
The Mitigation of CO Present in the Water–Gas Shift Reformate Gas over IR-TiO2 and IR-ZrO2 Catalysts


Abstract
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterisation
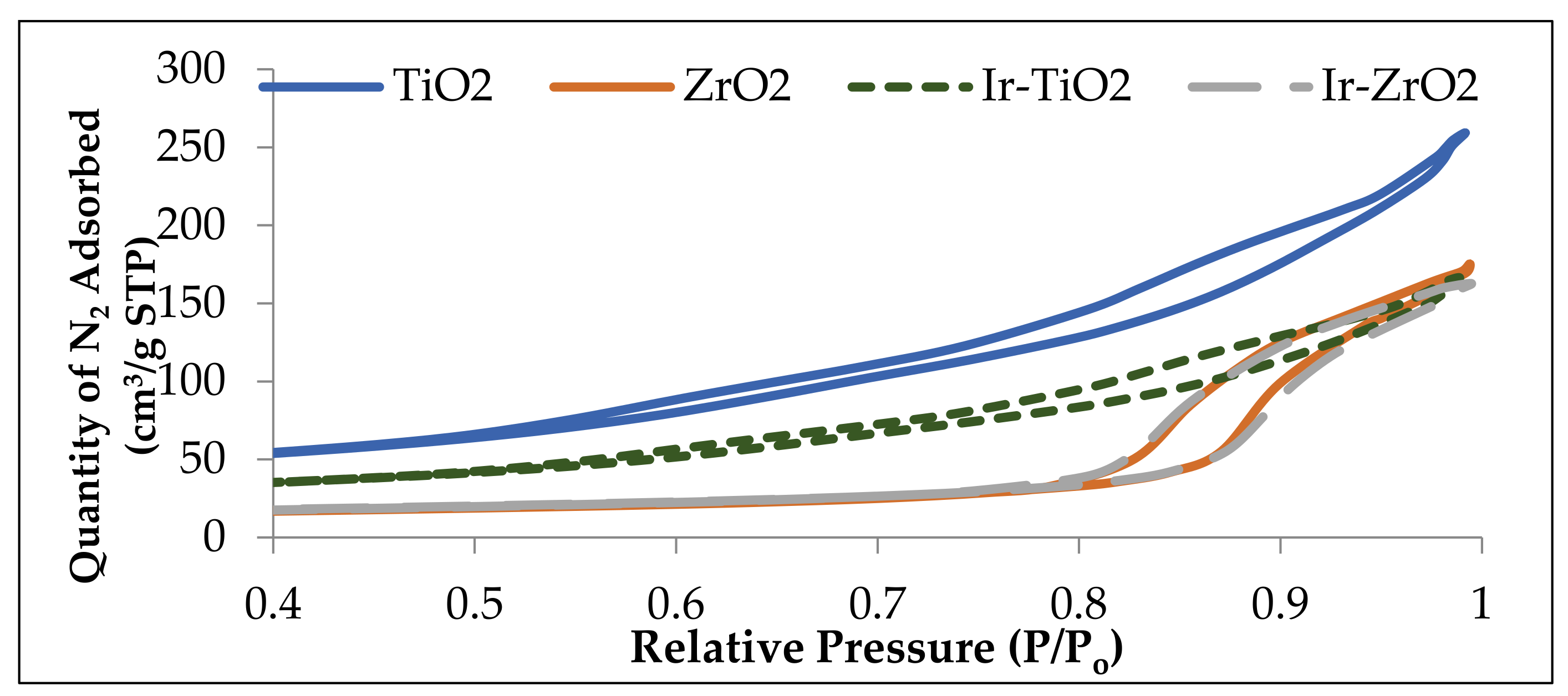
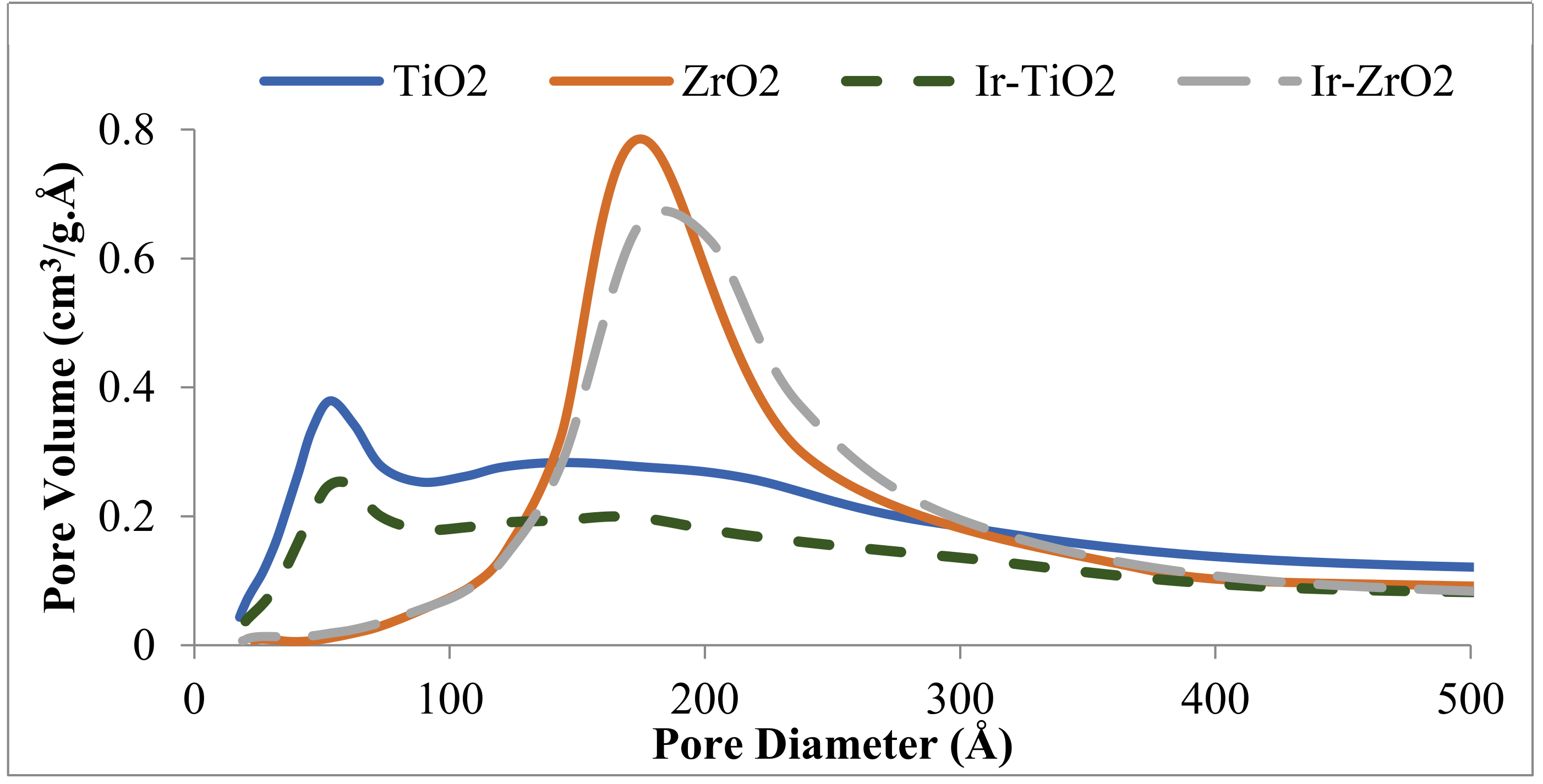
2.1.1. Physisorption and Elemental Analysis of the Materials
2.1.2. Powder X-ray Diffraction
2.1.3. X-ray Photon Spectroscopy
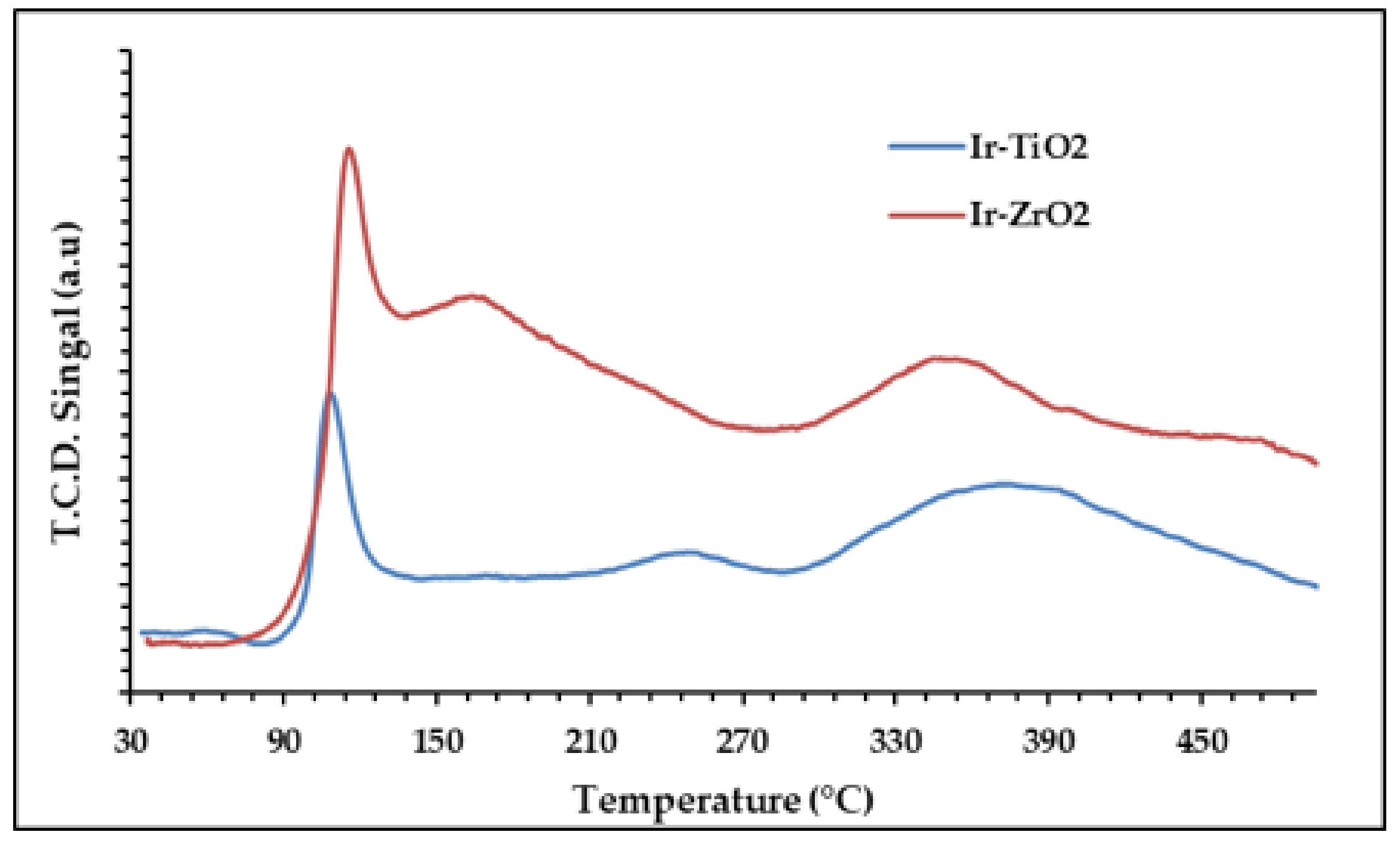
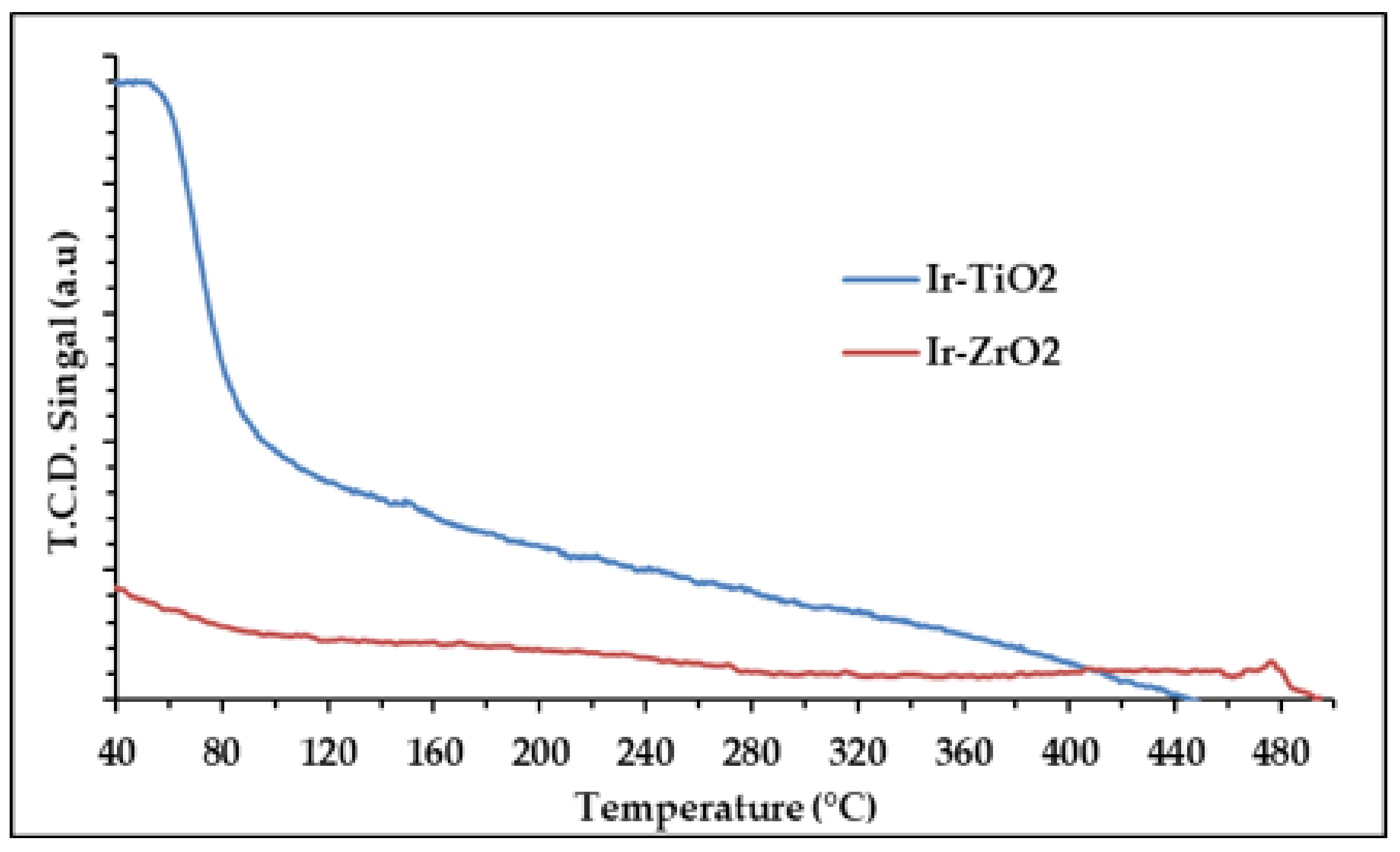
2.1.4. Temperature Programmed Studies
2.1.5. FTIR-CO
2.1.6. CO Chemisorption
2.1.7. Electron Microscopy
2.2. Catalytic Testing
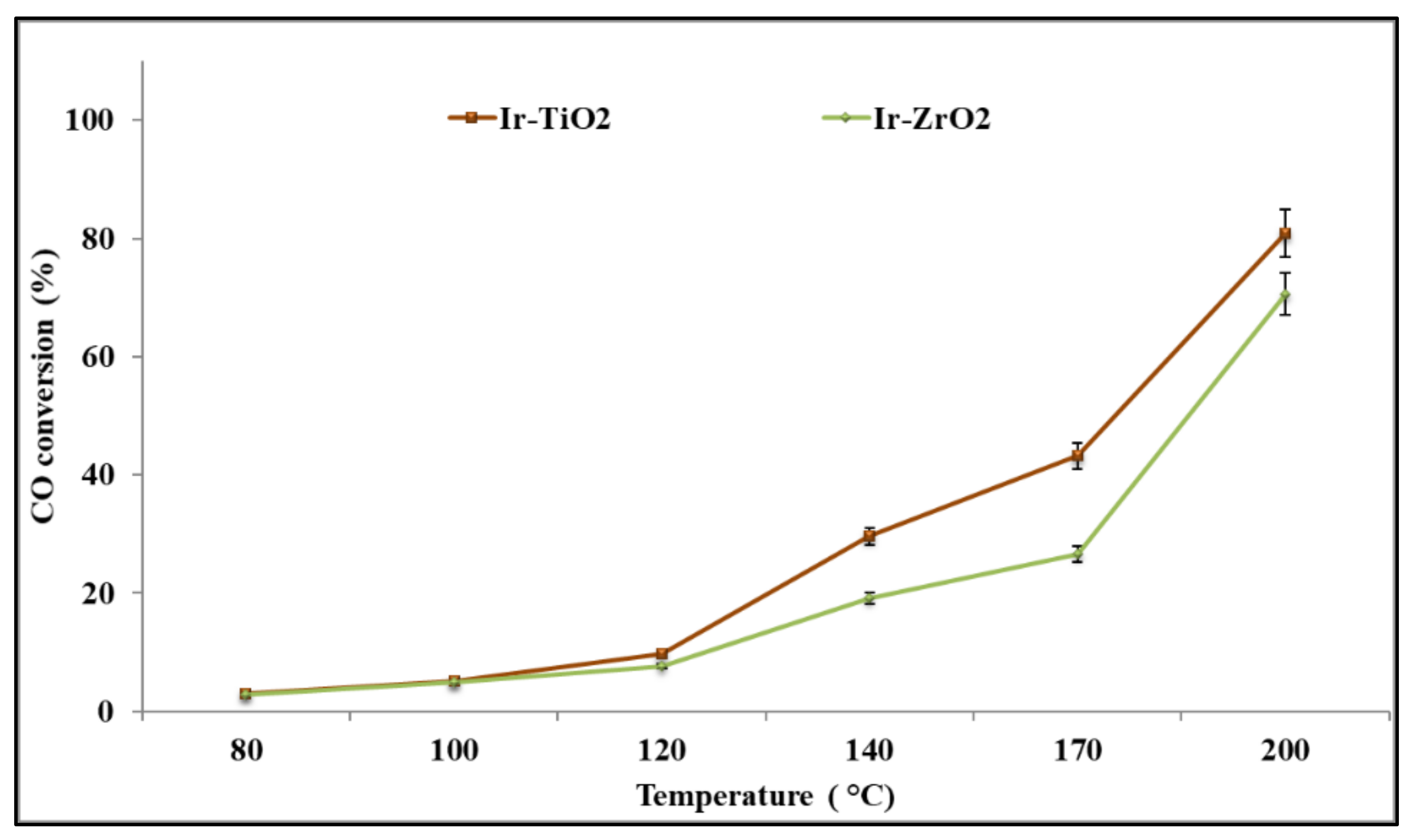
2.2.1. CO Oxidation
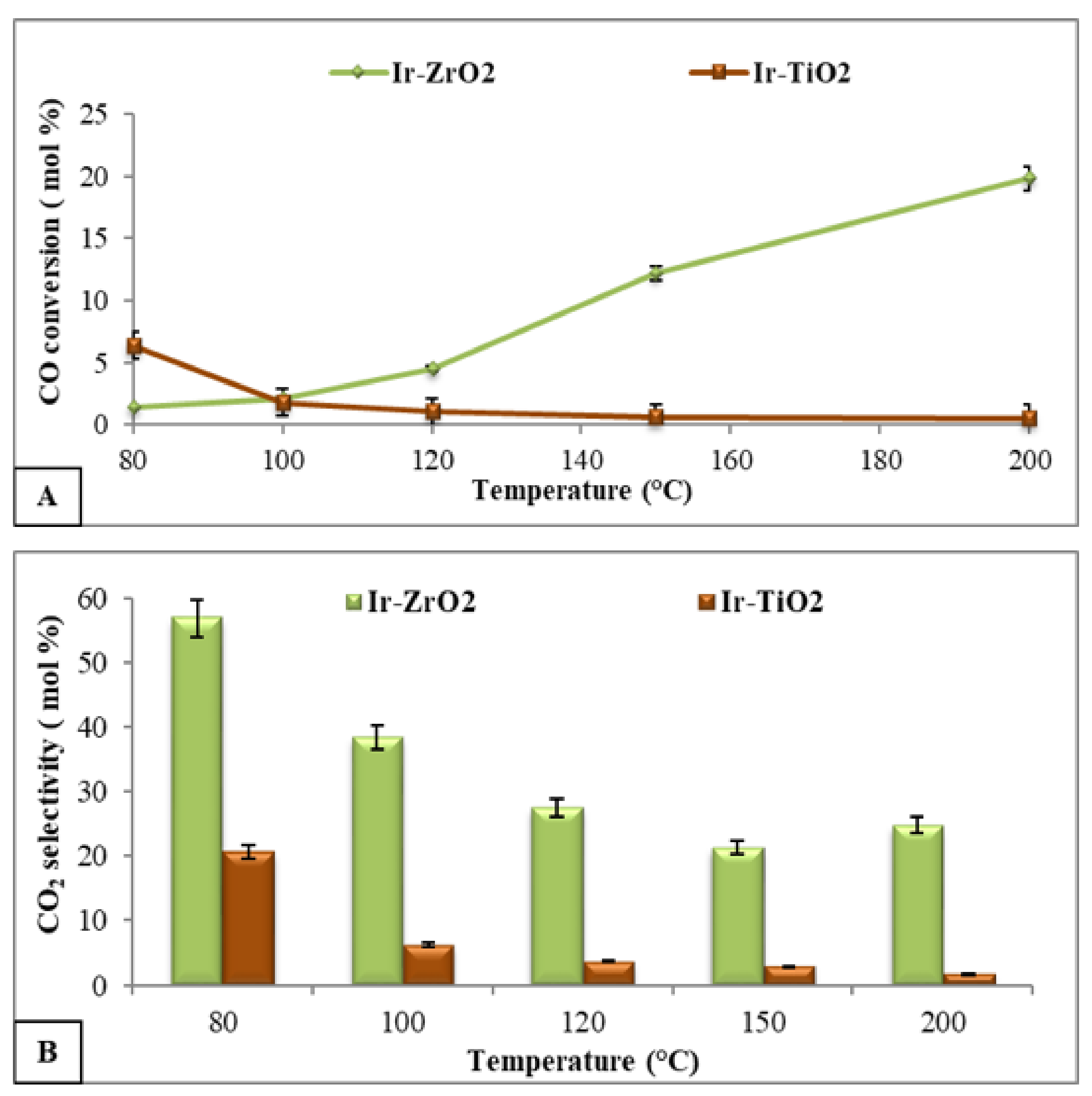
2.2.2. PROX Reaction
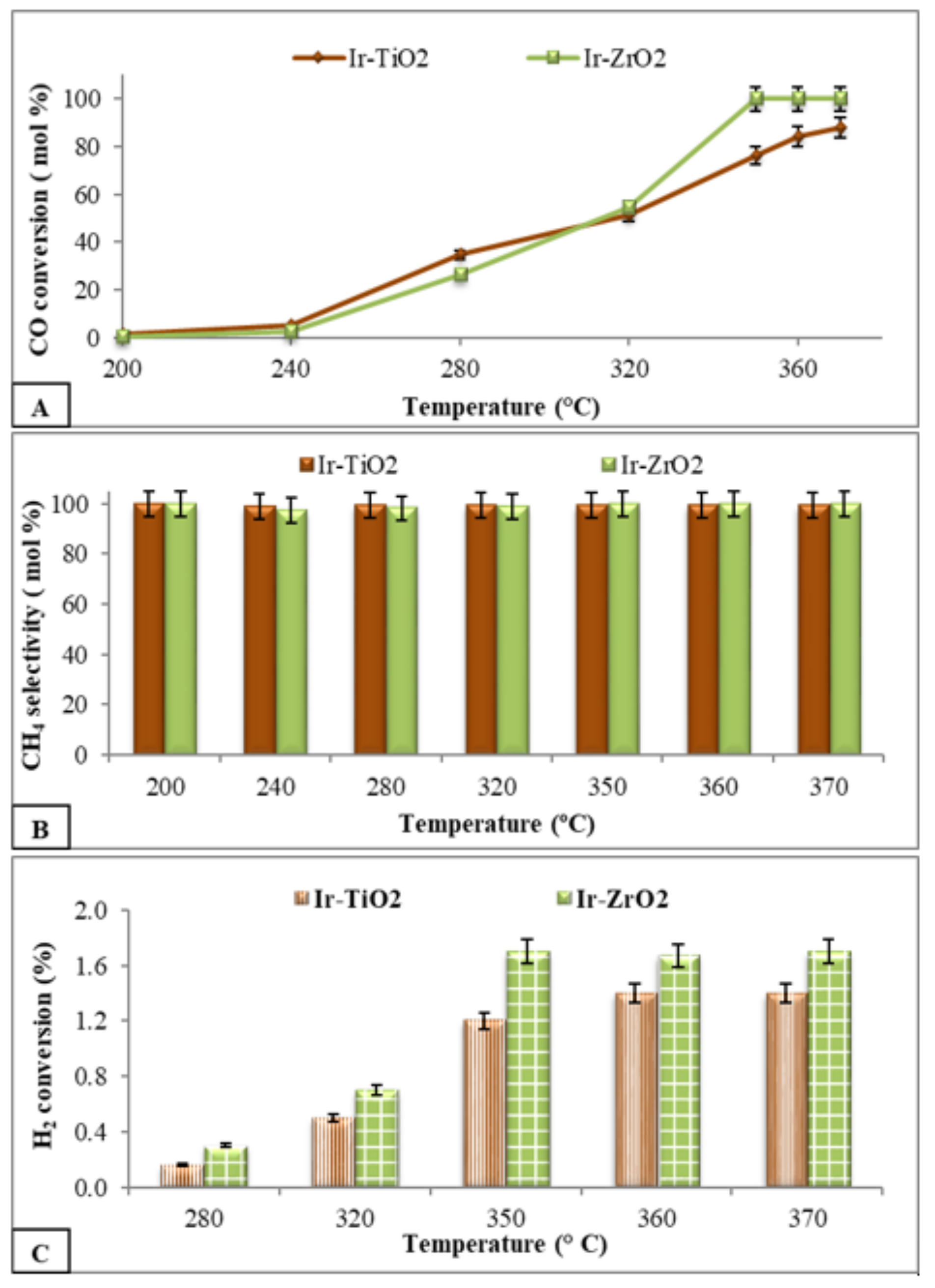
2.2.3. Hydrogenation Reactions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, L.; Yao, S.; Gao, R.; Liang, X.; Yu, Q.; Deng, Y.; Liu, J.; Peng, M.; Jiang, Z.; Li, S.; et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 2019, 14, 354–361. [Google Scholar] [CrossRef]
- Mohammedi, A.; Sahli, Y.; Ben Moussa, H. 3D investigation of the channel cross-section configuration effect on the power delivered by PEMFCs with straight channels. Fuel 2020, 263, 116713. [Google Scholar] [CrossRef]
- Moreno, M.; Baronetti, G.T.; Laborde, M.A.; Mariño, F.J. Kinetics of preferential CO oxidation in H2 excess (COPROX) over CuO/CeO2 catalysts. Int. J. Hydrogen Energy 2008, 33, 3538–3542. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported Ru catalysts. Appl. Catal. B 2009, 88, 470–478. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R.; Takagaki, A.; Sugawara, T.; Ted Oyama, S.; Satokawa, S. Effect of metal addition to Ru/TiO2 catalyst on selective CO methanation. Catal. Today 2014, 232, 16–21. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Zhang, Q.; Chmielewski, D.J.; Lauzze, K.C.; Inbody, M.A. Performance of CO preferential oxidation reactor with noble-metal catalyst coated on ceramic monolith for onboard fuel processing applications. Catal. Today 2005, 99, 271–283. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Hajduk, Š.; Ruiz-Zepeda, F.; Kovač, J.; Likozar, B.; Orel, Z.C. CeO2 and TiO2 support material effects on NH3 decomposition pathway mechanism over Cu–Zn catalysts. Fuel Process. Technol. 2021, 215, 106752. [Google Scholar] [CrossRef]
- Di, L.; Wu, G.; Dai, W.; Guan, N.; Li, L. Ru/TiO2 for the preferential oxidation of CO in H2-rich stream: Effects of catalyst pre-treatments and reconstruction of Ru sites. Fuel 2015, 143, 318–326. [Google Scholar] [CrossRef]
- Eckle, S.; Anfang, H.-G.; Behm, R.J. What drives the selectivity for CO methanation in the methanation of CO2-rich reformate gases on supported Ru catalysts? Appl. Catal. A 2011, 391, 325–333. [Google Scholar] [CrossRef]
- Mohamed, Z.; Dasireddy, V.D.B.C.; Singh, S.; Friedrich, H.B. The preferential oxidation of CO in hydrogen rich streams over platinum doped nickel oxide catalysts. Appl. Catal. B Environ. 2016, 180, 687–697. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.E.; Lee, H.C.; Choi, S.H.; Park, E.D. Active size-controlled Ru catalysts for selective CO oxidation in H2. Appl. Catal. B Environ. 2012, 127, 129–136. [Google Scholar] [CrossRef]
- Mariño, F.; Descorme, C.; Duprez, D. Noble metal catalysts for the preferential oxidation of carbon monoxide in the presence of hydrogen (PROX). Appl. Catal. B Environ. 2004, 54, 59–66. [Google Scholar] [CrossRef]
- Tada, S.; Minori, D.; Otsuka, F.; Kikuchi, R.; Osada, K.; Akiyama, K.; Satokawa, S. Effect of Ru and Ni ratio on selective CO methanation over Ru-Ni/TiO2. Fuel 2014, 129, 219–224. [Google Scholar] [CrossRef]
- Mohamed, Z.; Dasireddy, V.D.B.C.; Singh, S.; Friedrich, H.B. TiO2 and ZrO2 supported Ru catalysts for CO mitigation following the water-gas shift reaction. Int. J. Hydrogen Energy 2018, 43, 22291–22302. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B. Direct methanol production from mixed methane/H2O/N2O feedstocks over Cu–Fe/Al2O3 catalysts. Fuel 2021, 301, 121084. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, A.; Wang, X.; Zhang, T. Preferential oxidation of CO under excess H2 conditions over iridium catalysts. Int. J. Hydrogen Energy 2007, 32, 3880–3886. [Google Scholar] [CrossRef]
- Nguyen, T.-S.; Morfin, F.; Aouine, M.; Bosselet, F.; Rousset, J.-L.; Piccolo, L. Trends in CO oxidation and PROX performances of the platinum-group metals supported on ceria. Catal. Today 2015, 253, 106–114. [Google Scholar] [CrossRef]
- Doustkhah, E.; Assadi, M.H.N.; Komaguchi, K.; Tsunoji, N.; Esmat, M.; Fukata, N.; Tomita, O.; Abe, R.; Ohtani, B.; Ide, Y. In situ Blue titania via band shape engineering for exceptional solar H2 production in rutile TiO2. Appl. Catal. B Environ. 2021, 297, 120380. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Campos, C.; Torres, C.; Oportus, M.; Pena, M.A.; Fierro, J.L.G.; Reyes, P. Hydrogenation of substituted aromatic nitrobenzenes over 1% 1.0 wt.% Ir/ZrO2 catalyst: Effect of meta position and catalytic performance. Catal. Today 2013, 213, 93–100. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Neja, S.Š.; Blaž, L. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Yoshida, A.; Mori, Y.; Ikeda, T.; Azemoto, K.; Naito, S. Enhancement of catalytic activity of Ir/TiO2 by partially reduced titanium oxide in aerobic oxidation of alcohols. Catal. Today 2013, 203, 153–157. [Google Scholar] [CrossRef]
- Feng, W.; Wu, G.; Li, L.; Guan, N. Solvent-free selective photocatalytic oxidation of benzyl alcohol over modified TiO2. Green Chem. 2011, 13, 3265–3272. [Google Scholar] [CrossRef]
- Rojas, H.; Borda, G.; Reyes, P.; Martinez, J.J.; Valencia, J.; Fierro, J.L.G. Citral hydrogenation over Ir/TiO2 and Ir/TiO2/SiO2 catalysts. Catal. Today 2008, 133–135, 699–705. [Google Scholar] [CrossRef]
- Vicerich, M.A.; Oportus, M.; Benitez, V.M.; Reyes, P.; Pieck, C.L. Influence of Na content on the catalytic properties of Pt-Ir/Al2O3 catalysts for selective ring opening of decalin. Appl. Catal. A 2014, 480, 42–49. [Google Scholar] [CrossRef]
- Azzam, K.G.; Babich, I.V.; Seshan, K.; Lefferts, L. A bifunctional catalyst for the single-stage water–gas shift reaction in fuel cell applications. Part 2. Roles of the support and promoter on catalyst activity and stability. J. Catal. 2007, 251, 163–171. [Google Scholar] [CrossRef]
- Zhang, C.; He, H.; Tanaka, K.-i. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature. Appl. Catal. B Environ. 2006, 65, 37–43. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Christodoulakis, A.; Kondarides, D.I.; Boghosian, S. Particle size effects on the reducibility of titanium dioxide and its relation to the water–gas shift activity of Pt/TiO2 catalysts. J. Catal. 2006, 240, 114–125. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B. Activation and Decomposition of Methane over Cobalt-, Copper-, and Iron-Based Heterogeneous Catalysts for COx-Free Hydrogen and Multiwalled Carbon Nanotube Production. Energy Technol. 2017, 5, 1344–1355. [Google Scholar] [CrossRef]
- Hajduk, Š.; Dasireddy, V.D.B.C.; Likozar, B.; Dražić, G.; Orel, Z.C. COx-free hydrogen production via decomposition of ammonia over Cu–Zn-based heterogeneous catalysts and their activity/stability. Appl. Catal. B Environ. 2017, 211, 57–67. [Google Scholar] [CrossRef]
- Benvenutti, E.V.; Franken, L.; Moro, C.C.; Davanzo, C.U. FTIR Study of Hydrogen and Carbon Monoxide Adsorption on Pt/TiO2, Pt/ZrO2, and Pt/Al2O3. Langmuir 1999, 15, 8140–8146. [Google Scholar] [CrossRef]
- Pesty, F.; Steinrueck, H.-P.; Madey, T.E. Thermal stability of Pt films on TiO2(110): Evidence for encapsulation. Surf. Sci. 1995, 339, 83–95. [Google Scholar] [CrossRef]
- Hwang, K.-R.; Ihm, S.-K.; Park, S.-C.; Park, J.-S. Pt/ZrO2 catalyst for a single-stage water-gas shift reaction: Ti addition effect. Int. J. Hydrogen Energy 2013, 38, 6044–6051. [Google Scholar] [CrossRef]
- Mohamed, Z.; Dasireddy, V.D.B.C.; Singh, S.; Friedrich, H.B. Comparative studies for CO oxidation and hydrogenation over supported Pt catalysts prepared by different synthesis methods. Renew. Energy 2020, 148, 1041–1053. [Google Scholar] [CrossRef]
- Gao, Y.; Xie, K.; Mi, S.; Liu, N.; Wang, W.; Huang, W. Preferential oxidation of CO in a H2-rich stream over multi-walled carbon nanotubes confined Ru catalysts. Int. J. Hydrogen Energy 2013, 38, 16665–16676. [Google Scholar] [CrossRef]
- Amrousse, R.; Hori, K.; Fetimi, W. Iridium dispersion control in Ir/Al2O3-SiO2 catalysts by calcination temperature using chloroiridic acid as catalyst precursor. Catal. Commun. 2012, 27, 174–178. [Google Scholar] [CrossRef]
- Bourane, A.; Nawdali, M.; Bianchi, D. Heats of Adsorption of the Linear CO Species Adsorbed on a Ir/Al2O3 Catalyst Using in Situ FTIR Spectroscopy under Adsorption Equilibrium. J. Phys. Chem. B 2002, 106, 2665–2671. [Google Scholar] [CrossRef]
- Chen, P.; Lu, J.-Q.; Xie, G.-Q.; Hu, G.-S.; Zhu, L.; Luo, L.-F.; Huang, W.-X.; Luo, M.-F. Effect of reduction temperature on selective hydrogenation of crotonaldehyde over Ir/TiO2 catalysts. Appl. Catal. A 2012, 433–434, 236–242. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Paryjczak, T. Pt/ZrO2/TiO2 catalysts for selective hydrogenation of crotonaldehyde: Tuning the SMSI effect for optimum performance. App. Catal. A Gen. 2007, 320, 80–90. [Google Scholar] [CrossRef]
- Siddiquey, I.A.; Furusawa, T.; Sato, M.; Bahadur, N.M.; Uddin, M.N.; Suzuki, N. A rapid method for the preparation of silica-coated ZrO2 nanoparticles by microwave irradiation. Ceram. Int. 2011, 37, 1755–1760. [Google Scholar] [CrossRef]
- Bitter, J.H.; Seshan, K.; Lercher, J.A. The state of zirconia supported platinum catalysts for CO2/CH4 reforming. J. Catal. 1997, 171, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Doemoek, M.; Oszko, A.; Baan, K.; Sarusi, I.; Erdohelyi, A. Reforming of ethanol on Pt/Al2O3-ZrO2 catalyst. Appl. Catal. A 2010, 383, 33–42. [Google Scholar] [CrossRef]
- Coletta, V.C.; Gonçalves, R.V.; Bernardi, M.I.B.; Hanaor, D.A.H.; Assadi, M.H.N.; Marcos, F.C.F.; Nogueira, F.G.E.; Assaf, E.M.; Mastelaro, V.R. Cu-Modified SrTiO3 Perovskites Toward Enhanced Water–Gas Shift Catalysis: A Combined Experimental and Computational Study. ACS Appl. Energy Mater. 2021, 4, 452–461. [Google Scholar] [CrossRef]
- Okumura, M.; Masuyama, N.; Konishi, E.; Ichikawa, S.; Akita, T. CO Oxidation below Room Temperature over Ir/TiO2 Catalyst Prepared by Deposition Precipitation Method. J. Catal 2002, 208, 485–489. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Xia, G.-G.; Strohm, J.J.; Holladay, J.; Palo, D.R. Selective CO methanation catalysts for fuel processing applications. Appl. Catal. A 2007, 326, 213–218. [Google Scholar] [CrossRef]
- Sangeetha, P.; Zhao, B.; Chen, Y.-W. Au/CuOx−TiO2 Catalysts for Preferential Oxidation of CO in Hydrogen Stream. Ind. Eng. Chem. Res. 2010, 49, 2096–2102. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B.; Valand, J. Preferential oxidation of CO in H2/H2O/CO2 water–gas shift feedstocks over Cu-based carbon nanotubes-supported heterogeneous catalysts. Appl. Catal. B Environ. 2018, 237, 1044–1058. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Surface Area (m2/g) | Pore Volume (cm3/g) | Ir wt. (%) a |
|---|---|---|---|
| TiO2 | 151 | 0.35 | - |
| ZrO2 | 57 | 0.25 | - |
| Ir-TiO2 | 95 | 0.23 | 1.1 |
| Ir-ZrO2 | 50 | 0.23 | 1.1 |
| Catalyst | Binding Energy (eV) | Ir wt. % | |||
|---|---|---|---|---|---|
| Ir (4f 7/2) | O (1s) | Ti (2p 3/2) | Zr (3d 5/2) | ||
| Ir-TiO2 | 62.3 | 529.3 | 458.8 | - | 0.9 |
| 64.6 | 464.1 | - | |||
| Ir-ZrO2 | 62.3 | 534.3 | - | 184.3 | 1.0 |
| 64.8 | - | 188.1 | |||
| Catalysts | Properties | Temperature of Reduction | ||
|---|---|---|---|---|
| 200 °C | 370 °C | 500 °C | ||
| Ir-TiO2 | Metal dispersion (%) | 61.4 | 52.1 | 36.3 |
| Metallic surface area (m2/g metal) | 107.9 | 89.4 | 73.3 | |
| Crystallite size (nm) | 6.4 | 7.0 | 7.0 | |
| Chemisorption capacity (CO/Ir) | 0.49 | 0.31 | 0.24 | |
| Ir-ZrO2 | Metal dispersion (%) | 91.4 | 77.5 | 54.0 |
| Metallic surface area (m2/g metal) | 160.6 | 133.0 | 109.1 | |
| Crystallite size (nm) | 5.3 | 5.8 | 5.9 | |
| Chemisorption capacity (CO/Ir) | 0.69 | 0.44 | 0.35 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, Z.; Dasireddy, V.D.B.C.; Singh, S.; Friedrich, H.B. The Mitigation of CO Present in the Water–Gas Shift Reformate Gas over IR-TiO2 and IR-ZrO2 Catalysts. Catalysts 2021, 11, 1378. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111378
Mohamed Z, Dasireddy VDBC, Singh S, Friedrich HB. The Mitigation of CO Present in the Water–Gas Shift Reformate Gas over IR-TiO2 and IR-ZrO2 Catalysts. Catalysts. 2021; 11(11):1378. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111378
Chicago/Turabian StyleMohamed, Ziyaad, Venkata D. B. C. Dasireddy, Sooboo Singh, and Holger B. Friedrich. 2021. "The Mitigation of CO Present in the Water–Gas Shift Reformate Gas over IR-TiO2 and IR-ZrO2 Catalysts" Catalysts 11, no. 11: 1378. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111378