
Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− Dicarbollide Ligands

 , , and
, , and 
Abstract
:
1. Introduction
2. Results
2.1. Synthesis of B-Methylated nido-Carboranes
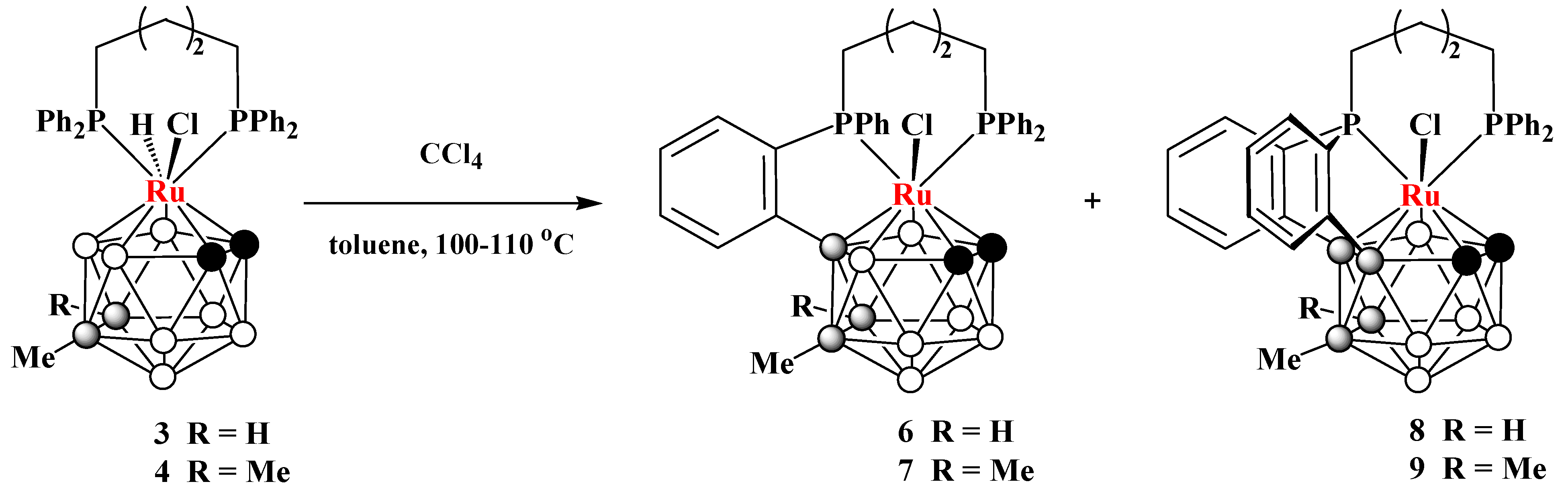
2.2. Synthesis of closo-Ruthenaboranes
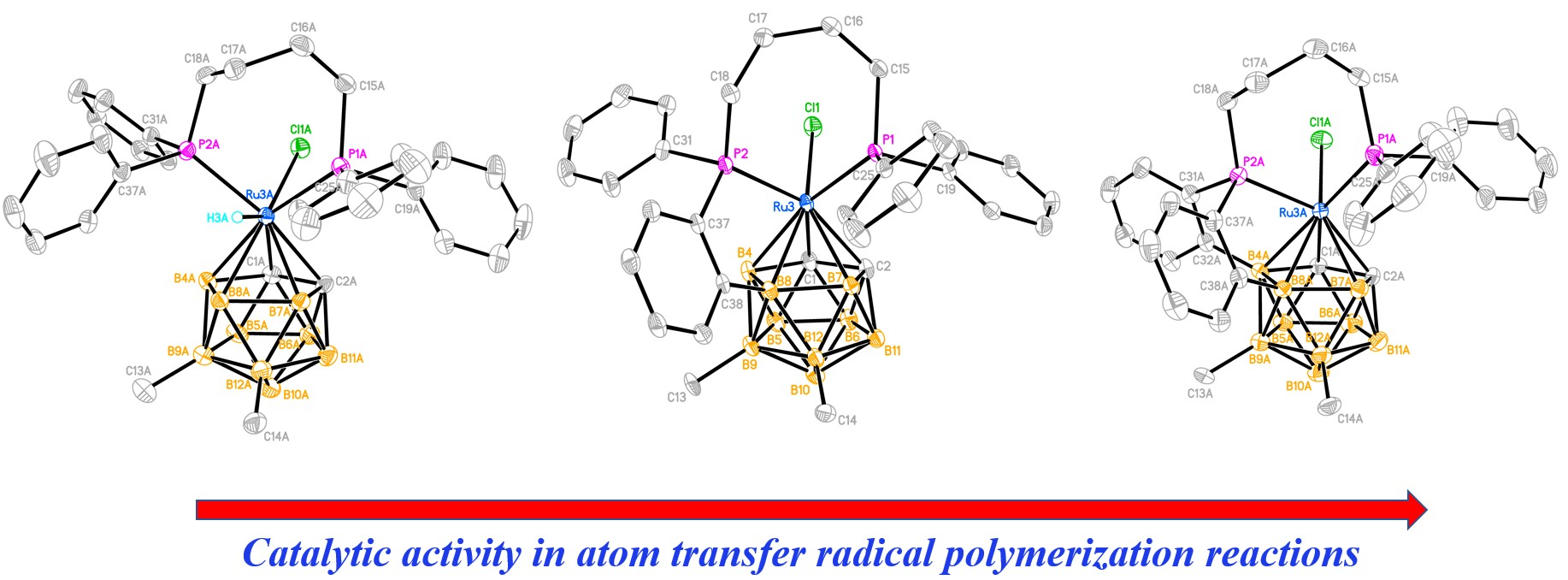
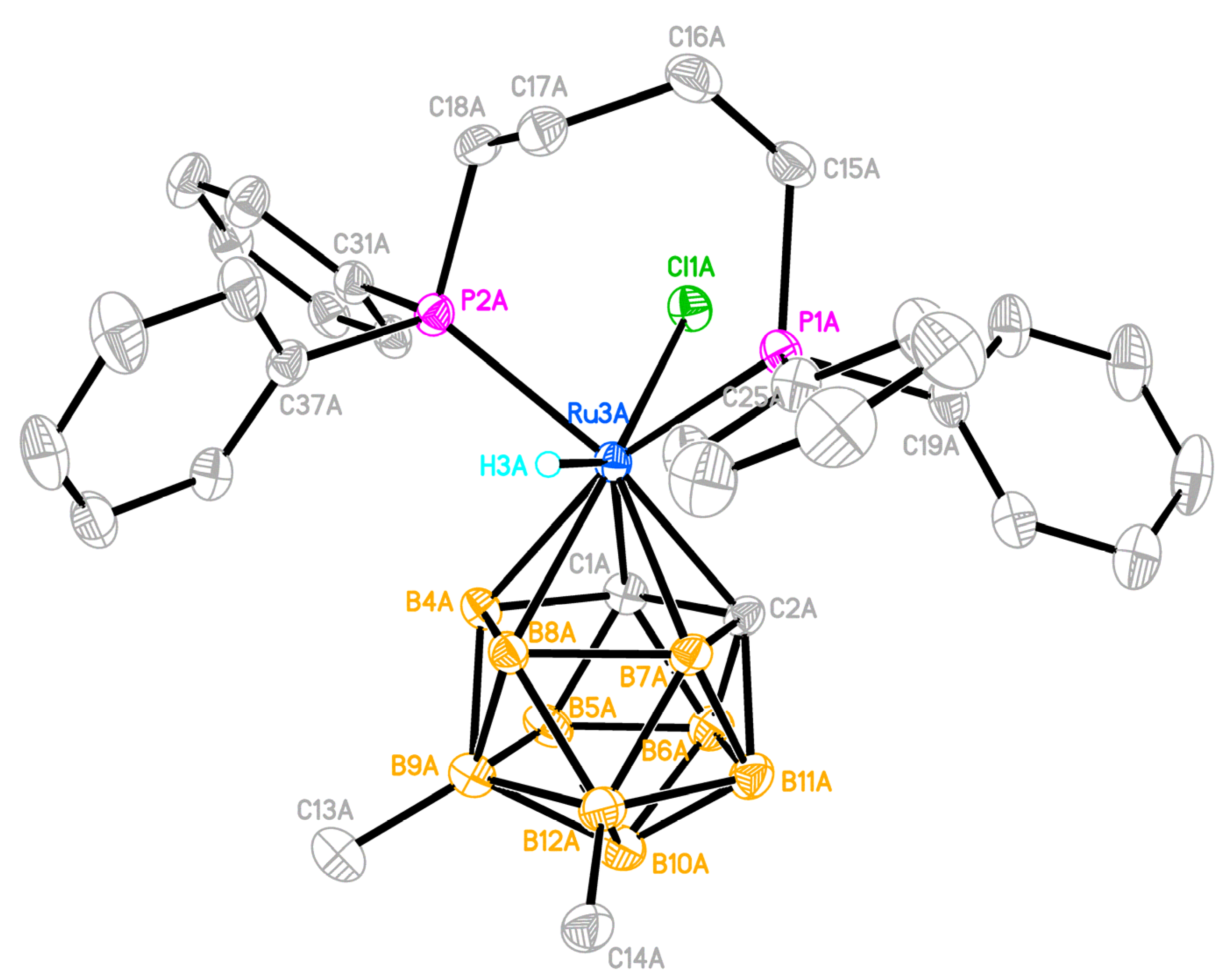
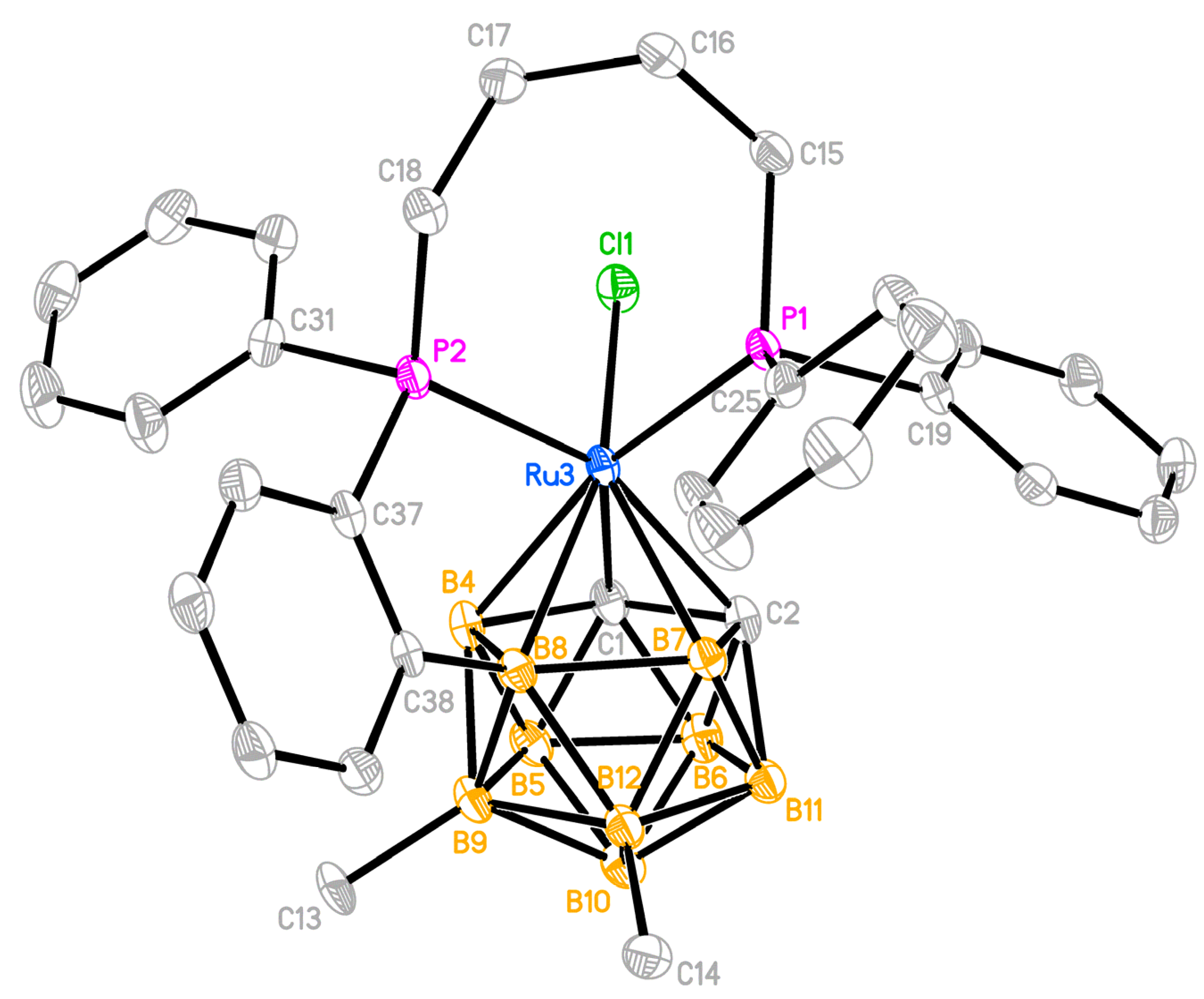
2.3. X-ray Diffraction Study of the Complexes
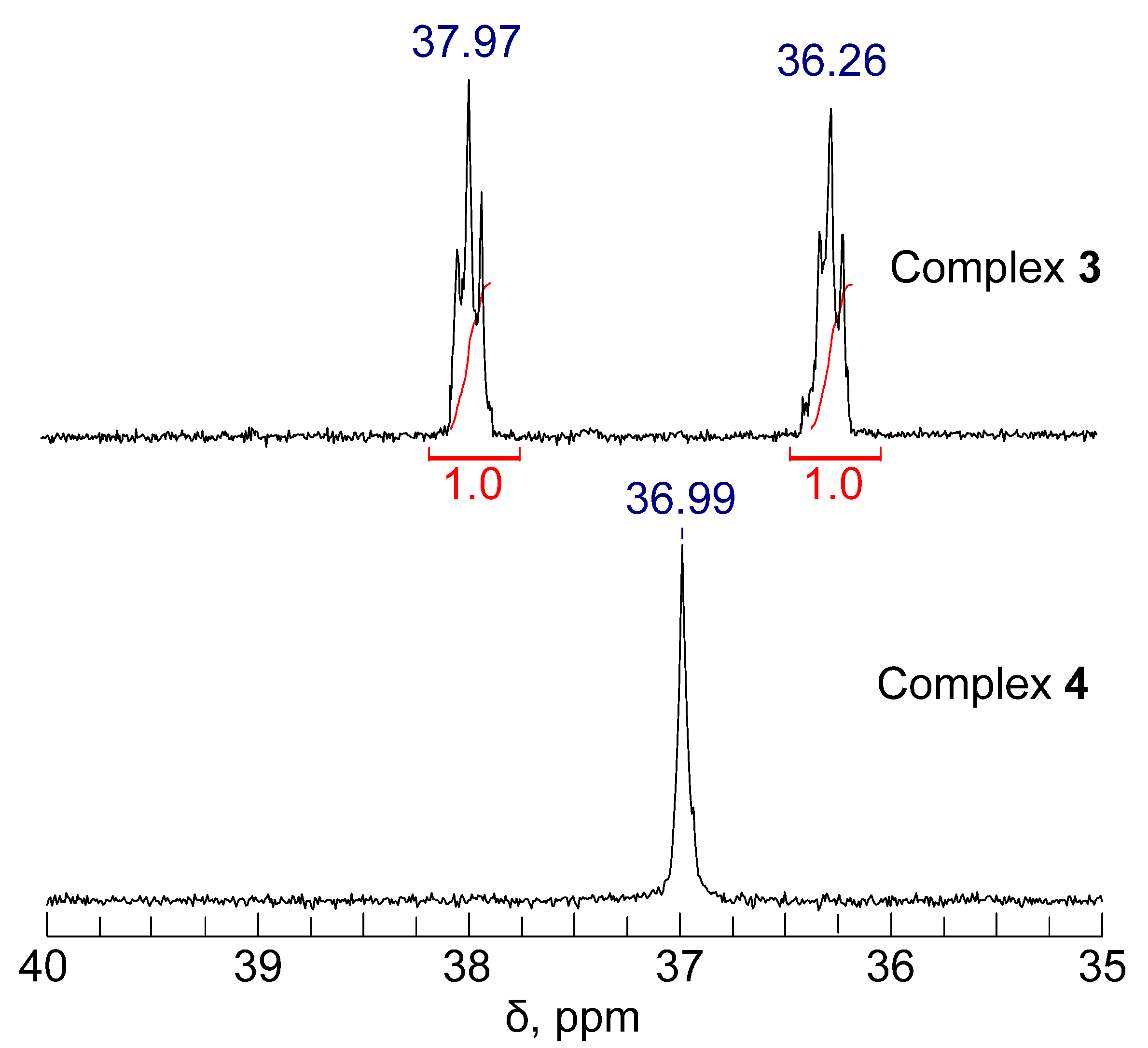
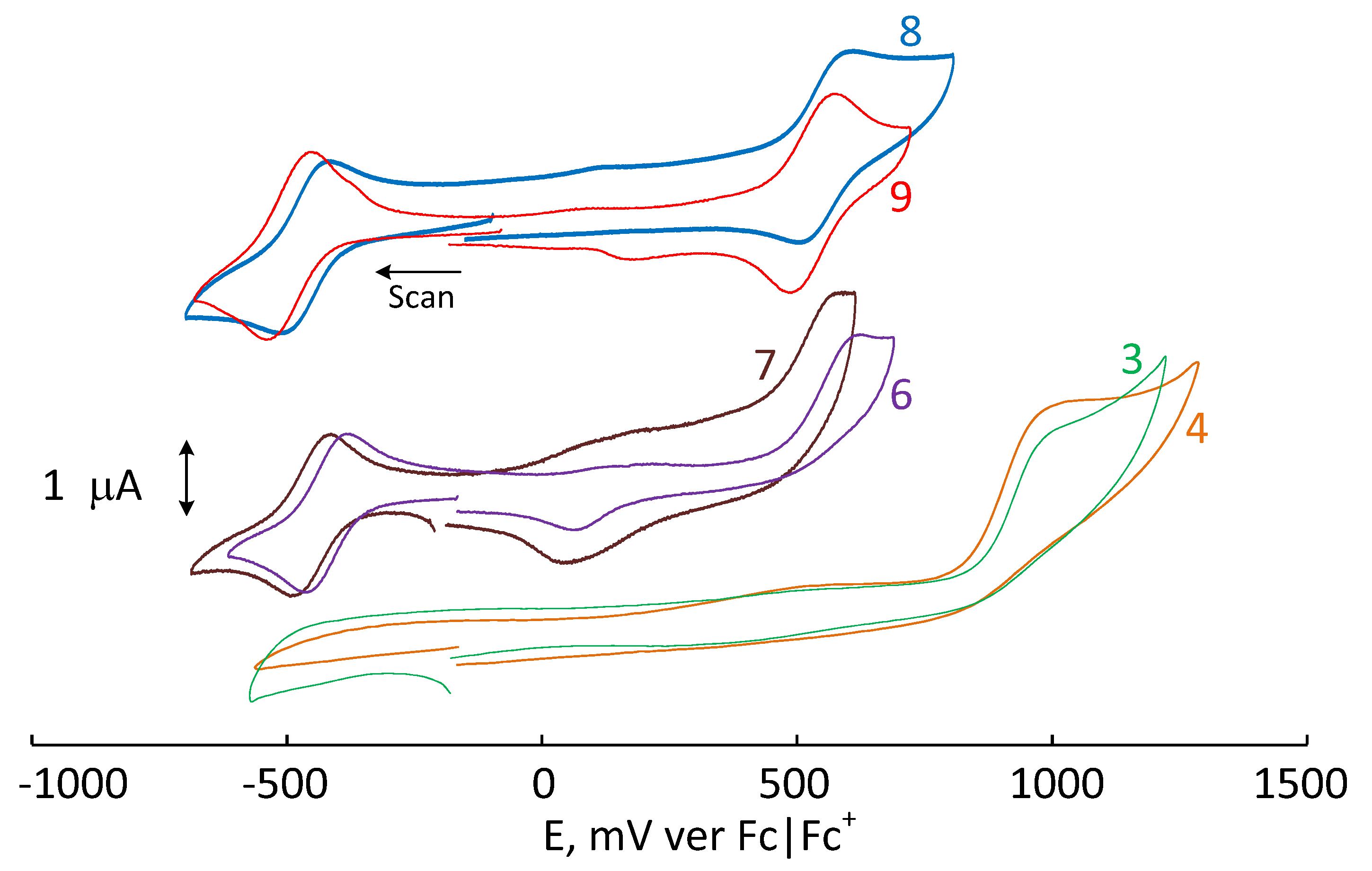
2.4. Electrochemical Studies
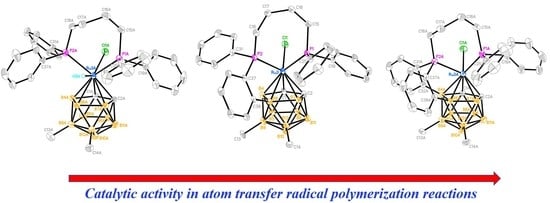
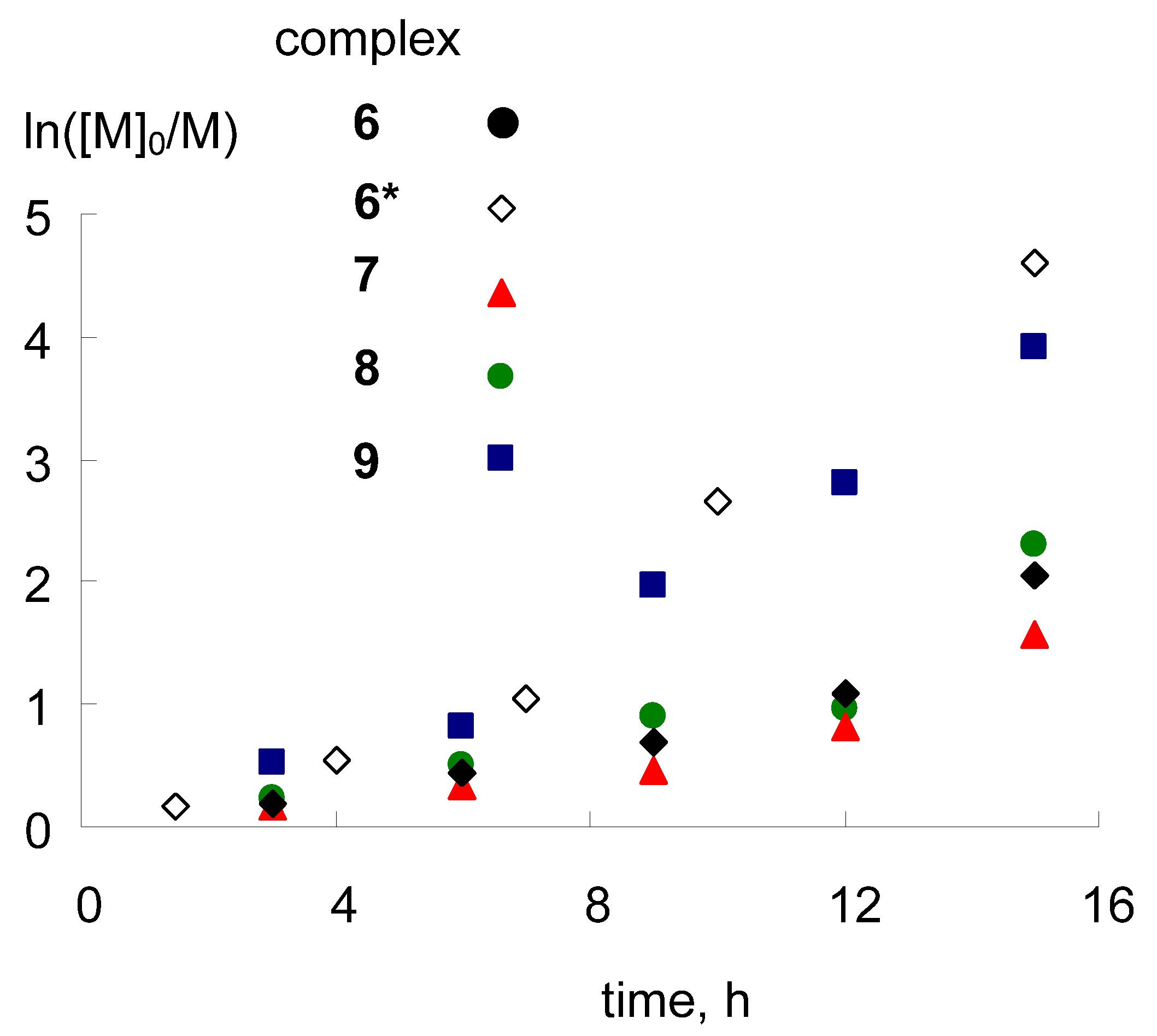
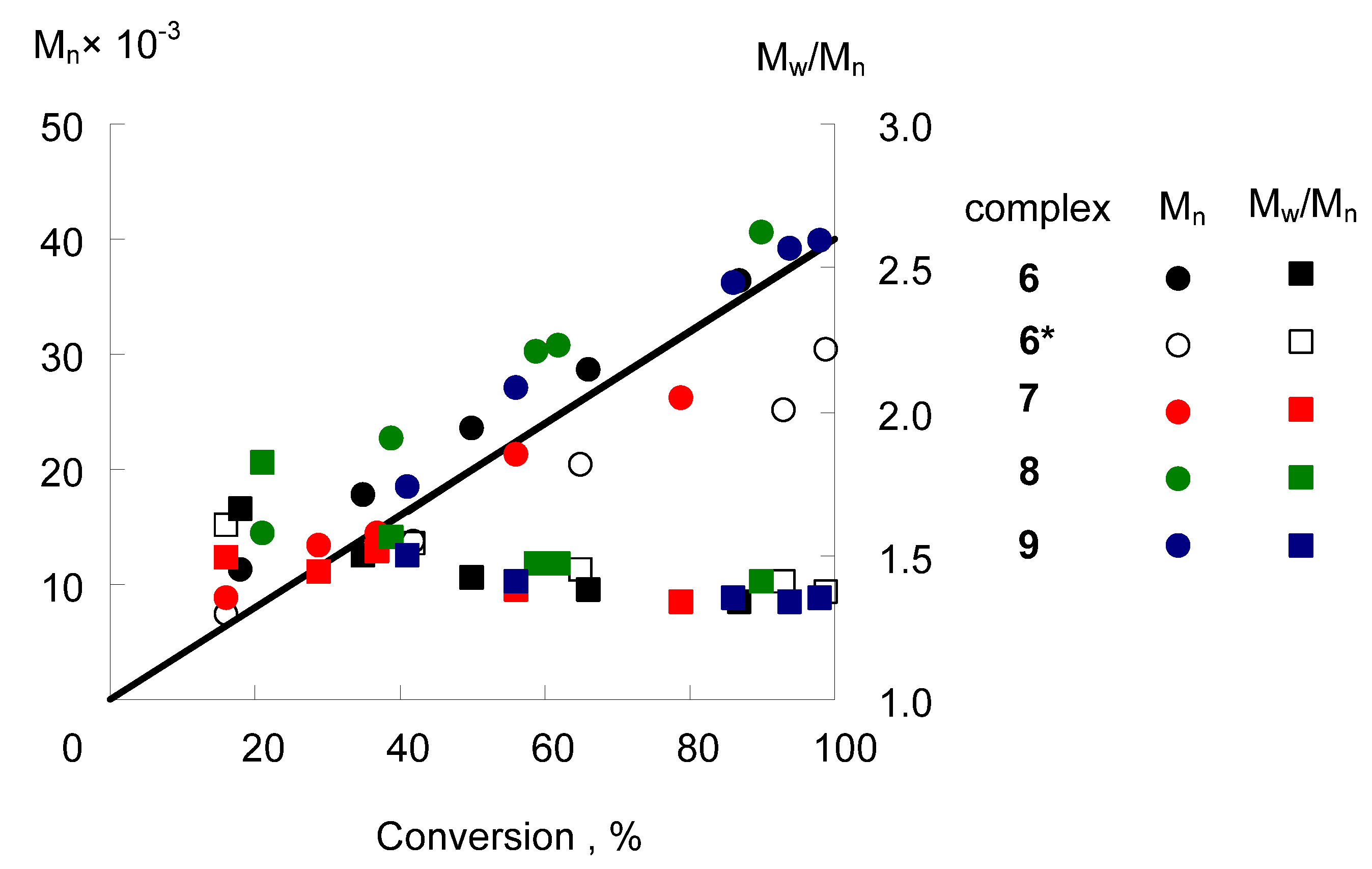
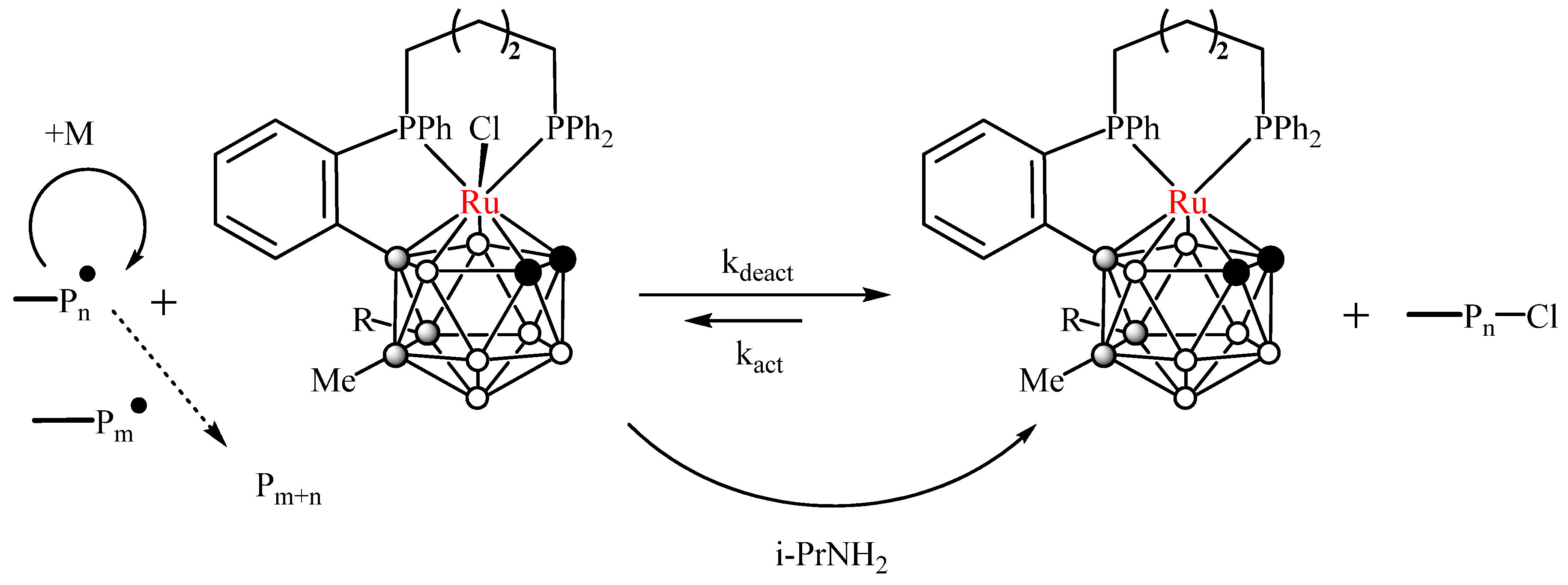
2.5. Catalytic Activity in Radical Polymerization
3. Experimental Part
3.1. Materials and Methods
3.2. Preparation of Carborane Ligands
3.3. Synthesis of Ruthenacarboranes
3.4. X-ray Diffraction Study
3.5. Polymerization Procedure
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grimes, R.N. Carboranes in catalysis. In Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 929–944. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and mixed-sandwich metallacarboranes in catalysis. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine—Volume 2—Boron in Catalysis; Hosmane, N.S., Eagling, R., Eds.; World Scientific Publishing Europe: London, UK, 2018; pp. 27–80. [Google Scholar] [CrossRef]
- Zhu, Y.; Hosmane, N.S. Carborane-based catalysts for polymerization of olefins. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine—Volume 2—Boron in Catalysis; Hosmane, N.S., Eagling, R., Eds.; World Scientific Publishing Europe: London, UK, 2018; pp. 117–134. [Google Scholar] [CrossRef]
- Tutusaus, O.; Delfosse, S.; Simal, F.; Demonceau, A.; Noels, A.F.; Núñez, R.; Viñas, C.; Teixidor, F. Half-sandwich ruthenium complexes for the controlled radical polymerisation of vinyl monomers. Inorg. Chem. Commun. 2002, 5, 941–945. [Google Scholar] [CrossRef]
- Grishin, I.D.; Kolyakina, E.V.; Cheredilin, D.N.; Chizhevskii, I.T.; Grishin, D.F. Specifics of radical polymerization of styrene and methyl methacrylate in the presence of ruthenium carborane complexes. Polym. Sci. A 2007, 49, 1079–1085. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Loginov, D.A. Rhoda- and iridacarborane halide complexes: Synthesis, structure and application in homogeneous catalysis. J. Organomet. Chem. 2020, 910, 121135. [Google Scholar] [CrossRef]
- Guerrero, I.; Kelemen, Z.; Viñas, C.; Romero, I.; Teixidor, F. Photosensitizers for the photooxidation of alcohols in water through single-electron-transfer processes. Chem. Eur. J. 2020, 26, 5027–5036. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.P.Y.; Parkinson, J.A.; Rosair, G.M.; Welch, A.J. Bis(phosphine)hydridorhodacarborane derivatives of 1,1′-bis(ortho-carborane) and their catalysis of alkene isomerization and the hydrosilylation of acetophenone. Inorg. Chem. 2020, 59, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Perveen, S.; Ouyang, Y.; Zhang, S.; Jiao, J.; He, G.; Nie, Y.; Li, P. Well-defined, versatile and recyclable half-sandwich nickelacarborane catalyst for selective carbene-transfer reactions. Chem. Eur. J. 2021, 27, 5754–5760. [Google Scholar] [CrossRef] [PubMed]
- Ribelli, T.G.; Fantin, M.; Daran, J.-C.; Augustine, K.F.; Poli, R.; Matyjaszewski, K. Synthesis and characterization of the most active copper atrp catalyst based on tris[(4-dimethylaminopyridyl)methyl]amine. J. Am. Chem. Soc. 2018, 140, 1525–1534. [Google Scholar] [CrossRef]
- Lorandi, F.; Matyjaszewski, K. Why do we need more active ATRP catalysts? Isr. J. Chem. 2019, 60, 108–123. [Google Scholar] [CrossRef] [Green Version]
- Llop, J.; Viñas, C.; Teixidor, F.; Victori, L.; Kivekäs, R.; Sillanpää, R. Redox potential modulation in mixed sandwich pyrrolyl/ dicarbollide complexes. Inorg. Chem. 2002, 41, 3347–3352. [Google Scholar] [CrossRef]
- Grishin, I.D.; Turmina, E.S.; D’yachihin, D.I.; Peregudova, S.M.; Chizhevsky, I.T.; Grishin, D.F. Ruthenium carborane complexes: A relationship between the structure, electrochemical properties, and reactivity in catalysis of polymerization processes. Russ. Chem. Bull. 2013, 62, 692–698. [Google Scholar] [CrossRef]
- Grimes, R.N. Icosahedral carboranes: 1,2-C2B10H12. In Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 283–502. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of 9,9′,12,12′-substituted cobalt bis(dicarbollide) derivatives. Russ. Chem. Bull. 2015, 64, 712–717. [Google Scholar] [CrossRef]
- Robertson, S.; Ellis, D.; Rosair, G.M.; Welch, A.J. Platination of [3-X-7,8-Ph2-7,8-nido-C2B9H8]2- (X = Et, F). Synthesis and characterisation of slipped and 1,2→1,7 isomerised products. J. Organomet. Chem. 2003, 680, 286–293. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Petrovskii, P.V. Synthesis of 5-organo-, 9-organo-, and 9,11-diorgano-nido-7,8-dicarbaundecaborane salts by cross-coupling. Russ. J. Gen. Chem. 2000, 70, 678–682. [Google Scholar]
- Cheredilin, D.N.; Balagurova, E.V.; Godovikov, I.A.; Solodovnikov, S.P.; Chizhevsky, I.T. Facile formation of exo-nido→closo-rearrangement products upon the replacement of PPh3 ligands with bis(diphenylphosphino)alkanes in “three-bridge” ruthenacarborane 5,6,10-[RuCl(PPh3)2]-5,6,10-(µ-H)3-10-H-exo-nido-7,8-C2B9H8. Russ. Chem. Bull. 2005, 54, 2535–2539. [Google Scholar] [CrossRef]
- Grishin, I.D.; Turmina, E.S.; D’yachihin, D.I.; Vinogradov, D.S.; Piskunov, A.V.; Smolyakov, A.F.; Dolgushin, F.M.; Chizhevsky, I.T.; Grishin, D.F. Efficient catalytic systems based on paramagnetic closo-ruthenacarboranes for the controlled synthesis of polymers. Russ. Chem. Bull. 2011, 60, 2375–2383. [Google Scholar] [CrossRef]
- Grishin, I.D.; D’yachihin, D.I.; Piskunov, A.V.; Dolgushin, F.M.; Smolyakov, A.F.; Il’in, M.M.; Davankov, V.A.; Chizhevsky, I.T.; Grishin, D.F. Carborane complexes of ruthenium(III): Studies on thermal reaction chemistry and the catalyst design for atom transfer radical polymerization of methyl methacrylate. Inorg. Chem. 2011, 50, 7574–7585. [Google Scholar] [CrossRef]
- Zimina, A.M.; Anufriev, S.A.; Derendyaeva, M.A.; Knyazeva, N.A.; Somov, N.V.; Malysheva, Y.B.; Sivaev, I.B.; Grishin, I.D. Ruthenium complexes of 5-MeC2B9-carborane ligand: Synthesis and application in polymerization catalysis. Doklady Chem. 2021, 498, 97–103. [Google Scholar] [CrossRef]
- Tang, H.; Radosz, M.; Shen, Y. CuBr2/N,N,N′,N′-tetra[(2-pyridal)methyl] ethylenediamine/tertiary amine as a highly active and versatile catalyst for atom-transfer radical polymerization via activator generated by electron transfer. Macromol. Rapid Commun. 2006, 27, 1127–1131. [Google Scholar] [CrossRef]
- Kwak, Y.; Matyjaszewski, K. ARGET ATRP of methyl methacrylate in the presence of nitrogen-based ligands as reducing agents. Polym. Int. 2009, 58, 242–247. [Google Scholar] [CrossRef]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef]
- Andrews, J.S.; Zayas, J.; Jones, M. 9-Iodo-o-carborane. Inorg. Chem. 1985, 24, 3715–3716. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiang, W.; Zinn, A.A.; Knobler, C.B.; Hawthorne, M.F. Facile electrophilic iodination of icosahedral carboranes. Synthesis of carborane derivatives with boron-carbon bonds via the palladium-catalyzed reaction of diiodocarboranes with Grignard reagents. Inorg. Chem. 1995, 34, 2095–2100. [Google Scholar] [CrossRef]
- Itatani, H.; Bailar, J.C. Homogenous catalysis in the reactions of olefinic substances. V. Hydrogenation of soybean oil methyl ester with triphenylphosphine and triphenylarsine palladium catalysts. J. Am. Oil Chem. Soc. 1967, 44, 147–151. [Google Scholar] [CrossRef]
- Jung, C.W.; Garrou, P.E.; Hoffman, P.R.; Caulton, K.G. Reexamination of the reactions of Ph2P(CH2)nPPh2 (n = 1–4) with RuCl2(PPh3)3. Inorg. Chem. 1984, 23, 726–729. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, NJ, USA, 2009. [Google Scholar]
- Gal’chenko, G.L.; Tamm, N.B.; Pavlovich, V.K.; Ol’shevskaya, V.A.; Zakharkin, L.I. 9-Methyl-o-carbaborane(12). Synthesis and determination of thermochemical characteristics. Organomet. Chem. USSR 1990, 3, 203–206. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
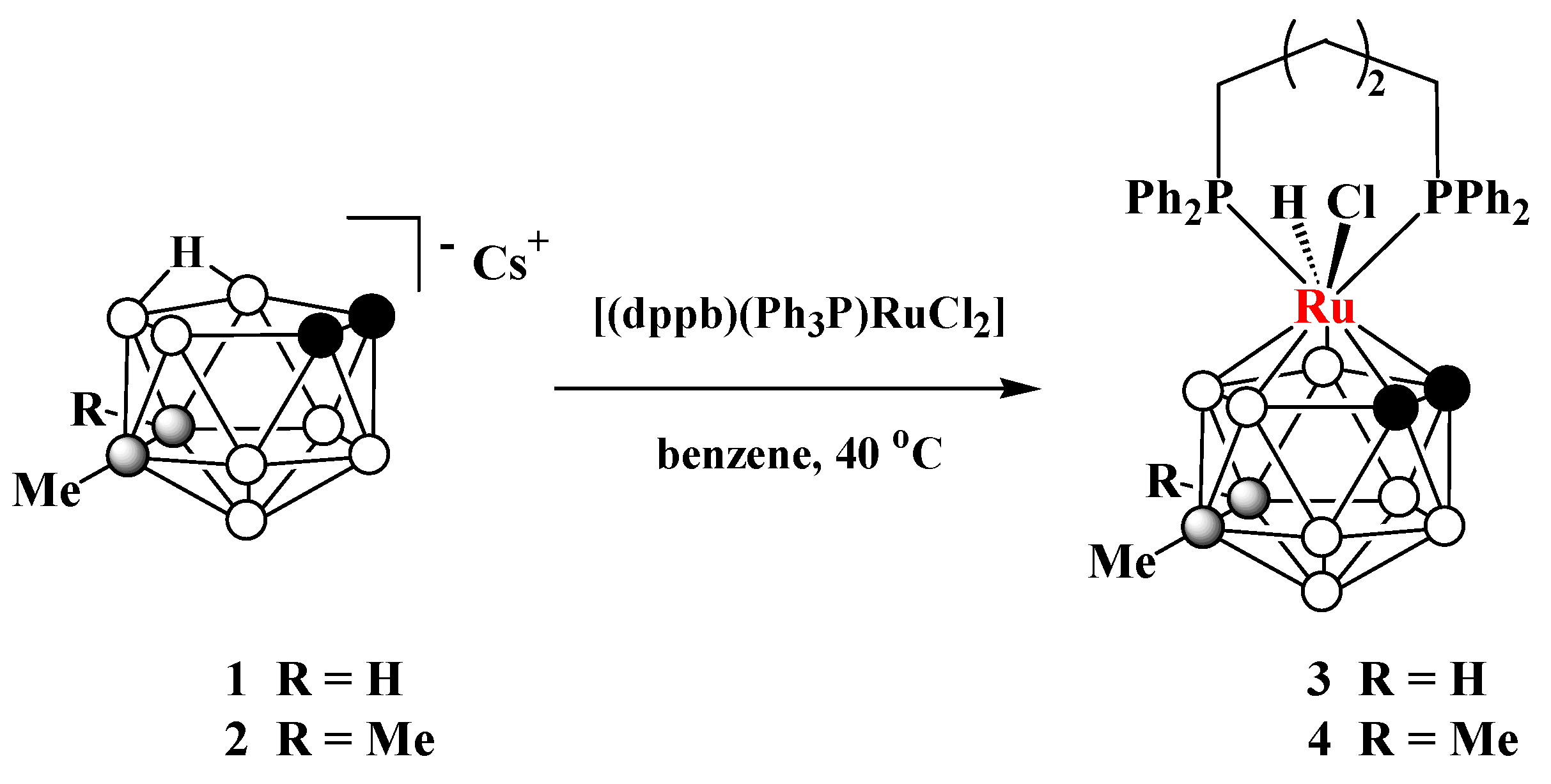{kind=link}
{kind=link}
{kind=link}
| Parameter | Compound | ||||
|---|---|---|---|---|---|
| 5 [21] | 3 [20] | 4(A) | 7 | 9(A) | |
| Bond lengths, Ǻ | |||||
| Ru-P(1) | 2.3670(5) | 2.3701(6) | 2.3620(8) | 2.3688(8) | 2.3509(9) |
| Ru-P(2) | 2.3305(5) | 2.3382(6) | 2.3429(9) | 2.2954(8) | 2.2703(9) |
| Ru-H | 1.51(3) | 1.53(3) | 1.50(4) | ||
| Ru-Cl | 2.4463(5) | 2.4384(5) | 2.4278(8) | 2.3740(8) | 2.3864(8) |
| Ru-C(1) | 2.258(3) | 2.220(2) | 2.281(3) | 2.240(3) | 2.202(3) |
| Ru-C(2) | 2.229(2) | 2.198(2) | 2.229(3) | 2.256(3) | 2.246(3) |
| Ru-B(4) | 2.315(2) | 2.296(2) | 2.322(3) | 2.251(3) | 2.249(4) |
| Ru-B(7) | 2.230(2) | 2.265(2) | 2.224(4) | 2.232(3) | 2.225(4) |
| Ru-B(8) | 2.282(2) | 2.298(2) | 2.266(4) | 2.291(3) | 2.260(4) |
| C(1)-C(2) | 1.624(3) | 1.624(3) | 1.624(3) | 1,601(4) | 1,617(5) |
| B(9)-B(12) | 1.789(4) | 1.798(4) | 1.814(6) | 1.800(5) | 1.818(8) |
| B(8)-B(12) | 1.805(3) | 1.812(4) | 1.818(5) | 1.825(5) | 1.825(7) |
| B(8)-B(9) | 1.804(4) | 1.810(3) | 1.816(6) | 1.836(5) | 1.833(7) |
| B(10)-B(12) | 1.782(4) | 1.791(3) | 1.798(6) | 1.776(5) | 1.801(9) |
| B(10)-B(9) | 1.776(3) | 1.783(4) | 1.796(5) | 1.776(5) | 1.783(8) |
| B(8)-C(38) | 1.587(4) | 1.595(5) | |||
| B(4)-C(32) | 1.586(5) | ||||
| Valence angles, deg. | |||||
| Cl-Ru-H | 136(1) | 136.40(1) | 138.5(14) | ||
| P-Ru-P | 102.51(2) | 102.28(2) | 102.14(3) | 90.11(3) | 91.08(3) |
| P(1)-Ru-Cl | 80.48(2) | 80.48(2) | 80.36(3) | 90.24(3) | 90.04(3) |
| P(2)-Ru-Cl | 83.12(2) | 83.16(2) | 82.84(3) | 95.68(3) | 89.02(3) |

| Transition | Compound | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 10 [13] | 6 | 7 | 12 [13] | 11 [13] | 8 | 9 | ||
| M−/M | Epa, mV | - | - | −316 | −383 | −417 | −425 | −358 | −419 | −454 |
| Epc, mV | - | - | −393 | −463 | −487 | −496 | −428 | −510 | −540 | |
| E1/2, mV | - | - | −354 | −423 | −452 | −461 | −393 | −464 | −497 | |
| M/M+ | Epa, mV | 1010 | 960 | 683 | 600 | 576 | 554 | 627 | 601 | 571 |
| Epc, mV | - | - | - | - | - | - | 551 | 509 | 488 | |
| E1/2, mV | - | - | - | - | - | - | 589 | 554 | 530 | |
| Complex | Conversion | Mn × 10−3 | Mw/Mn |
|---|---|---|---|
| 6 | 66 | 28.6 | 1.38 |
| 7 | 56 | 21.2 | 1.38 |
| 8 | 62 | 30.7 | 1.47 |
| 9 | 94 | 39.2 | 1.34 |
| Identification Code | 4 | 7 | 9 |
|---|---|---|---|
| CCDC No | 2117788 | 2117789 | 2117790 |
| Empirical formula | C32H44B9ClP2Ru | C32H41B9ClP2Ru | C32H39B9ClP2Ru CH2Cl2 |
| Molecular weight | 724.42 | 721.40 | 804.31 |
| Crystal size (mm) | 0.16 × 0.10 × 0.06 | 0.22 × 0.16 × 0.02 | 0.16 × 0.12 × 0.04 |
| Temperature (K) | 150(2) | 100(2) | 100(2) |
| Crystal system | monoclinic | monoclinic | monoclinic |
| Space group | P21/c | P21/c | P21/n |
| a (Å) | 16.8803(10) | 13.9369(18) | 11.0895(3) |
| b (Å) | 20.5774(12) | 12.4716(16) | 18.9868(5) |
| c (Å) | 20.9312(12) | 20.085(3) | 34.9643(10) |
| β (deg) | 106.242(2) | 93.786(4) | 90.0597(10) |
| V (Å3) | 6980.3(7) | 3483.5(8) | 7361.9(3) |
| Z | 8 | 4 | 8 |
| Dcalcd (g.cm–3) | 1.379 | 1.376 | 1.451 |
| linear absorption μ (cm–1) | 6.41 | 6.42 | 7.56 |
| Tmin/Tmax | 0.904/0.963 | 0.913/0.987 | 0.898/0.970 |
| 2θmax (deg) | 56 | 60 | 56 |
| Reflections collected | 90,541 | 70,986 | 70,362 |
| Independent reflections (Rint) | 16,854 (0.0878) | 10,170 (0.0659) | 17,778 (0.0618) |
| Observed reflections (I > 2σ(I)) | 11,813 | 8543 | 13,228 |
| Number of parameters | 900 | 440 | 938 |
| R1 (on F for I > 2σ(I)) a | 0.0419 | 0.0570 | 0.0474 |
| wR2 (on F2 for all data) b | 0.0918 | 0.0985 | 0.1088 |
| GOOF | 1.024 | 1.177 | 1.029 |
| Largest diff. peak/hole (e Å−3) | 0.506/−0.710 | 1.257/−1.175 | 1.163/−1.081 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, I.D.; Zimina, A.M.; Anufriev, S.A.; Knyazeva, N.A.; Piskunov, A.V.; Dolgushin, F.M.; Sivaev, I.B. Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− Dicarbollide Ligands. Catalysts 2021, 11, 1409. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111409
Grishin ID, Zimina AM, Anufriev SA, Knyazeva NA, Piskunov AV, Dolgushin FM, Sivaev IB. Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− Dicarbollide Ligands. Catalysts. 2021; 11(11):1409. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111409
Chicago/Turabian StyleGrishin, Ivan D., Anastasiya M. Zimina, Sergey A. Anufriev, Nadezhda A. Knyazeva, Alexander V. Piskunov, Fedor M. Dolgushin, and Igor B. Sivaev. 2021. "Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− Dicarbollide Ligands" Catalysts 11, no. 11: 1409. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111409