
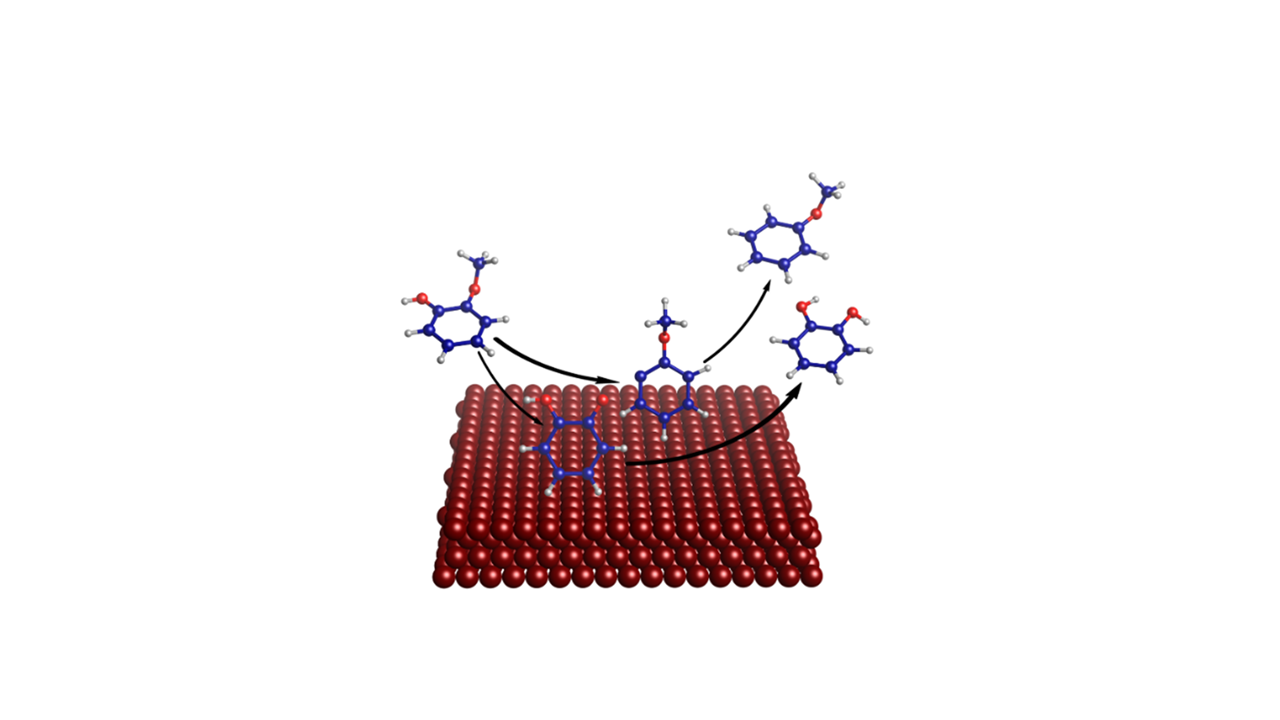
Mechanism of Guaiacol Hydrodeoxygenation on Cu (111): Insights from Density Functional Theory Studies


Abstract
:
1. Introduction
2. Computational Details
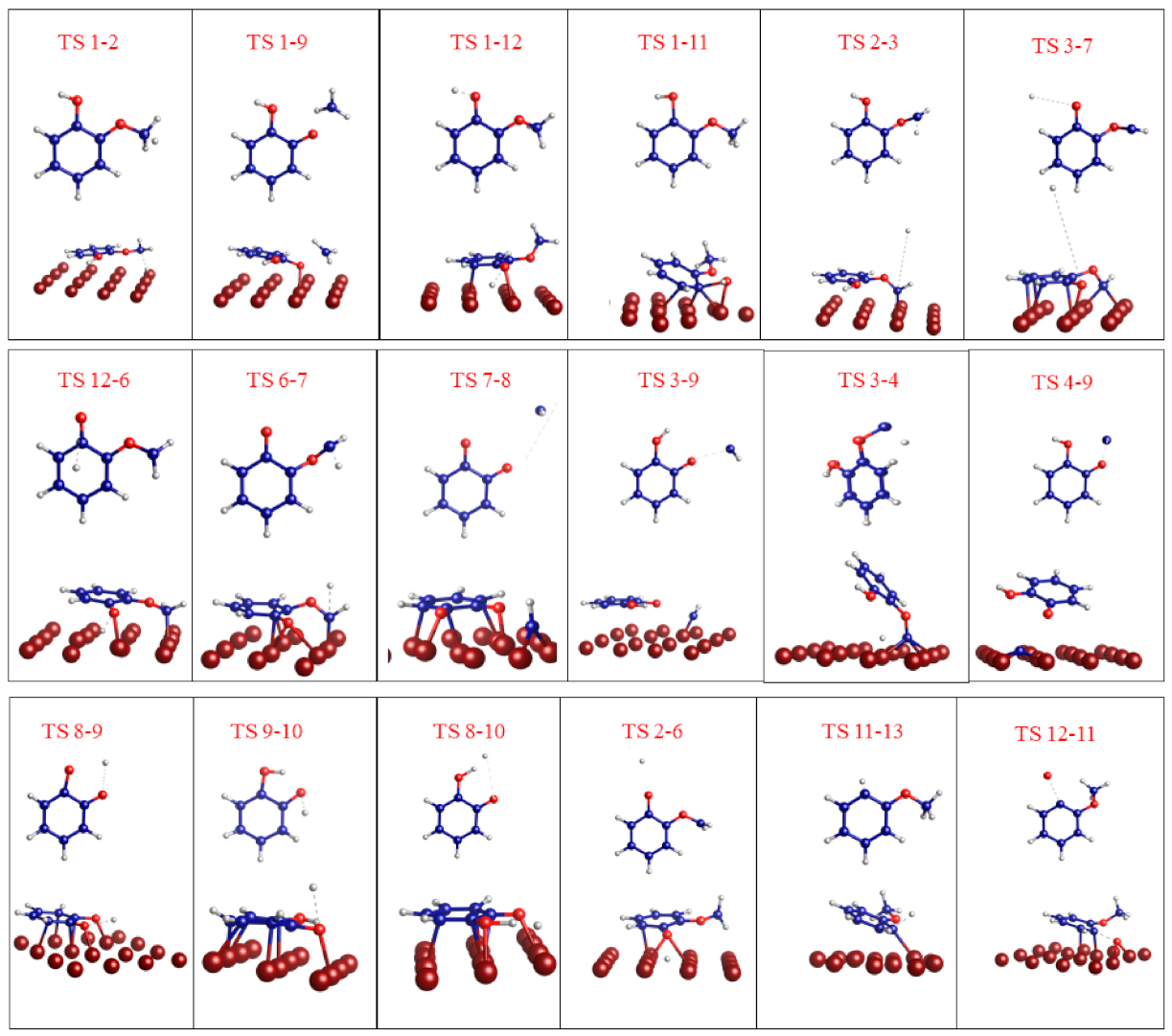
3. Results and Discussion
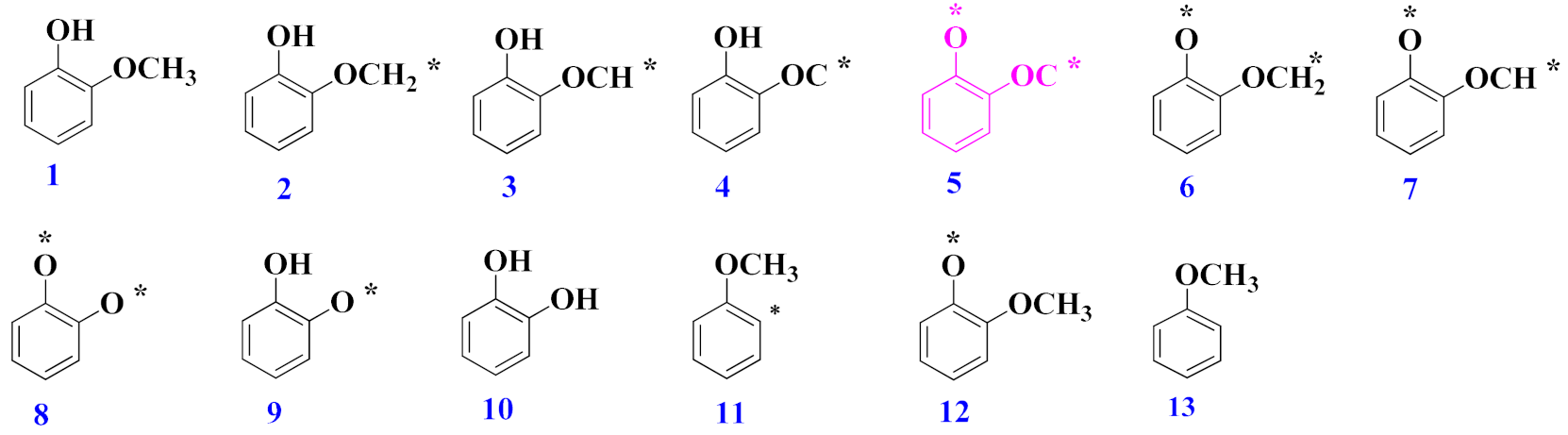
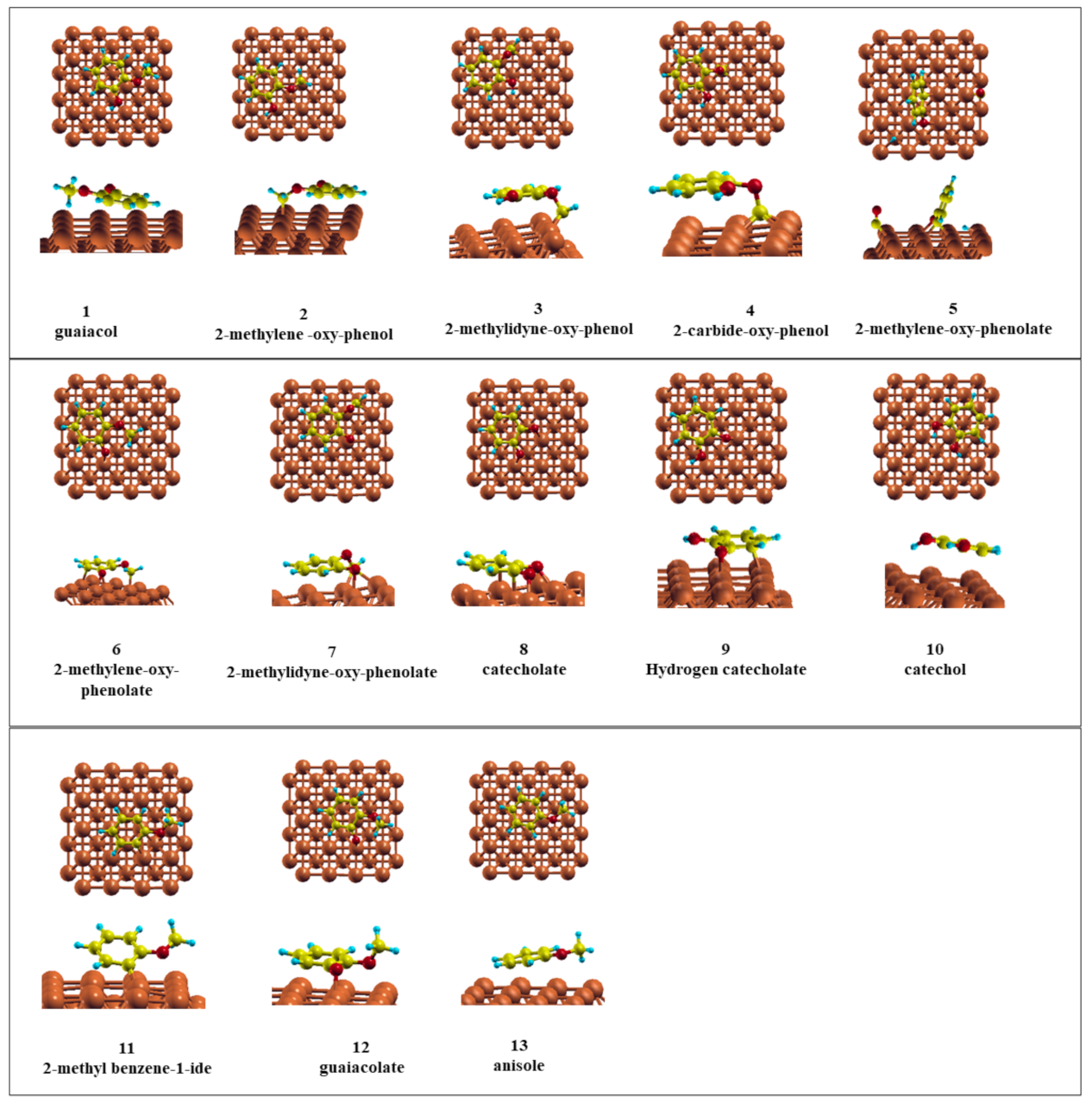
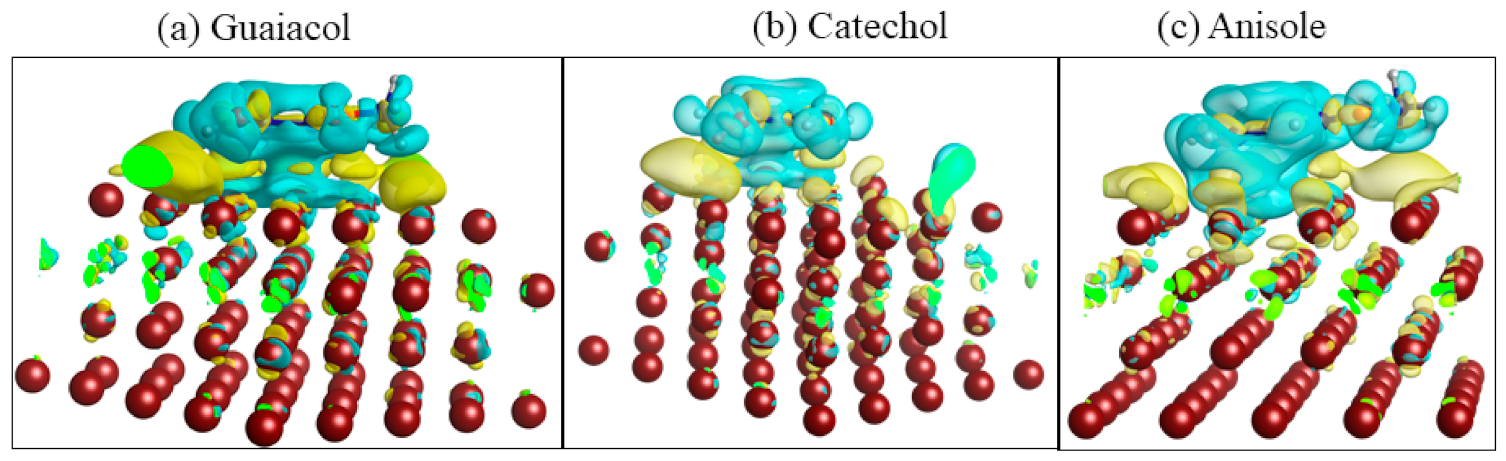
3.1. Adsorption of Desired Compounds and Their Charge Density Difference Plots
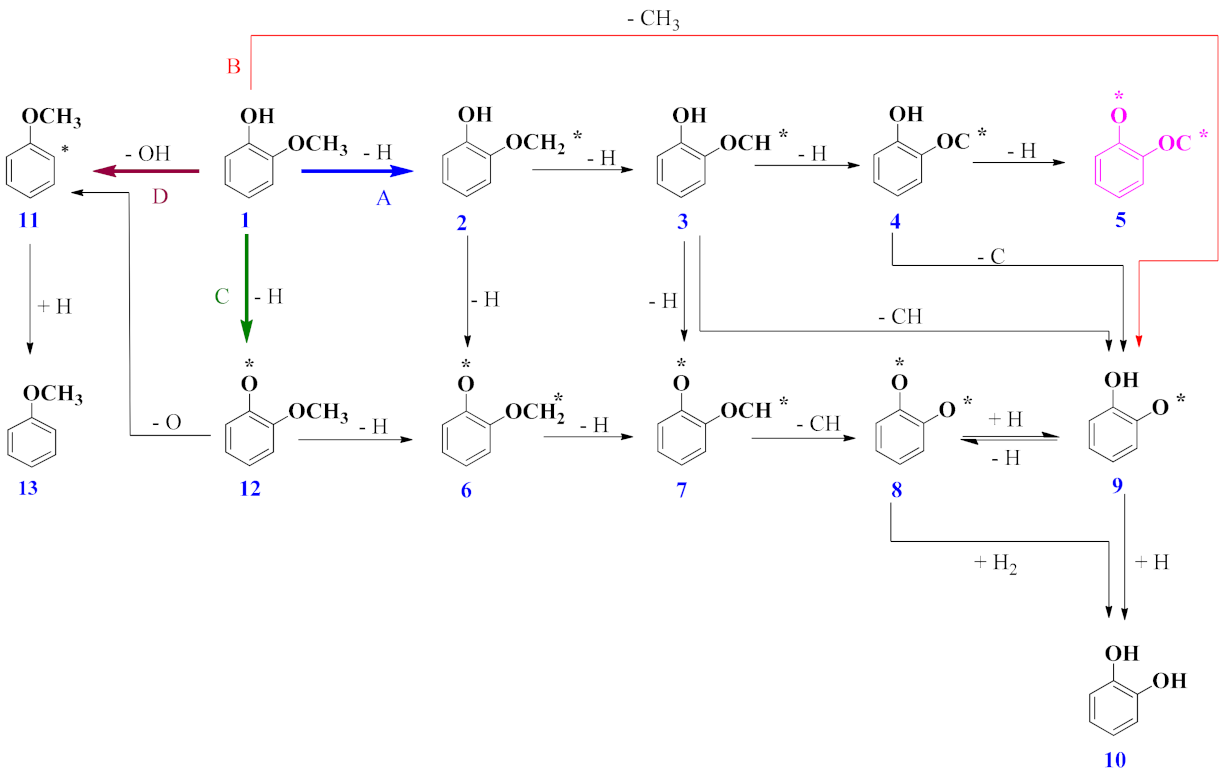
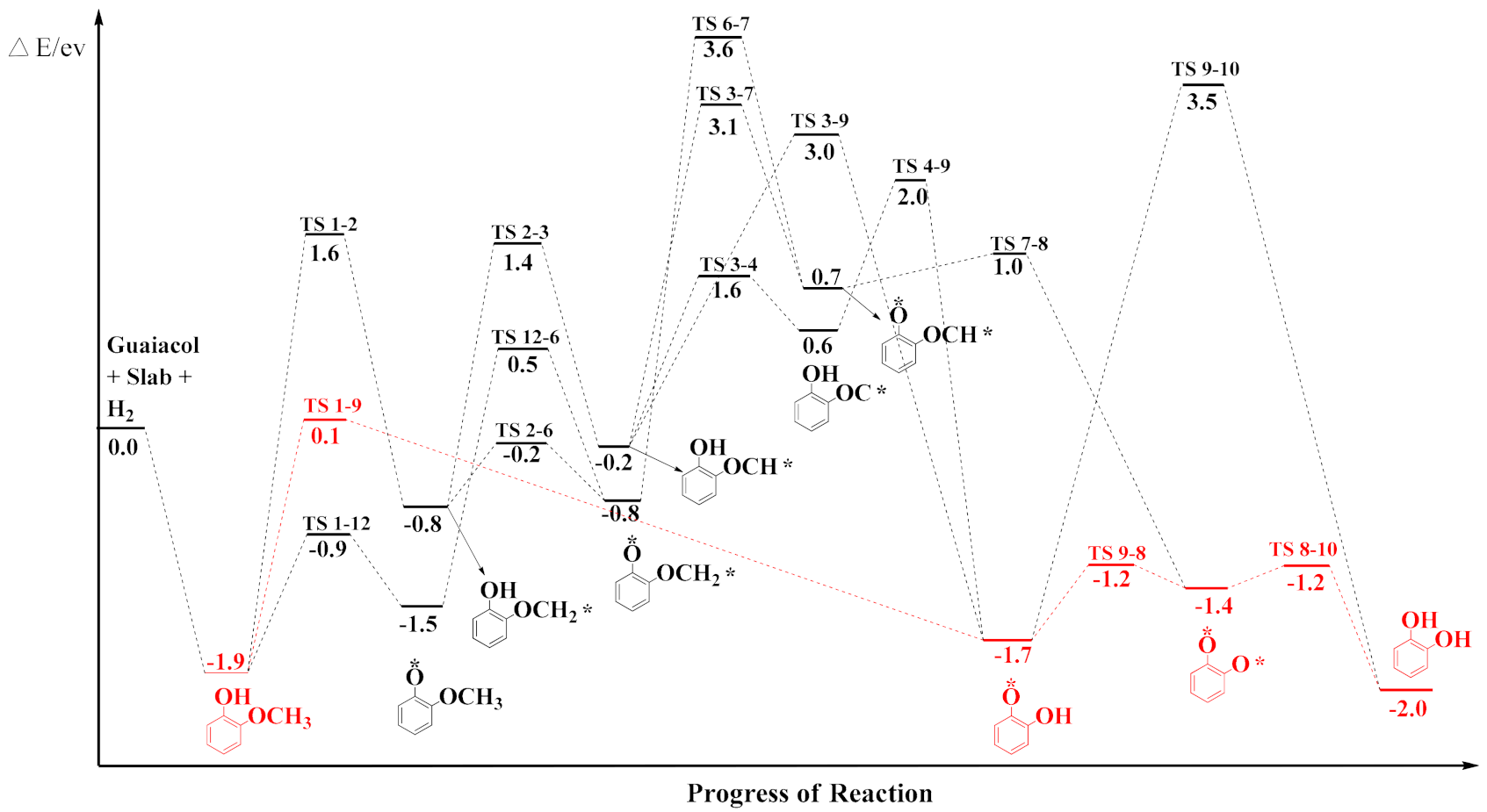
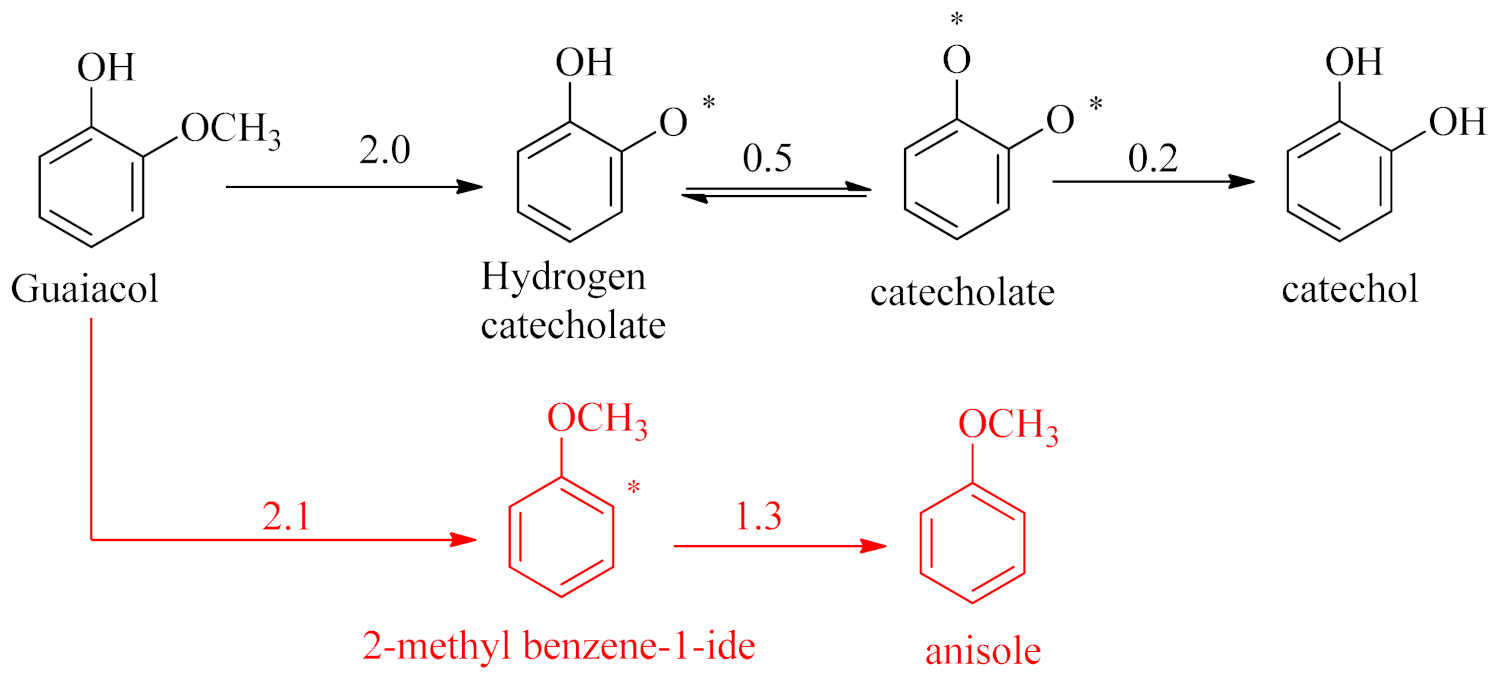
3.2. Mechanism of Catechol Formation
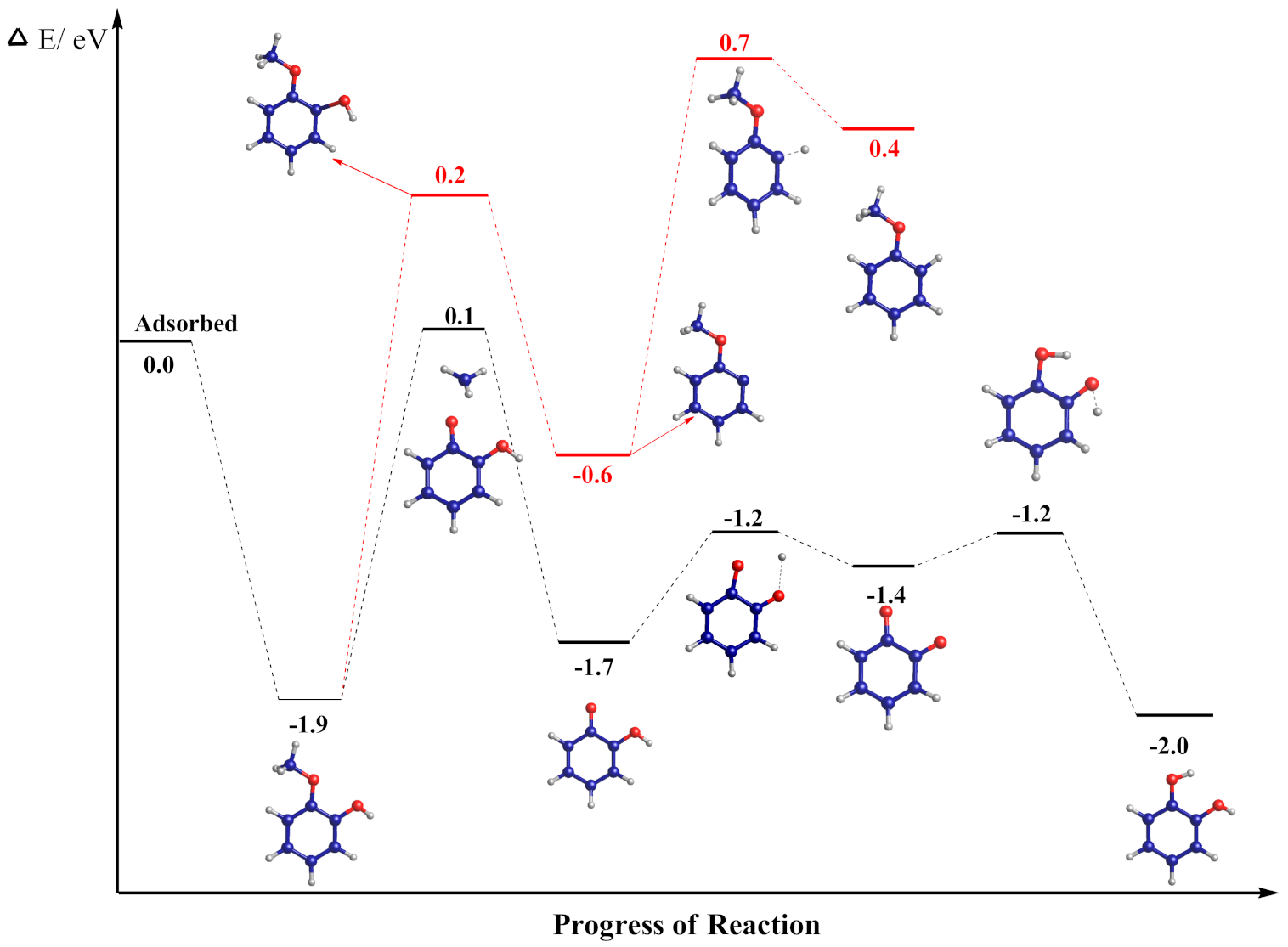
3.3. Mechanism of Anisole Formation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Kunkes, E.L.; Simonetti, D.A.; West, R.M.; Serrano-Ruiz, J.C.; Gärtner, C.A.; Dumesic, J.A. Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes. Science 2008, 322, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A Gen. 2009, 358, 150–156. [Google Scholar] [CrossRef]
- He, Z.; Wang, X.; Laurent, E.; Delmon, B. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28–52. [Google Scholar] [CrossRef]
- Wildschut, J.; Mahfud, F.H.; Venderbosch, R.H.; Heeres, H.J. Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts. Ind. Eng. Chem. Res. 2009, 48, 10324–10334. [Google Scholar] [CrossRef]
- Kumaniaev, I.; Samec, J.S.M. Adsorption isotherms of lignin-derived compounds on a palladium catalyst. Ind. Eng. Chem. Res. 2019, 58. [Google Scholar] [CrossRef]
- Paone, E.; Espro, C.; Pietropaolo, R.; Mauriello, F. Selective arene production from transfer hydrogenolysis of benzyl phenyl ether promoted by a co-precipitated Pd/Fe3O4 catalyst. Catal. Sci. Technol. 2016, 6, 7937–7941. [Google Scholar] [CrossRef]
- Dai, G.; Zhu, Y.; Yang, J.; Pan, Y.; Wang, G.; Reubroycharoen, P.; Wang, S. Mechanism study on the pyrolysis of the typical ether linkages in biomass. Fuel 2019, 249, 146–153. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Murzin, D.Y. Hydrodeoxygenation of lignin-derived phenols: From fundamental studies towards industrial applications. Catalysts 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Wang, Y.; He, M.; Zhao, C. Precise oxygen scission of lignin derived aryl ethers to quantitatively produce aromatic hydrocarbons in water. Green Chem. 2015, 18, 433–441. [Google Scholar] [CrossRef]
- Liu, G.; Robertson, A.W.; Li, M.M.-J.; Kuo, W.C.H.; Darby, M.T.; Muhieddine, M.H.; Lin, Y.-C.; Suenaga, K.; Stamatakis, M.; Warner, J.H.; et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9, 810–816. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Genest, A.; Borgna, A.; Rösch, N. Hydrodeoxygenation of guaiacol over Ru(0001): A DFT study. ACS Catal. 2014, 4, 4178–4188. [Google Scholar] [CrossRef]
- Nimmanwudipong, T.; Aydin, C.; Lu, J.; Runnebaum, R.C.; Brodwater, K.C.; Browning, N.D.; Block, D.E.; Gates, B.C. Selective hydrodeoxygenation of guaiacol catalyzed by platinum supported on magnesium oxide. Catal. Lett. 2012, 142, 1190–1196. [Google Scholar] [CrossRef]
- Wan, H.; Chaudhari, R.V.; Subramaniam, B. Catalytic hydroprocessing of p-cresol: Metal, solvent and mass-transfer effects. Top. Catal. 2012, 55, 129–139. [Google Scholar] [CrossRef]
- Jin, S.; Guan, W.; Tsang, C.-W.; Yan, D.Y.S.; Chan, C.-Y.; Liang, C. Enhanced hydroconversion of lignin-derived oxygen-containing compounds over bulk nickel catalysts though Nb2o5 modification. Catal. Lett. 2017, 147, 2215–2224. [Google Scholar] [CrossRef]
- Zhao, C.; Kou, Y.; Lemonidou, A.A.; Li, X.; Lercher, J.A. Hydrodeoxygenation of bio-derived phenols to hydrocarbons using RANEY® Ni and Nafion/SiO2catalysts. Chem. Commun. 2010, 46, 412–414. [Google Scholar] [CrossRef] [Green Version]
- Ohta, H.; Kobayashi, H.; Hara, K.; Fukuoka, A. Hydrodeoxygenation of phenols as lignin models under acid-free conditions with carbon-supported platinum catalysts. Chem. Commun. 2011, 47, 12209–12211. [Google Scholar] [CrossRef] [Green Version]
- Girgis, M.J.; Gates, B.C. Reactivities, reaction networks, and kinetics in high-pressure catalytic hydroprocessing. Ind. Eng. Chem. Res. 1991, 30, 2021–2058. [Google Scholar] [CrossRef]
- Lesnard, H.; Bocquet, M.-L.; Lorente, N. Dehydrogenation of aromatic molecules under a scanning tunneling microscope: Pathways and inelastic spectroscopy simulations. J. Am. Chem. Soc. 2007, 129, 4298–4305. [Google Scholar] [CrossRef] [PubMed]
- Mittendorfer, F.; Hafner, J. Hydrogenation of benzene on Ni(111)A DFT study. J. Phys. Chem. B 2002, 106, 13299–13305. [Google Scholar] [CrossRef]
- Morin, C.; Simon, D.; Sautet, P. Density-functional study of the adsorption and vibration spectra of benzene molecules on PT(111). J. Phys. Chem. B 2003, 107, 2995–3002. [Google Scholar] [CrossRef]
- Honkela, M.L.; Björk, J.; Persson, M. Computational study of the adsorption and dissociation of phenol on Pt and Rh surfaces. Phys. Chem. Chem. Phys. 2012, 14, 5849. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Johnson, D.M.; Wang, J.; Liu, S.; Zhang, S. Measuring the regional availability of forest biomass for biofuels and the potential of GHG reduction. Energies 2018, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Site, L.D.; Alavi, A.; Abrams, C.F. Adsorption energies and geometries of phenol on the (111) surface of nickel: Anab initiostudy. Phys. Rev. B 2003, 67, 1–3. [Google Scholar] [CrossRef]
- Bonalumi, N.; Vargas, A.; Ferri, D.; Baiker, A. Catalytic chiral metal surfaces generated by adsorption ofO-phenyl derivatives of cinchonidine. J. Phys. Chem. C 2007, 111, 9349–9358. [Google Scholar] [CrossRef]
- Yang, J.; Williams, C.L.; Ramasubramaniam, A.; Dauenhauer, P.J. Aqueous-phase hydrodeoxygenation of highly oxygenated aromatics on platinum. Green Chem. 2014, 16, 675–682. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V.; Lovell, E.; Kan, T.; Weldekidan, H.; He, J.; Dastjerdi, B.; Scott, J. Bio-oil upgrading with catalytic pyrolysis of biomass using Copper/zeolite-Nickel/zeolite and Copper-Nickel/zeolite catalysts. Bioresour. Technol. 2019, 279, 404–409. [Google Scholar] [CrossRef]
- Ben, H.; Ferguson, G.A.; Mu, W.; Pu, Y.; Huang, F.; Jarvis, M.; Biddy, M.; Deng, Y.; Ragauskas, A.J. Hydrodeoxygenation by deuterium gas–A powerful way to provide insight into the reaction mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 19138–19142. [Google Scholar] [CrossRef]
- Jenkins, S.J. Aromatic adsorption on metals via first-principles density functional theory. Proc. R. Soc. A Math. Phys. Eng. Sci. 2009, 465, 2949–2976. [Google Scholar] [CrossRef]
- Badawi, M.; Paul, J.-F.; Payen, E.; Romero, Y.; Richard, F.; Brunet, S.; Popov, A.; Kondratieva, E.; Gilson, J.-P.; Mariey, L.; et al. Hydrodeoxygenation of phenolic compounds by sulfided (Co) Mo/Al2O3 catalysts, a combined experimental and theoretical study. Oil Gas Sci. Technol. Rev. de l’IFP 2013, 68, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Gu, G.H.; Mullen, C.A.; Boateng, A.A.; Vlachos, D.G. Guaiacol hydrodeoxygenation mechanism on PT(111): Insights from density functional theory and linear free energy relations. ChemSusChem 2014, 8, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Greeley, J.P.; Nørskov, J.K.; Mavrikakis, M. Electronicstructure andcatalysis onmetalsurfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Runnebaum, R.C.; Nimmanwudipong, T.; Block, D.E.; Gates, B.C. Catalytic conversion of compounds representative of lignin-derived bio-oils: A reaction network for guaiacol, anisole, 4-methylanisole, and cyclohexanone conversion catalysed by Pt/γ-Al2O3. Catal. Sci. Technol. 2012, 2, 113–118. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Erxn/eV | Ea/eV | TS |
|---|---|---|---|
| C6H4(OH)(OCH2) → C6H4(OH)(OCH) + H | −0.22 | 2.16 | TS 2–3 |
| C6H4(OH)(OCH) → C6H4(O)(OCH) + H | 0.74 | 3.34 | TS 3–7 |
| C6H4(O)(OCH3) → C6H4(O)(OCH2) + H | −0.75 | 1.98 | TS 12–6 |
| C6H4(O)(OCH2) → C6H4(O)(OCH) + H | 0.74 | 4.39 | TS 6–7 |
| C6H4(O)(OCH) → C6H4(O)(O) + CH | −1.43 | 0.33 | TS 7–8 |
| C6H4(OH)(OCH) → C6H4(OH)(O) + CH | −1.66 | 3.03 | TS 3–9 |
| C6H4(OH)(OCH) → C6H4(OH)(OC) + H | 0.57 | 0.64 | TS 3–4 |
| C6H4(OH)(OC) → C6H4(OH)(O) + C | −1.70 | 1.39 | TS 4–9 |
| C6H4(O)(O) + H → C6H4(O)(OH) | −1.66 | 0.17 | TS 8–9 |
| C6H4(O)(OH) + H → C6H4(OH)(OH) | −2.03 | 5.17 | TS 9–10 |
| C6H4(O)(O) + H2 → C6H4(OH)(OH) | −2.03 | 0.22 | TS 8–10 |
| C6H4(OH)(OCH2) → C6H4(O)(OCH2) + H | −0.75 | 0.60 | TS 2–6 |
| C6H4(OCH3) + H → C6H5(OCH3) | 0.41 | 1.25 | TS 11–13 |
| C6H4(O)(OCH3) → C6H4(OCH3) + O | −0.61 | 2.65 | TS 12–11 |
| C6H4(OH)(OCH3) → C6H4(OH)(OCH2) + H | −0.78 | 3.50 | TS 1–2 |
| C6H4(OH)(OCH3) → C6H4(OH)(O) + CH3 | −1.70 | 1.97 | TS 1–9 |
| C6H4(OH)(OCH3) → C6H4(O)(OCH3) + H | −1.50 | 0.99 | TS 1–12 |
| C6H4(OH)(OCH3) → C6H4(OCH3) + OH | −0.61 | 2.07 | TS 1–11 |
| Compound | Eads/eV | d (Cu–C)/Å |
|---|---|---|
| Guaiacol | −1.90 | 2.61 |
| Catechol | −2.18 | 2.59 |
| Anisole | −0.72 | 2.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konadu, D.; Kwawu, C.R.; Tia, R.; Adei, E.; de Leeuw, N.H. Mechanism of Guaiacol Hydrodeoxygenation on Cu (111): Insights from Density Functional Theory Studies. Catalysts 2021, 11, 523. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11040523
Konadu D, Kwawu CR, Tia R, Adei E, de Leeuw NH. Mechanism of Guaiacol Hydrodeoxygenation on Cu (111): Insights from Density Functional Theory Studies. Catalysts. 2021; 11(4):523. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11040523
Chicago/Turabian StyleKonadu, Destiny, Caroline Rosemyya Kwawu, Richard Tia, Evans Adei, and Nora Henriette de Leeuw. 2021. "Mechanism of Guaiacol Hydrodeoxygenation on Cu (111): Insights from Density Functional Theory Studies" Catalysts 11, no. 4: 523. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11040523