 °C for 4 h with a heating rate of 5 °C/min. The TiO2 was crushed and sieved to make 40–60 mesh granules. The 1.75 g/L platinum precursor solution was prepared by dissolving 5.25 mg PtCl4 in 3 mL deionized water, and it was dropwise absorbed by 2.625 g TiO2 granules under continuous stirring. The wet granules were dried at 120 °C for 6 h and then calcined at 400 °C for 4 h with a ramping rate of 5 °C/min to obtain 0.1 wt.% Pt/TiO2 catalyst.
Reductive pretreatment of Pt/TiO2 catalyst was performed in a tube furnace in 2 vol.% H2/Ar and in 2 vol.% CO/N2 at 200 °C for 2 h to obtain Pt/TiO2-H2 and Pt/TiO2-CO catalysts, respectively. After pretreatments, the catalysts were swept by N2 for 15 min to remove the physically adsorbed gas molecules.
4.3. Catalysts Characterization
Powder X-ray diffraction (XRD) patterns were collected on a Bruker D8 X-ray diffractometer (XRD) using Cu Kα radiation (λ = 1.54178 Å). Micro-Raman spectra of catalysts were generated by a LabRAM ARAMIS micro-Raman spectrometer (HORIBA Scientifics, Kyoto, Japan) with an excitation laser of 532 nm. N2 physisorption of the granular samples was conducted on a V-sorb 2800 P surface area and porosimetry analyzer (Gold APP Instruments), before which the samples were outgassed in vacuum at 120 °C for 2 h. High-resolution transmission electron microscopy (HRTEM) patterns were collected on an FEI (Hillsboro, OR, USA) Talos F200S and analyzed by the Gatan Digital Micrograph program. X-ray photoelectron spectra (XPS) were analyzed by a Thermo Fisher Scientific (Waltham, MA, USA) K-alpha, with the binding energy calibrated to C 1s at 284.8 eV.
The O2 temperature-programmed desorption (O2-TPD), H2 temperature-programmed reduction (H2-TPR), and CO pulse chemisorption were performed on an AutoChem II 2920 automated chemisorption analyzer equipped with a thermal conductivity detector (TCD) detector. A total of 0.1 g granular catalyst was packed into the reactor of the chemisorption analyzer. The pretreatment was performed in a He atmosphere at 200 °C for 60 min for the O2-TPD, H2-TPR, and the CO pulse chemisorption measurements. The O2 adsorption was conducted at 50 °C for 60 min in 10 vol.% O2/He flow at a rate of 50 mL/min, and then the temperature program was carried out in He flow from 50 °C to 600 °C at a heating rate of 10 °C/min. A total of 10 vol.% H2/Ar was used in H2-TPR experiment, with a temperature ramping rate of 10 °C/min from 50 °C to 450 °C. The CO pulse chemisorption was carried out at room temperature to estimate the Pt diameter of the Pt/TiO2 catalyst, and prior to the CO chemisorption, the sample was reduced by H2/Ar at 450 °C for 30 min.
The in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) for CO adsorption (i.e., CO + O2) and CO oxidation (i.e., CO/O2) was performed using a Thermo Fisher Nicolet iS20 Fourier transform infrared (FTIR) spectrometer equipped with a Mercury–Cadmium–Telluride (MCT) detector. Each spectrum was recorded for 8 scans at a resolution of 8 cm−1. The sample was loaded into the Harrick reaction cell with ZnSe windows and purged at 200 °C in 50 mL/min N2 for 30 min. A background spectrum was recorded at 200 °C before in situ DRIFTS measurements of the CO adsorption. A total of 2 vol.% CO/N2 was passed through the catalyst bed for 10 min, followed by N2 purging for 15 min, and by O2 feeding for 6 min. In the CO oxidation DRIFTS measurements, the background spectra of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO under N2 flow were recorded at 150 °C, 135 °C, and 90 °C, respectively. As the reaction gas (0.7 vol.% CO, 16 vol.% O2, balanced with N2) passed through the catalyst bed, the spectra were recorded at a temperature interval of 15 °C. For each temperature, 5 min was maintained to reach the equilibrium.
4.4. Catalytic Performance Tests
The CO oxidation was carried out in a stainless-steel tubular reactor (ID = 15 mm) encased by a tubular furnace with a thermocouple inserted in the reactor above the catalyst bed. A total of 2.5 g catalyst (40–60 mesh) was packed into the fixed bed reactor to generate a 17 mm-high catalyst bed. The gas hourly space velocity (GHSV) was 32,000/h as the flow rate of the reaction gases was 1600 mL/min. The large GHSV rules out the mass transfer limitation. Typically, the components of the reaction gases were 0.7 vol.% CO, 16 vol.% O2, 0 vol.%/ 0.01 vol.% NO, and N2 for balance. The reactant gases were modulated by the mass flow controller and pre-mixed in a chamber before feeding into the reactor. The catalytic performance was evaluated by analyzing the exhaust gases using a GASMET, DX4000 FTIR gas analyzer.
5. Conclusions
The Pt/TiO2 surface composition and textural properties were significantly modified after the reduction treatment in H2 and CO at 200 °C. Reduction pretreatment improved the catalytic performance of Pt/TiO2 in low-temperature oxidation by facilitating the formation of metallic Pt sites that can adsorb CO effectively. The unpretreated Pt/TiO2 with insufficient metallic Pt sites adsorbed CO on Pt2+, which participate in the oxidation reaction at elevated temperatures. The carbonate species on the unpretreated catalyst is hard to convert to CO2, leading to low activity. CO pretreatment is more effective than H2 pretreatment to trigger the reduction of Pt2+ to Pt0 with low coordination numbers, which exhibits the most remarkable CO adsorption and oxidation property at low temperatures. The metallic Pt sites triggered by the CO reduction are more stable upon oxygen exposure than H2, without transiting to Pt2+. The copious metallic Pt sites on the CO-pretreated Pt catalyst facilitate the production of active oxygen species (e.g., O2−ads). The CO adsorbed on metallic Pt sites participate in low-temperature CO oxidation with the surface oxygen species via the L–H mechanism, presenting bicarbonate as reactive intermediates. As a result, the Pt/TiO2-CO catalyst exhibited high activity with complete oxidation of CO at 150 °C. Thus, it is tentatively concluded that Pt on TiO2 is more activated with thermal pretreatment in an atmosphere of CO than H2.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal11111280/s1, Figure S1: XPS narrow spectra of (a) O 1s and (b) Ti 2p of Pt/TiO2, Pt/TiO2-H2 and Pt/TiO2-CO catalysts. Figure S2: The deconvoluted peaks of the IR spectra obtained after CO adsorption and N2 purging at 24 min for (a) Pt/TiO2, (b) Pt/TiO2-H2 and (c) Pt/TiO2-CO. Figure S3: The plots of CO-Pt0 and CO-Pt2+ peaks intensity to time in CO adsorption–desorption–oxidation. Figure S4. The peak intensity of CO2 in the in situ DRIFT spectra of the CO adsorption–desorption–oxidation for Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts at 200 °C. Figure S5. The peak intensity of CO2, the integrated peak area of carboxylate and bicarbonate as a function of temperature for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO. Figure S6: (a) Influence of NO on CO conversion as a function of reaction temperatures, and (b) apparent activation energy (Ea) calculated by the Arrhenius equation of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts (R = 8.314 × 10−3 kJ/(mol∙K)). Table S1: Crystal sizes of TiO2 calculated by the Debye–Scherrer equation from the XRD patterns of Pt/TiO2, Pt/TiO2-H2 and Pt/TiO2-CO catalysts. Table S2: Raman shift and full width at half maximum (FWHM) of TiO2 Eg vibration mode obtained from the Raman spectra of Pt/TiO2, Pt/TiO2-H2 and Pt/TiO2-CO catalysts. Table S3: Desorption temperature during O2-TPD of oxygen species and H2 consumption in H2-TPR experiment. Table S4: Vibration mode assignments of IR bands on DRIFTS of Pt/TiO2, Pt/TiO2-H2 and Pt/TiO2-CO catalysts. Table S5: The T100 and T50 in the CO oxidation experiment over Pt/TiO2-CO and Pt/TiO2-CO oxidized in 2 vol. % O2/N2 for 2 h at 200 °C.
Author Contributions
Conceptualization, K.H.; Formal analysis, Q.W., K.H.; Funding acquisition, K.H., Q.W.; Investigation, K.H.; Methodology, K.H., Q.W.; Visualization, Q.W.; Writing—original draft, K.H.; Writing—review and editing, Q.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Changsha science and technology program project funds, grant number kh2005005, the Hunan Special funds for the innovative province construction, grant number 2020GK4055, the Research Start-up Grants-Institute of Zhejiang University-Quzhou, and the Key R&D Program of Hunan province, grant number 2022SK2065.
Data Availability Statement
The data presented in this study are available within the article or supplementary material.
Acknowledgments
The authors thank the China Minmetals Corporation for the financial support from Sci-Tech Innovation project.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Nan, B.; Fu, Q.; Yu, J.; Shu, M.; Zhou, L.-L.; Li, J.; Wang, W.-W.; Jia, C.-J.; Ma, C.; Chen, J.-X.; et al. Unique structure of active platinum-bismuth site for oxidation of carbon monoxide. Nat. Commun. 2021, 12, 3342. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.-X.; Allard, L.F.; Lee, S.; Liu, J.; Li, H.; Wang, J.; Wang, J.; Oh, S.H.; Li, W.; et al. Surpassing the single-atom catalytic activity limit through paired Pt-O-Pt ensemble built from isolated Pt1 atoms. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 2017, 358, 1419–1423. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Liu, X.; Zhu, T.; Tian, M. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. 2021, 28, 24847–24871. [Google Scholar] [CrossRef]
- Tripathi, A.; Hareesh, C.; Sinthika, S.; Andersson, G.; Thapa, R. CO oxidation on Pt based binary and ternary alloy nanocatalysts: Reaction pathways and electronic descriptor. Appl. Surf. Sci. 2020, 528, 146964. [Google Scholar] [CrossRef]
- Lin, J.; Wang, X.; Zhang, T. Recent progress in CO oxidation over Pt-group-metal catalysts at low temperatures. Cuihua Xuebao/Chinese J. Catal. 2016, 37, 1805–1813. [Google Scholar] [CrossRef]
- Newton, M.A.; Ferri, D.; Smolentsev, G.; Marchionni, V.; Nachtegaal, M. Room-temperature carbon monoxide oxidation by oxygen over Pt/Al2O3 mediated by reactive platinum carbonates. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Beniya, A.; Higashi, S.; Ohba, N.; Jinnouchi, R.; Hirata, H.; Watanabe, Y. CO oxidation activity of non-reducible oxide-supported mass-selected few-atom Pt single-clusters. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Guzman, J.; Carrettin, S.; Fierro-Gonzalez, J.C.; Hao, Y.; Gates, B.C.; Corma, A. CO oxidation catalyzed by supported gold: Cooperation between gold and nanocrystalline rare-earth supports forms reactive surface superoxide and peroxide species. Angew. Chem.-Int. Ed. 2005, 44, 4778–4781. [Google Scholar] [CrossRef]
- Gerrard, A.L.; Weaver, J.F. Kinetics of CO oxidation on high-concentration phases of atomic oxygen on Pt(111). J. Chem. Phys. 2005, 123, 224703. [Google Scholar] [CrossRef]
- Jan, A.; Shin, J.; Ahn, J.; Yang, S.; Yoon, K.J.; Son, J.-W.; Kim, H.; Lee, J.-H.; Ji, H.-I. Promotion of Pt/CeO2 catalyst by hydrogen treatment for low-temperature CO oxidation. RSC Adv. 2019, 9, 27002–27012. [Google Scholar] [CrossRef] [Green Version]
- DeRita, L.; Resasco, J.; Dai, S.; Boubnov, A.; Thang, H.V.; Hoffman, A.S.; Ro, I.; Graham, G.W.; Bare, S.R.; Pacchioni, G.; et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 2019, 18, 746–751. [Google Scholar] [CrossRef]
- Bailón-García, E.; Carrasco-Marín, F.; Pérez-Cadenas, A.F.; Maldonado-Hódar, F.J. Influence of the pretreatment conditions on the development and performance of active sites of Pt/TiO2 catalysts used for the selective citral hydrogenation. J. Catal. 2015, 327, 86–95. [Google Scholar] [CrossRef]
- Gänzler, A.M.; Casapu, M.; Vernoux, P.; Loridant, S.; Aires, F.J.C.S.; Epicier, T.; Betz, B.; Hoyer, R.; Grunwaldt, J.D. Tuning the Structure of Platinum Particles on Ceria In Situ for Enhancing the Catalytic Performance of Exhaust Gas Catalysts. Angew. Chem.-Int. Ed. 2017, 56, 13078–13082. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Moemen, A.; Abdel-Mageed, A.M.; Bansmann, J.; Parlinska-Wojtan, M.; Behm, R.J.; Kučerová, G. Deactivation of Au/CeO2 catalysts during CO oxidation: Influence of pretreatment and reaction conditions. J. Catal. 2016, 341, 160–179. [Google Scholar] [CrossRef]
- Oh, S.; Ha, H.; Choi, H.; Jo, C.; Cho, J.; Choi, H.; Ryoo, R.; Kim, H.Y.; Park, J.Y. Oxygen activation on the interface between Pt nanoparticles and mesoporous defective TiO2 during CO oxidation. J. Chem. Phys. 2019, 151, 234716. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Su, X.; Duan, H.; Liang, B.; Huang, Y.; Zhang, T. Catalytic performance of the Pt/TiO2 catalysts in reverse water gas shift reaction: Controlled product selectivity and a mechanism study. Catal. Today 2017, 281, 312–318. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Bañares, M.A.; Weng, L.-T.; Gu, Q.; Price, J.; Han, W.; Yeung, K.L. A Novel Approach to High-Performance Aliovalent-Substituted Catalysts—2D Bimetallic MOF-Derived CeCuOx Microsheets. Small 2019, 15, 1903525. [Google Scholar] [CrossRef]
- Kȩpiński, L.; Wołcyrz, M. Microstructure of Pd/CeO2 catalyst: Effect of high temperature reduction in hydrogen. Appl. Catal. A Gen. 1997, 150, 197–220. [Google Scholar] [CrossRef]
- Apopei, P.; Catrinescu, C.; Teodosiu, C.; Royer, S. Mixed-phase TiO2 photocatalysts: Crystalline phase isolation and reconstruction, characterization and photocatalytic activity in the oxidation of 4-chlorophenol from aqueous effluents. Appl. Catal. B Environ. 2014, 160–161, 374–382. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, L.; Su, B.; Xu, J.; Ding, Z.; Wang, S. Unravelling the Promotional Effect of La2O3 in Pt/La-TiO2 Catalysts for CO2 Hydrogenation. Chem.-A Eur. J. 2020, 26, 517–523. [Google Scholar] [CrossRef]
- Macino, M.; Barnes, A.J.; Althahban, S.M.; Qu, R.; Gibson, E.K.; Morgan, D.J.; Freakley, S.J.; Dimitratos, N.; Kiely, C.J.; Gao, X.; et al. Tuning of catalytic sites in Pt/TiO2 catalysts for the chemoselective hydrogenation of 3-nitrostyrene. Nat. Catal. 2019, 2, 873–881. [Google Scholar] [CrossRef]
- Motin, A.M.; Haunold, T.; Bukhtiyarov, A.V.; Bera, A.; Rameshan, C.; Rupprechter, G. Surface science approach to Pt/carbon model catalysts: XPS, STM and microreactor studies. Appl. Surf. Sci. 2018, 440, 680–687. [Google Scholar] [CrossRef]
- Li, N.; Chen, Q.-Y.; Luo, L.-F.; Huang, W.-X.; Luo, M.-F.; Hu, G.-S.; Lu, J.-Q. Kinetic study and the effect of particle size on low temperature CO oxidation over Pt/TiO2 catalysts. Appl. Catal. B Environ. 2013, 142–143, 523–532. [Google Scholar] [CrossRef]
- Benkoula, S.; Sublemontier, O.; Patanen, M.; Nicolas, C.; Sirotti, F.; Naitabdi, A.; Gaie-Levrel, F.; Antonsson, E.; Aureau, D.; Ouf, F.-X.; et al. Water adsorption on TiO2 surfaces probed by soft X-ray spectroscopies: Bulk materials vs. isolated nanoparticles. Sci. Rep. 2015, 5, 15088. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, W.; Zhou, Z.; Huang, Q.; Liu, Y.; Duan, Q. Hydroxyl groups attached to Co2+ on the surface of Co3O4: A promising structure for propane catalytic oxidation. Catal. Sci. Technol. 2020, 10, 2573–2582. [Google Scholar] [CrossRef]
- Kim, G.J.; Lee, S.M.; Hong, S.C.; Kim, S.S. Active oxygen species adsorbed on the catalyst surface and its effect on formaldehyde oxidation over Pt/TiO2 catalysts at room temperature; Role of the Pt valence state on this reaction? RSC Adv. 2018, 8, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Leung, D.Y.C. Complete elimination of indoor formaldehyde over supported Pt catalysts with extremely low Pt content at ambient temperature. J. Catal. 2011, 280, 60–67. [Google Scholar] [CrossRef]
- Simonsen, S.B.; Wang, Y.; Jensen, J.O.; Zhang, W. Coarsening of carbon black supported Pt nanoparticles in hydrogen. Nanotechnology 2017, 28, 475710. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Du, X.; Zhu, Y.; Ren, S.; Li, J. Catalytic oxidation of chlorobenzene over Pd-TiO2 /Pd-Ce/TiO2 catalysts. Catalysts 2020, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Neumann, S.; Gutmann, T.; Buntkowsky, G.; Paul, S.; Thiele, G.; Sievers, H.; Bäumer, M.; Kunz, S. Insights into the reaction mechanism and particle size effects of CO oxidation over supported Pt nanoparticle catalysts. J. Catal. 2019, 377, 662–672. [Google Scholar] [CrossRef]
- Hoang, S.; Guo, Y.; Binder, A.J.; Tang, W.; Wang, S.; Liu, J. (Jimmy); Liu, H.; Lu, X.; Wang, Y.; Ding, Y.; et al. Activating low-temperature diesel oxidation by single-atom Pt on TiO2 nanowire array. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, H.; Tanaka, K. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature. Appl. Catal. B Environ. 2006, 65, 37–43. [Google Scholar] [CrossRef]
- Ke, J.; Zhu, W.; Jiang, Y.; Si, R.; Wang, Y.J.; Li, S.C.; Jin, C.; Liu, H.; Song, W.G.; Yan, C.H.; et al. Strong Local Coordination Structure Effects on Subnanometer PtOx Clusters over CeO2 Nanowires Probed by Low-Temperature CO Oxidation. ACS Catal. 2015, 5, 5164–5173. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, G.J.; Lee, S.H.; Hwang, I.H.; Hong, S.C.; Kim, S.S. Catalytic Performance of Ce0.6Y0.4O2-Supported Platinum Catalyst for Low-Temperature Water-Gas Shift Reaction. ACS Omega 2018, 3, 3156–3163. [Google Scholar] [CrossRef] [Green Version]
- DeRita, L.; Dai, S.; Lopez-Zepeda, K.; Pham, N.; Graham, G.W.; Pan, X.; Christopher, P. Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150–14165. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhao, Y.; Lee, S.; Yang, S.; Liu, J.; Giannakakis, G.; Li, M.; Ouyang, M.; Wang, D.; Sykes, E.C.H.; et al. High-loading single Pt atom sites [Pt-O(OH)x] catalyze the CO PROX reaction with high activity and selectivity at mild conditions. Sci. Adv. 2020, 6, eaba3809. [Google Scholar] [CrossRef]
- Liu, A.; Liu, X.; Liu, L.; Pu, Y.; Guo, K.; Tan, W.; Gao, S.; Luo, Y.; Yu, S.; Si, R.; et al. Getting Insights into the Temperature-Specific Active Sites on Platinum Nanoparticles for CO Oxidation: A Combined in Situ Spectroscopic and ab Initio Density Functional Theory Study. ACS Catal. 2019, 9, 7759–7768. [Google Scholar] [CrossRef]
- Chernyshova, I.V.; Somasundaran, P.; Ponnurangam, S. On the origin of the elusive first intermediate of CO2 electroreduction. Proc. Natl. Acad. Sci. USA 2018, 115, E9261–E9270. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cao, T.; Gao, Y.; Li, D.; Xiong, F.; Huang, W. Probing surface structures of CeO2, TiO2, and Cu2O nanocrystals with CO and CO2 chemisorption. J. Phys. Chem. C 2016, 120, 21472–21485. [Google Scholar] [CrossRef]
- Li, J.-J.; Zhu, B.-L.; Wang, G.-C.; Liu, Z.-F.; Huang, W.-P.; Zhang, S.-M. Enhanced CO catalytic oxidation over an Au–Pt alloy supported on TiO2 nanotubes: Investigation of the hydroxyl and Au/Pt ratio influences. Catal. Sci. Technol. 2018, 8, 6109–6122. [Google Scholar] [CrossRef]
- Kim, G.J.; Kwon, D.W.; Hong, S.C. Effect of Pt Particle Size and Valence State on the Performance of Pt/TiO2 Catalysts for CO Oxidation at Room Temperature. J. Phys. Chem. C 2016, 120, 17996–18004. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Haruta, M. Role of water in CO oxidation on gold catalysts. Catal. Lett. 2014, 144, 1475–1486. [Google Scholar] [CrossRef]
Figure 1.
(a) XRD patterns, (b) micro-Raman spectra, and (c) N2 adsorption–desorption isotherm of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts.
Figure 1.
(a) XRD patterns, (b) micro-Raman spectra, and (c) N2 adsorption–desorption isotherm of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts.

Figure 2.
Up: Schematic diagram of the Pt/TiO2 pretreated in H2 and CO; Down: (a–c) XPS narrow spectra of Pt 4f for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts; and (d–f) high-resolution, high-angle annular dark-field (HAADF) STEM images, EDS-mapping and size distribution of (d) Pt/TiO2, (e) Pt/TiO2-H2, and (f) Pt/TiO2-CO catalysts.
Figure 2.
Up: Schematic diagram of the Pt/TiO2 pretreated in H2 and CO; Down: (a–c) XPS narrow spectra of Pt 4f for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts; and (d–f) high-resolution, high-angle annular dark-field (HAADF) STEM images, EDS-mapping and size distribution of (d) Pt/TiO2, (e) Pt/TiO2-H2, and (f) Pt/TiO2-CO catalysts.

Figure 3.
(a) O2-TPD and (b) H2-TPR profiles of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts.

Figure 4.
(a) CO conversion as a function of reaction temperatures, and (b) apparent activation energy (Ea) calculated by the Arrhenius equation of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts (R = 8.314 × 10−3 kJ/(mol∙K)). (Note: 2.5 g catalyst, 0.7 vol.% CO, 16 vol.%O2 in N2, GHSV is 32,000/h).
Figure 4.
(a) CO conversion as a function of reaction temperatures, and (b) apparent activation energy (Ea) calculated by the Arrhenius equation of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts (R = 8.314 × 10−3 kJ/(mol∙K)). (Note: 2.5 g catalyst, 0.7 vol.% CO, 16 vol.%O2 in N2, GHSV is 32,000/h).

Figure 5.
(a–c) Adsorbed CO, and (d–f) Contour map of in situ DRIFT spectra in the CO adsorption–desorption–oxidation and in situ DRIFT spectra for (a,d) Pt/TiO2, (b,e) Pt/TiO2-H2, and (c,f) Pt/TiO2-CO catalysts at 200 °C. (Note: In situ DRIFT study was carried in 2 vol.%CO for 10 min, in N2 for 15 min, and in O2 for 6 min.)
Figure 5.
(a–c) Adsorbed CO, and (d–f) Contour map of in situ DRIFT spectra in the CO adsorption–desorption–oxidation and in situ DRIFT spectra for (a,d) Pt/TiO2, (b,e) Pt/TiO2-H2, and (c,f) Pt/TiO2-CO catalysts at 200 °C. (Note: In situ DRIFT study was carried in 2 vol.%CO for 10 min, in N2 for 15 min, and in O2 for 6 min.)

Figure 6.
The carbonaceous species presented on in situ DRIFT spectra in the CO adsorption–desorption–oxidation for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts at 200 °C. (Note: In situ DRIFT study was carried in 2 vol.%CO for 10 min, in N2 for 15 min, and in O2 for 6 min.)
Figure 6.
The carbonaceous species presented on in situ DRIFT spectra in the CO adsorption–desorption–oxidation for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts at 200 °C. (Note: In situ DRIFT study was carried in 2 vol.%CO for 10 min, in N2 for 15 min, and in O2 for 6 min.)

Figure 7.
In situ DRIFT spectra in the CO oxidation for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts; and IR intensity of (d) Pt0-CO and (e) Pt2+-CO as a function of temperatures.
Figure 7.
In situ DRIFT spectra in the CO oxidation for (a) Pt/TiO2, (b) Pt/TiO2-H2, and (c) Pt/TiO2-CO catalysts; and IR intensity of (d) Pt0-CO and (e) Pt2+-CO as a function of temperatures.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Surface species concentrations (based on XPS peak area) and porous properties of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts.
Table 1.
Surface species concentrations (based on XPS peak area) and porous properties of Pt/TiO2, Pt/TiO2-H2, and Pt/TiO2-CO catalysts.
| Catalysts | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) | Pt0/(Pt0 + Pt2+) | Oads/(Olatt + Oads) | Ti3+/(Ti3+ + Ti4+) |
|---|---|---|---|---|---|---|
| Pt/TiO2 | 62.2 | 0.3 | 14.2 | 16.7% | 12.5% | 4.5% |
| Pt/TiO2-H2 | 65.8 | 0.3 | 13.6 | 23.7% | 16.5% | 3.8% |
| Pt/TiO2-CO | 65.0 | 0.3 | 14.9 | 46.9% | 17.0% | 4.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
He, K.; Wang, Q. Activation of Pt Nanoclusters on TiO2 via Tuning the Metallic Sites to Promote Low-Temperature CO Oxidation. Catalysts 2021, 11, 1280. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111280
AMA Style
He K, Wang Q. Activation of Pt Nanoclusters on TiO2 via Tuning the Metallic Sites to Promote Low-Temperature CO Oxidation. Catalysts. 2021; 11(11):1280. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111280
Chicago/Turabian StyleHe, Kailin, and Qingyue Wang. 2021. "Activation of Pt Nanoclusters on TiO2 via Tuning the Metallic Sites to Promote Low-Temperature CO Oxidation" Catalysts 11, no. 11: 1280. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111280
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.