
Design of Iridium N-Heterocyclic Carbene Amino Acid Catalysts for Asymmetric Transfer Hydrogenation of Aryl Ketones


Department of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(6), 671; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11060671
Submission received: 24 April 2021
/
Revised: 20 May 2021
/
Accepted: 21 May 2021
/
Published: 24 May 2021
(This article belongs to the Special Issue 10th Anniversary of Catalysts: Molecular Catalysis)
Abstract
:A series of chiral complexes of the form Ir(NHC)2(aa)(H)(X) (NHC = N-heterocyclic carbene, aa = chelated amino acid, X = halide) was synthesized by oxidative addition of α-amino acids to iridium(I) bis-NHC compounds and screened for asymmetric transfer hydrogenation of ketones. Following optimization of the reaction conditions, NHC, and amino acid ligands, high enantioselectivity was achieved when employing the Ir(IMe)2(l-Pro)(H)(I) catalyst (IMe = 1,3-dimethylimidazol-2-ylidene), which asymmetrically reduces a range of acetophenone derivatives in up to 95% enantiomeric excess.
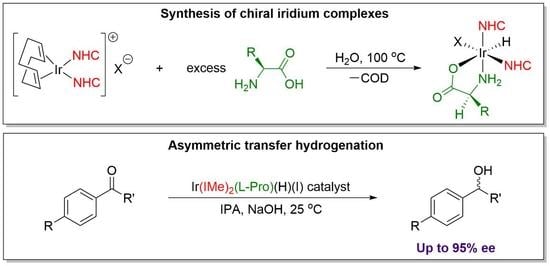
1. Introduction
The production of optically pure alcohols is is an essential transformation in organic synthesis, with particular relevance in the pharmaceutical, agrochemical, and fine chemical industries [1]. A common and practical approach to these compounds in academia and industry is the enantioselective reduction of ketones to chiral alcohols using homogeneous transition metal catalysts (Scheme 1) [2]. The synthesis of an optically pure alcohol via addition of H2 across the C=O bond of a prochiral ketone in the presence of a chiral catalyst is known as asymmetric hydrogenation. This process is referred to as asymmetric transfer hydrogenation (ATH) when it is carried out using a hydrogen source other than H2, typically in the presence of a suitable hydrogen donor molecule and base.
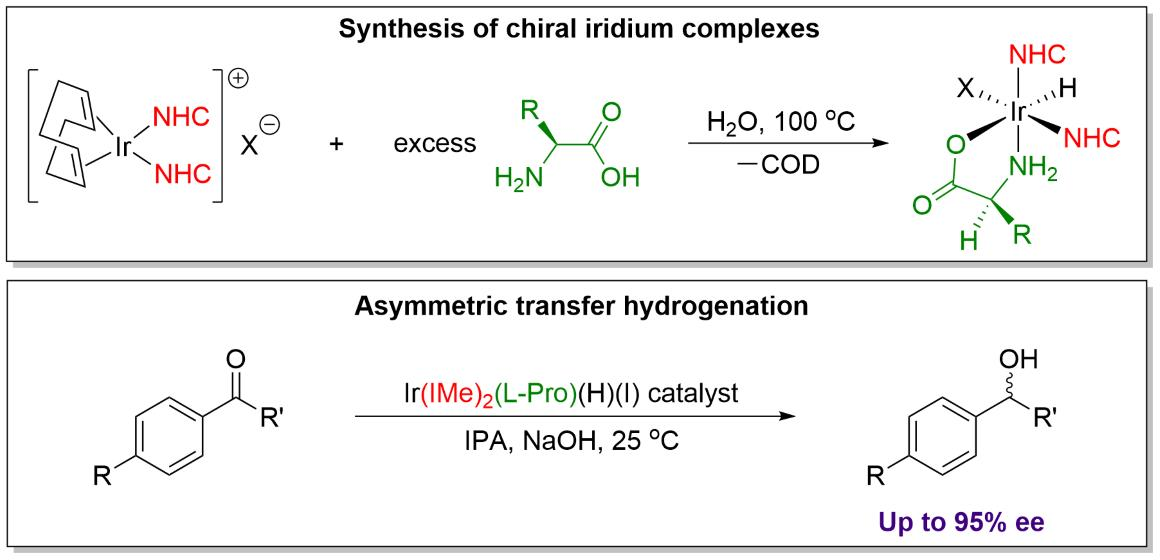
The major breakthrough in asymmetric transfer hydrogenation came in 1995 from Noyori and coworkers, with the synthesis of a ruthenium(II) arene catalyst featuring a chiral mono-tosylated DPEN ligand (Figure 1) [3,4,5,6]. The catalyst is highly active for enantioselective reduction of a range of aromatic ketones, generating the chiral alcohol products in up to 99% enantiomeric excess (ee). Noyori’s system operates through an outer-sphere transfer hydrogenation mechanism in which the substrate does not bind directly to the metal center, with hydrogen transfer instead occurring via a highly ordered cyclic transition state between the catalyst and substrate [7,8,9,10,11]. This concept of a “metal-ligand bifunctional catalyst” [12,13,14,15,16] is widely used today in a vast number of hydrogenation systems, and numerous related chiral catalysts and ligands have been investigated for ATH. Many successful contemporary ATH catalysts often employ noble metals such as ruthenium, rhodium, and iridium in combination with a variety of chiral ligands, the most common being derived from diamines, amino alcohols, and diphosphines [17,18,19,20,21,22,23].
While ATH is well-studied, ligands frequently used in these transformations, such as TsDPEN or chiral amino alcohols, are often synthetically challenging and not particularly cost-effective. Since many successful ATH catalysts make use of rather expensive noble metals, the incorporation of cheaper chiral ligands would be highly desirable. Amino acids are an intriguing class of ligand candidates due to their vast array of structural diversity and relatively low cost (∼$1 per gram). A number of studies have demonstrated the promising potential of transition metal catalysts featuring amino acid ligands for asymmetric transfer hydrogenation [24,25,26,27,28,29,30,31,32], including rhodium and iridium Cp* amino acid catalysts previously developed by the Merola group [33,34].
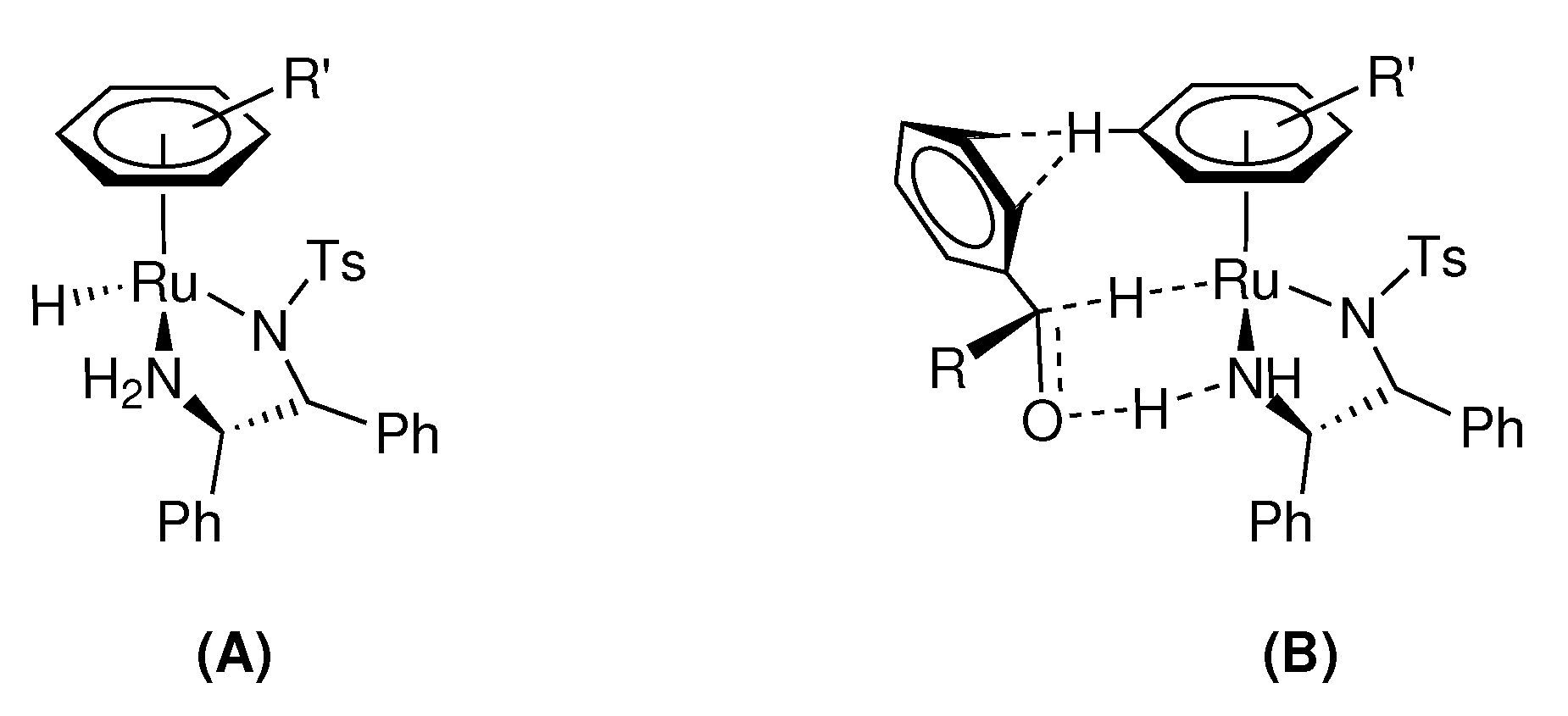
N-Heterocyclic carbenes (NHCs) have also emerged as ligands of interest for asymmetric transformations due to their their strong σ-donor properties, inertness, and vast structural tunabilility [35,36], allowing for the design of various unique chiral scaffolds [37,38,39]. Although noble metal NHC complexes generally display excellent activity for catalytic transfer hydrogenation, examples of highly enantioselective catalysts of this type remain relatively scarce [2,40,41,42,43]. Iridium NHC complexes have been extensively studied for ATH of ketones; however, the majority of these catalysts employ synthetically challenging chiral NHC ligands that generate the alcohol products in only low to moderate enantiomeric excess (Figure 2) [44,45,46,47,48,49,50,51,52].
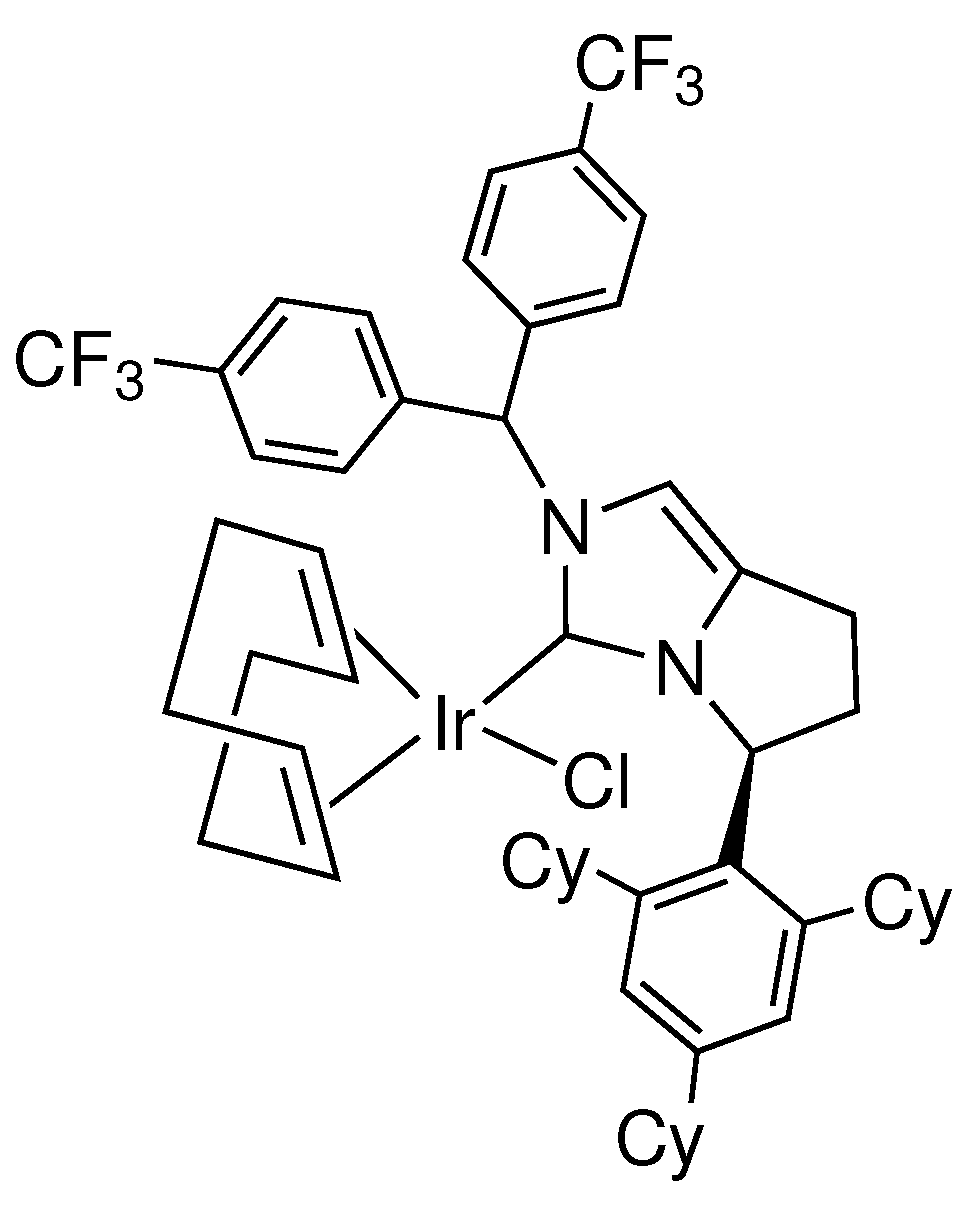
A rare example of a highly enantioselective noble metal NHC catalyst for ATH of ketones was reported by Yoshida et al. in 2015 [53]. The system incorporated a series of chiral bicyclic NHC ligands featuring a variety of different substituents. Optimal results were achieved when employing the catalyst variant shown in Figure 3, which reduces a variety of acetophenone derivatives in as high as 97% enantiomeric excess. Similar to many of the chiral ligands that have previously been investigated for asymmetric catalysis, preparation of the NHC precursors requires a multistep synthetic pathway in combination with several chromatographic separation techniques.
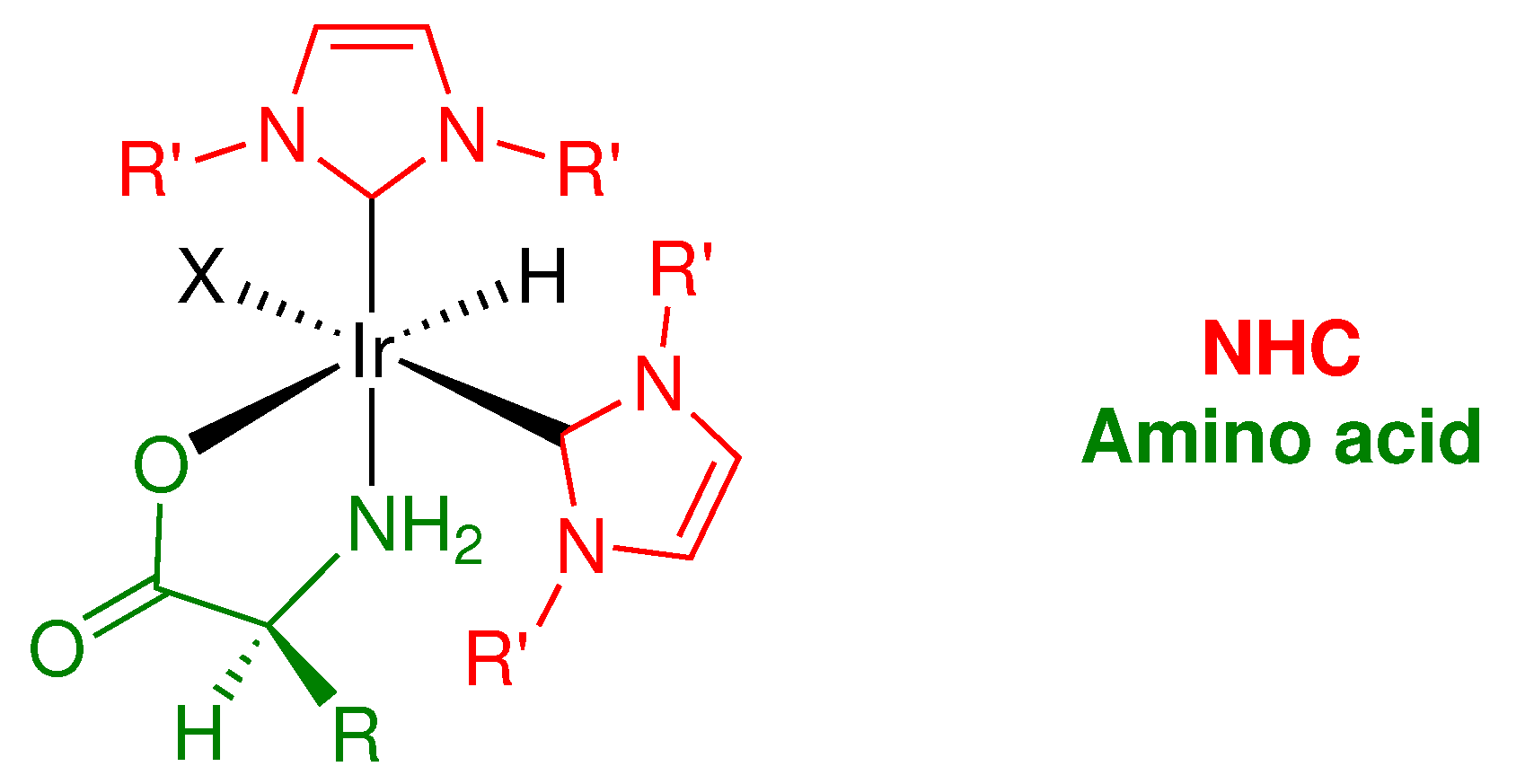
Given how frequently transition metal catalysts featuring NHC ligands are reported in contemporary literature, the lack of noble metal NHC catalysts displaying practical enantioselectivity for ATH of ketones is rather surprising. Although there are many examples of highly enantioselective direct asymmetric hydrogenation catalysts of this type [38], transfer hydrogenation is typically preferred due to operational simplicity, as it eliminates the need for for a pressured vessel of H2 gas in the experimental setup, and often uses inexpensive, readily available chemicals as the hydrogen donor instead. In this article, we report an iridium NHC catalytic system that gives good to excellent enantioselectivity for a range of aryl ketone substrates. The catalysts designed in this work have all the benefits typically provided by strongly σ-donating NHC spectator ligands, but incorporate simple, synthetically accessible NHC scaffolds in combination with with “off-the-shelf” chiral α-amino acid ligands, rather than costly or synthetically challenging chiral ligands usually employed in asymmetric transformations (Figure 4). This consequently allows for easy fine-tuning and optimization of the catalytic system for asymmetric transfer hydrogenation of ketones.
2. Results and Discussion
2.1. Synthesis
Iridium COD bis-NHC complexes (COD = 1,5-cyclooctadiene) were found to undergo oxidative addition with a range of α-amino acids, generating complexes of the form Ir(NHC)2(aa)(H)(X), where aa = chelated amino acid and X = halide (Scheme 2). A Schlenk flask was charged with the desired iridium COD bis-NHC complex, amino acid, and water. Upon heating the mixture to 100 °C, both reagents fully dissolved, and the solution was allowed to react for a period of 12 h, over which time the color changed from bright orange to light yellow. Following organic workup, the products were isolated as off-white solids in variable yields (Table 1).
2.2. Characterization
2.2.1. X-ray Crystallography
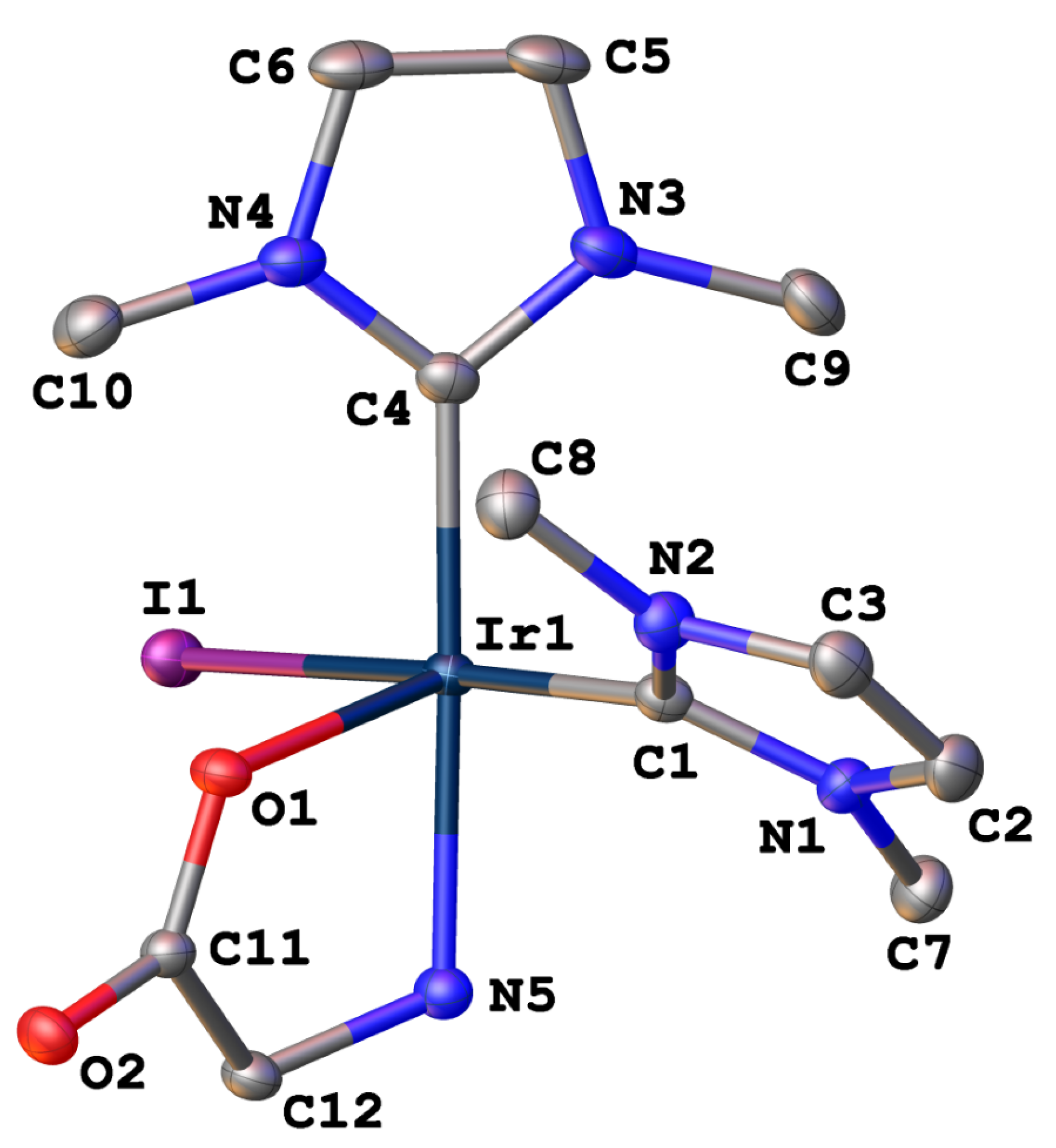
Early in our studies, a yellow single-crystal of Ir(IMe)2(Gly)(H)(I) suitable for X-ray diffraction was collected from an NMR sample in CDCl3, providing insight into the chemical connectivity of these complexes. The solved crystal structure of the glycine complex is shown in Figure 5, and selected bond lengths and angles are provided in Table 2. The complex crystallized in the triclinic space group P (#2). The geometry around the iridium center in pseudo-octahedral, with the largest angle (C4–Ir1–O1) having a deviation of 11.52° from the ideal 90° due to the bidentate nature of the amino acid ligand. The Ir1–C1 bond length (NHC trans to the iodide) is 1.986 Å, while the Ir1–C4 bond length (NHC trans to the chelated nitrogen) is 2.004 Å, giving an average Ir–NHC bond distance of 1.995 Å. Both Ir–NHC bond lengths in Ir(IMe)2(Gly)(H)(I) fall within the range of similar complexes that have been published in the literature, with a search of the Cambridge Structural Database (CSD) giving 75 hits for previously reported octahedral IrIII bis-NHC complexes, which feature Ir–NHC bond lengths ranging from 1.910–2.116 Å.
Though hydride ligands in noble metal complexes are often difficult to detect by single-crystal X-ray diffraction, a hydride resonance is observed far upfield in the 1H NMR spectra of Ir(IMe)(Gly)(H)(I) at −24.33 ppm and it is apparent that based on the octahedral geometry of the crystal structure, the hydride is located trans to the chelated oxygen of the amino acid. This aligns the hydride and amine protons in a fashion that should be well-suited for outer-sphere transfer hydrogenation, although this crystal structure is not necessarily representative of the structural orientation in solution. The NHC ligands in Ir(IMe)(Gly)(H)(I) appear to be rotated in a particular orientation to minimize steric interactions, with the N1 and N3 substituents of the NHC ligands directed towards the hydride face of the metal. This likely has ramifications in employing these complexes as catalysts for ATH, as longer chain or bulkier N-substituents may obstruct the approach or alignment of an incoming substrate. It should be noted that four unique resonances arising from the NHC methyl groups are observed in the 1H NMR spectra of these complexes, indicating restricted rotation about the Ir–NHC bonds.
2.2.2. Stereochemistry
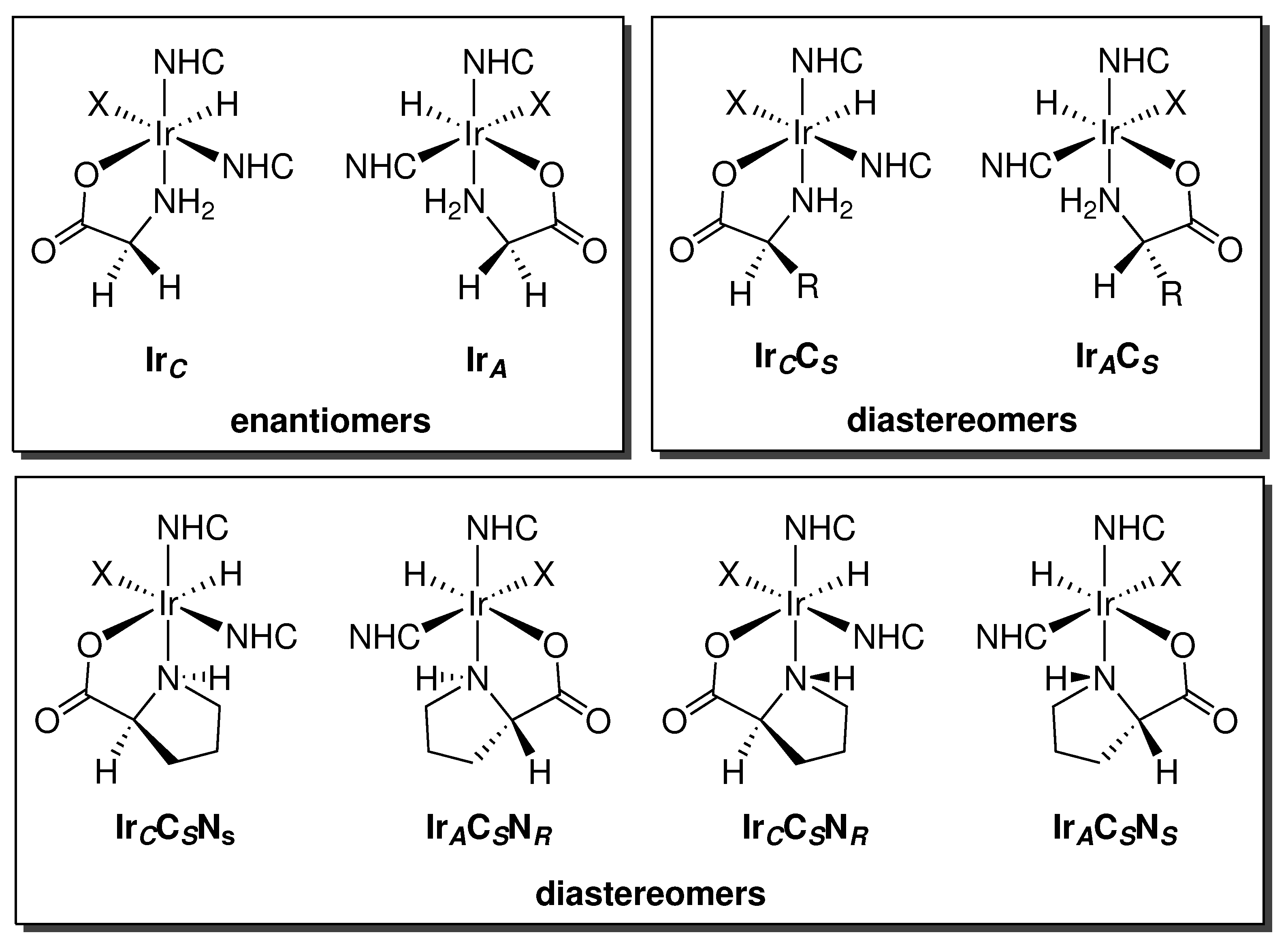
Using the crystal structure of the glycine complex as a template for chemical connectivity, it is apparent that upon oxidative addition of the amino acid, an octahedral stereogenic metal center is formed. The simplest amino acid glycine, which is achiral, produces two metal enantiomers having absolute configurations of C or A at the iridium center. When homochiral primary amino acids are used, two metal diastereomers of the form IrCCS or IrACS are generated, where S represents the configuration at the α-carbon of the amino acid. Amino acids containing secondary amines, such as proline, have an additional center of chirality at the nitrogen. This leads to the potential formation of four possible diastereomeric products: IrCCSNS, IrCCSNR, IrACSNS, and IrACSNR (Figure 6).
2.2.3. 1H NMR Spectroscopy
The hydride resonances in the 1H NMR spectra (DMSO-d6) of these complexes are observed far upfield beyond −23 ppm. As expected, only one hydride resonance is observed in the spectra of the glycine complex, as the two metal enantiomers are indistinguishable by 1H NMR spectroscopy. Complexes featuring homochiral primary amino acids (Ala, Val, and Phg) display two hydride resonances, confirming the formation of two metal diastereomers (Figure 7). The diastereomeric ratios were determined by integration of these two resonances in order to distinguish between the major and minor isomers. The 1H NMR spectra are significantly more complicated upon the incorporation of secondary amino acids (Aze, Pro, Pip), with numerous hydride resonances observed beyond −24 ppm. This is attributable to the formation of two additional metal diastereomers arising from the extra chiral center on the nitrogen as expected, but also from the presence of other structural isomers. The number and ratios of the hydride resonances in the secondary amino acid complexes were found to change significantly over time in different NMR solvents, suggesting various exchange processes are occurring.
2.3. Asymmetric Transfer Hydrogenation of Ketones
2.3.1. Initial Catalytic Studies
Catalytic activity for transfer hydrogenation was initially investigated using the substrate acetophenone. Three catalysts were screened in these preliminary studies, all having the same NHC ligand (IMe), but different amino acids (Gly, l-Ala, l-Pro). Trials were carried out in water employing sodium formate as the hydrogen donor, and in isopropyl alcohol with sodium hydroxide base (Scheme 3). After 24 h, the catalyst was extracted from the reaction mixture, and catalytic activity was assessed via 1H NMR spectroscopy to observe any significant conversion of acetophenone to 1-phenylethanol. Reactions carried out in water displayed only moderate activity, reducing acetophenone in under 60% conversion after 24 h. Trials in IPA were more promising, particularly the catalyst containing the secondary amino acid l-proline (Table 3).
Since these catalysts were active for transfer hydrogenation in the NMR studies, catalytic trials in IPA were conducted again, this time screening for asymmetric reduction. A wider scope of catalysts were employed, all having the same NHC ligand (IMe), but a variety of primary and secondary amino acids. Conversion and enantiomeric excess were quantified after 24 h by chiral gas chromatography. As expected, the catalyst containing the achiral amino acid glycine generated a racemic mixture of the 1-phenylethanol product. While catalysts featuring l-Ala, l-Val, l-Phg, l-Aze, and l-Pip all displayed poor enantioselectivity, the l-proline variant reduced acetophenone in 68% ee of the R-enantiomer. Incorporation of d-proline generated the opposite configuration of the the alcohol product (S) in the same enantiomeric excess (Table 4).
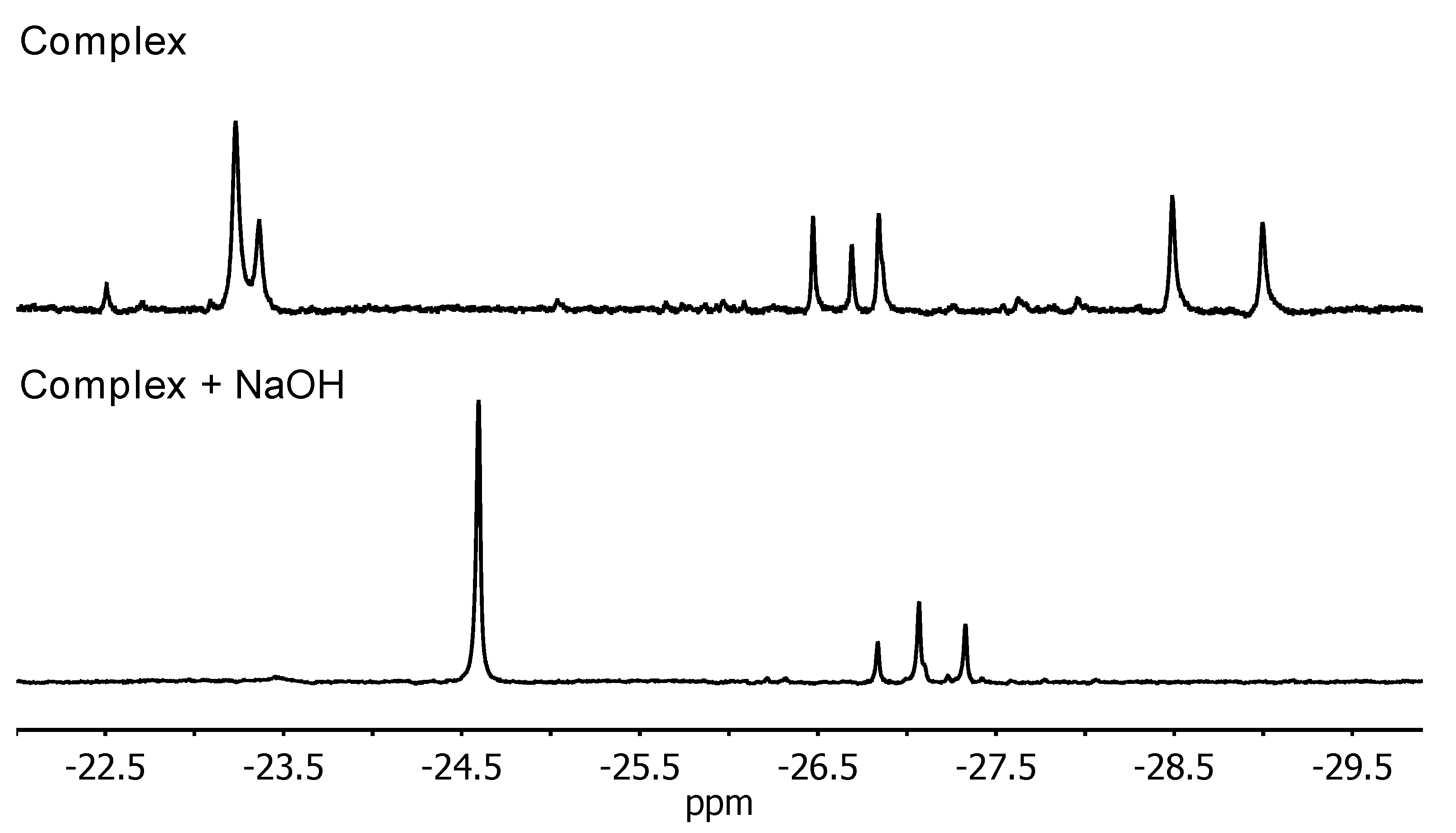
In order to gain insight into the active catalytic species, room temperature 1H NMR spectra (IPA, DMSO-d6 insert) of Ir(IMe)2(l-Pro)(H)(I) were acquired before and after the addition of base (Figure 8). As mentioned previously, a variety of hydride resonances were observed in the spectrum of just the initial complex. Upon adding 2 equivalents of NaOH, there was a notable change in the hydride region. One major resonance emerged at −24.60 ppm, accompanied by three minor resonances at −26.84, −27.07 and −27.33 ppm. This may indicate the presence of four potentially active catalytic species as suggested earlier, presumably generated by dissociation of the iodide anion. The downfield portion of the spectrum still featured numerous overlapping resonances, and the precise structures of these species could not be elucidated from this experiment.
2.3.2. Optimization: Temperature
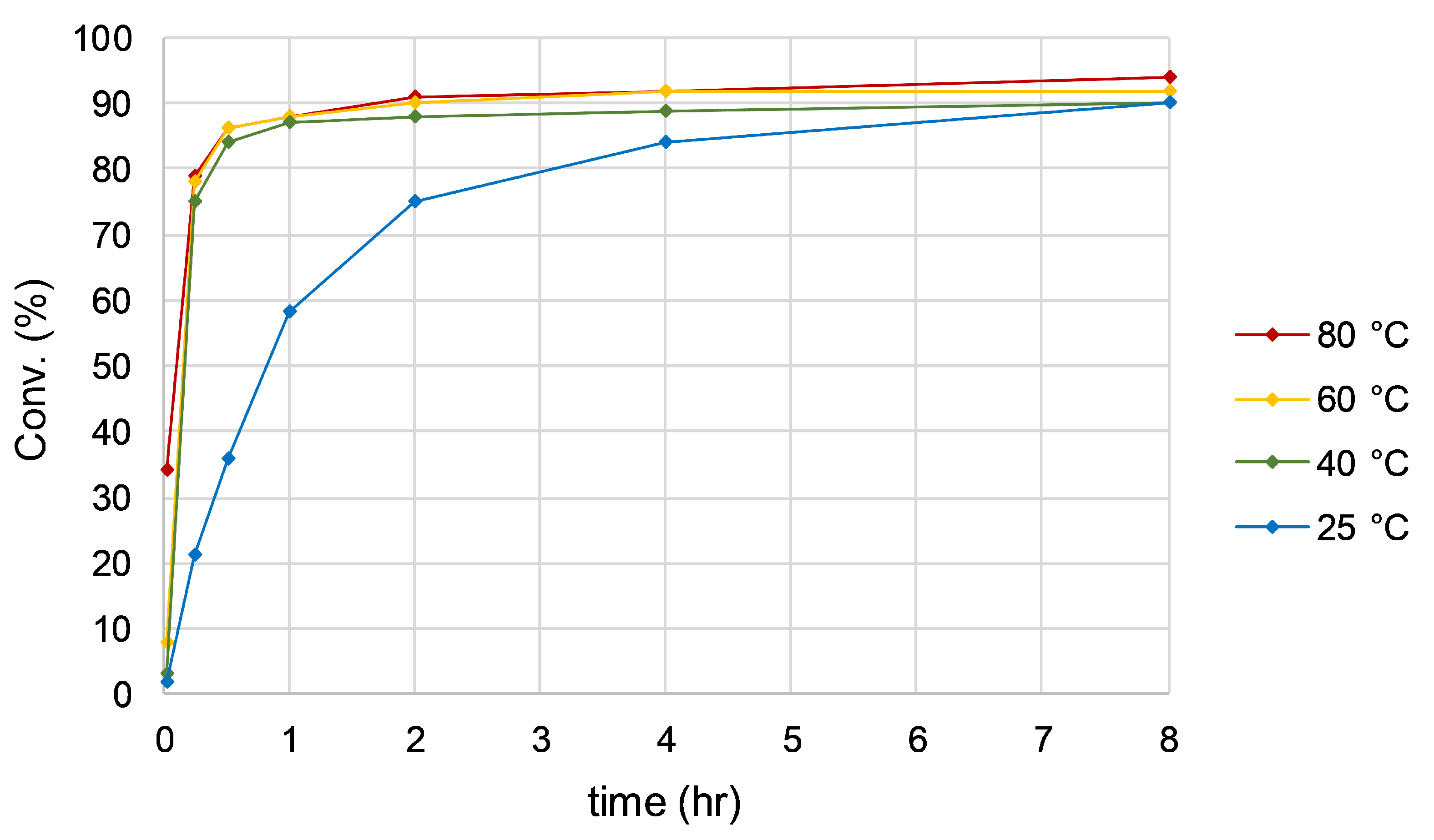
Catalytic trials were carried out at temperatures of 25, 40, 60, and 80 °C using 2.5 mol% of Ir(IMe)2(l-Pro)(H)(I) and 2 equivalents of NaOH in isopropyl alcohol. All reactions were monitored by chiral GC at specific time increments over a period of 48 h in order to quantify conversion (Figure 9) and enantiomeric excess (Figure 10) over the course of the catalysis. No significant induction period was observed for any of the trials, with the catalysis beginning immediately upon addition of base. This was attributed to the presence of a hydride on the complex prior to catalysis initiation. As expected, catalytic activity is higher at elevated temperatures, giving initial turnover frequencies as high as 1029 h−1 at 80 °C, compared to 57 h−1 at 25 °C. Conversion began to level off over time in all trials, implying the active catalyst could be degrading.
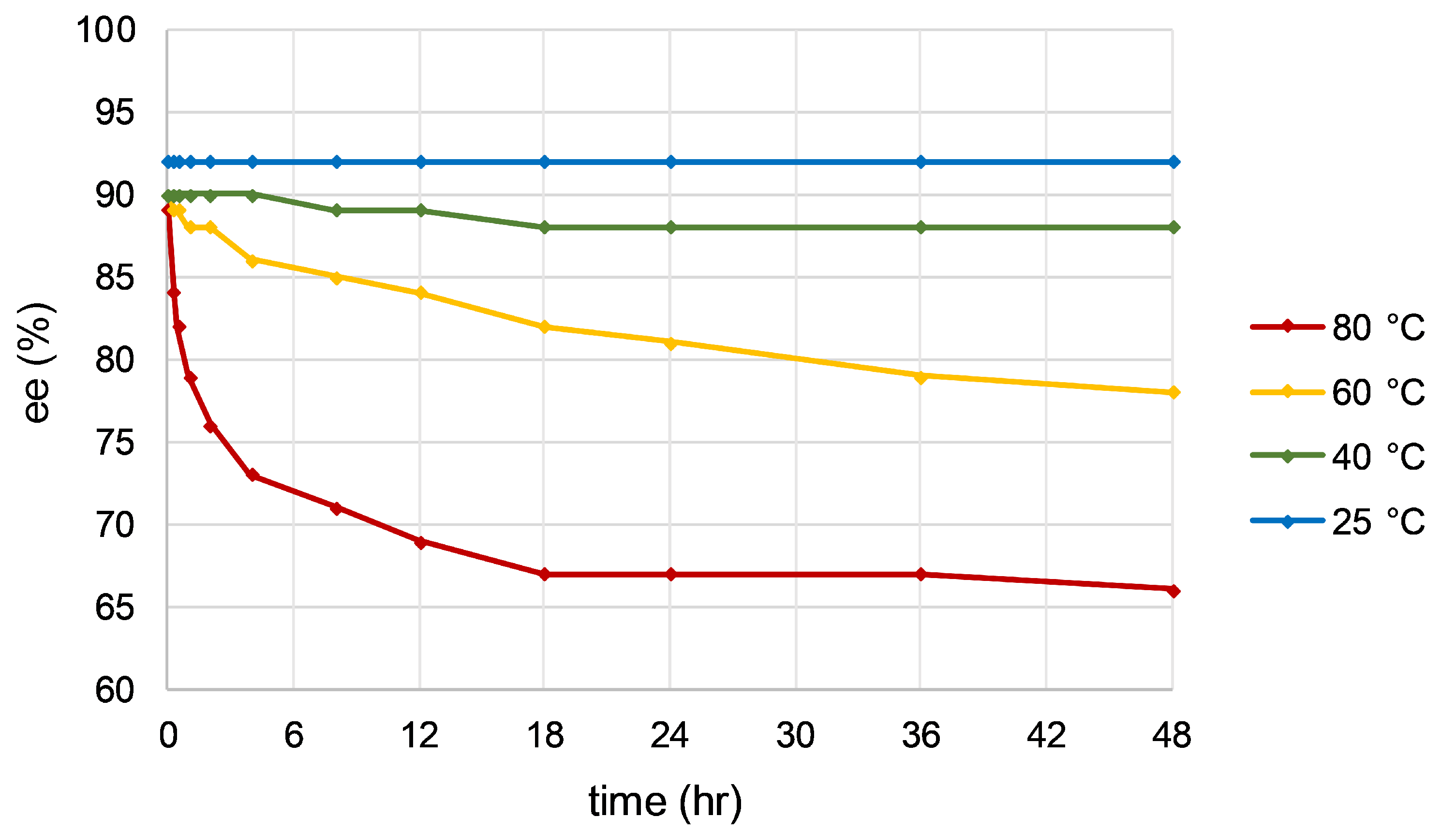
Optimal enantioselectivity was achieved when carrying out the catalysis at 25 °C, with generation of R-1-phenylethanol in 92% ee, a notable increase from the 68% ee observed in the initial ATH studies. Enantiomeric excess of the product gradually decreased over the course of the reaction at higher temperatures, most notably at 80 °C, where the ee fell from 89% to 66% after 48 h. The phenomenon is often attributed to a higher energy metal diastereomeric transition state becoming relatively more favorable at elevated temperatures, though it is possible higher temperatures facilitate product racemization by an achiral metal species generated via catalyst degradation.
2.3.3. Optimization: Base
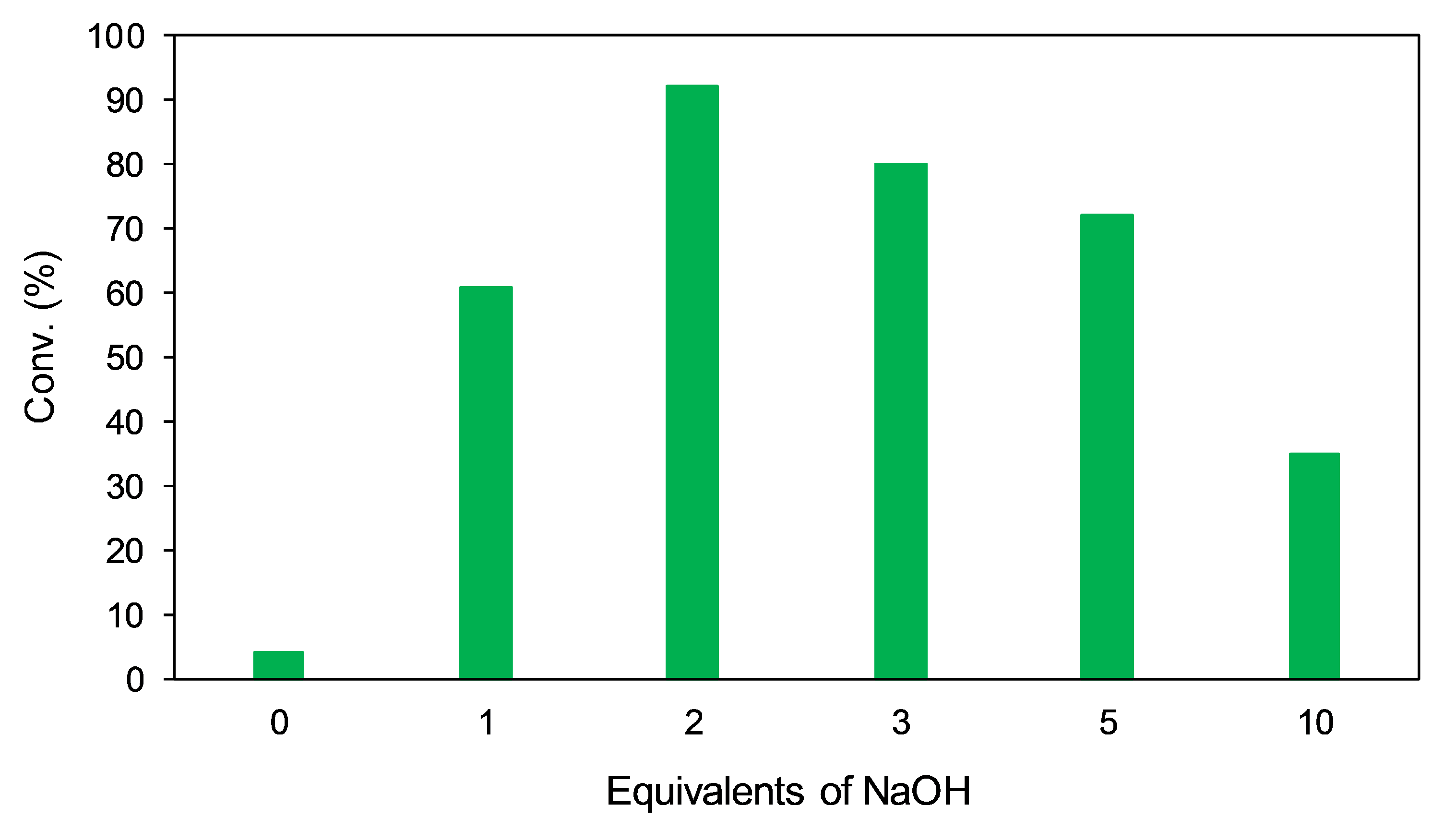
A range of different bases was screened using 2.5 mol% of the Ir(IMe)2(l-Pro)(H)(I) catalyst in IPA at 25 °C over a period of 24 h. Practically no substrate conversion was observed when employing the weak bases NaCO, NaHCO, or HCOONa. This is likely attributable to slower regeneration of the active catalyst, as weaker bases are typically less efficient in promoting the oxidation of IPA to acetone by the metal center via β-hydride elimination. Using KOH in lieu of NaOH gave no notable enhancement in activity. Trials were then carried out using different amounts of NaOH to probe any effects on conversion or enantioselectivity (Figure 11). No notable substrate conversion occurred in the absence of base, while 1 equivalent of NaOH gave just over 60% conversion. Conversion steadily declined when employing any amount over 2 equivalents of NaOH, indicating excess hydroxide may poison the catalyst. Varying the amount of base did not significantly affect enantioselectivity.
2.3.4. Optimization: Catalyst Loading
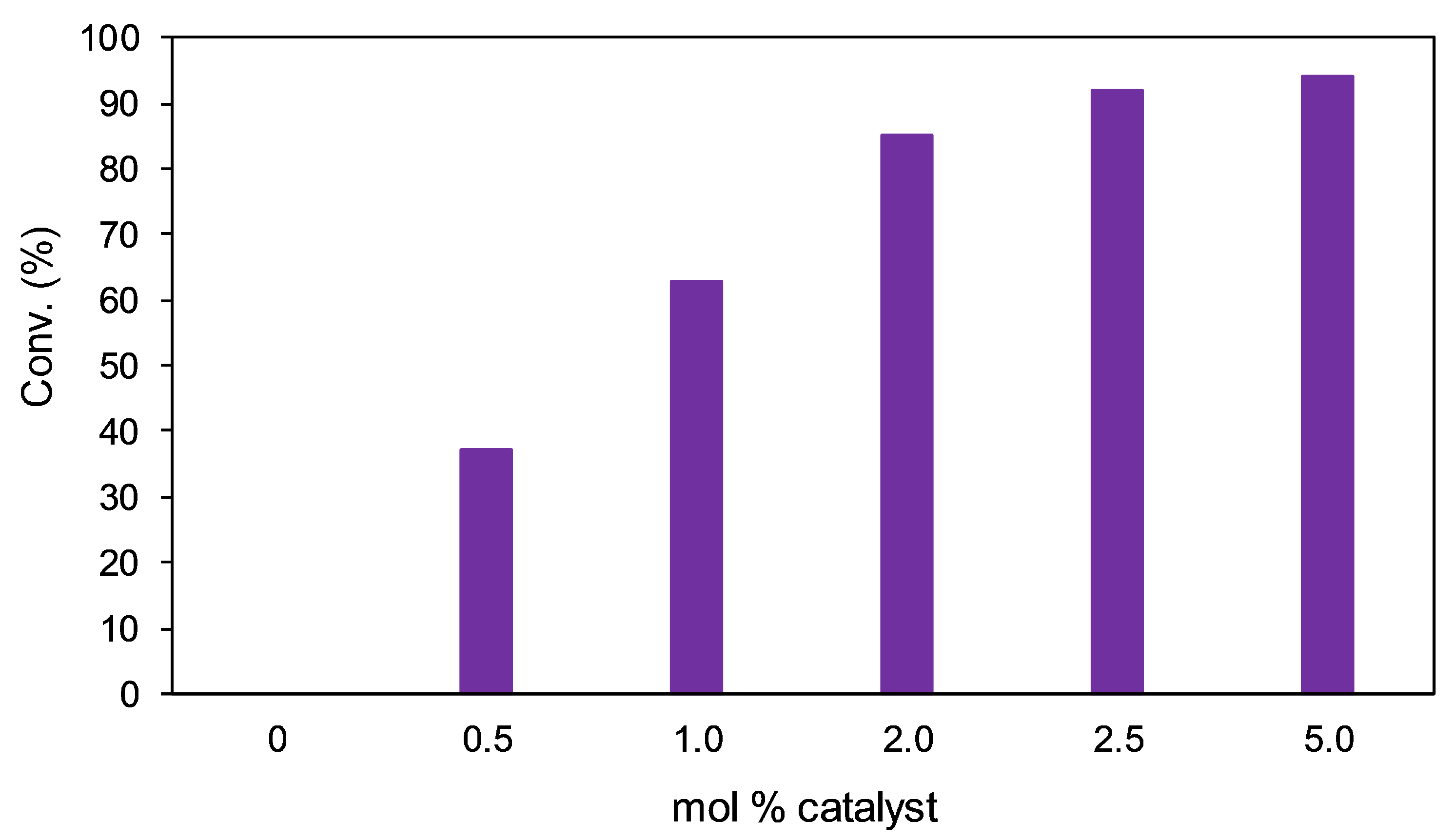
Trials were carried out at different catalyst loadings of Ir(IMe)2(l-Pro)(H)(I) with 2 equivalents of NaOH in IPA at 25 °C over a period of 24 h (Figure 12). As expected, no conversion was observed when the catalyst was omitted from the reaction. Conversion was found to steadily increase with catalyst loading in the other trials, although there was not a notable enhancement in activity when going from 2.5 to 5 mol% of catalyst. Enantioselectivity remained consistent regardless of the catalyst loading, with all five trials generating R-1-phenylethanol in 92% ee.
2.3.5. Optimized Results: Catalyst Scope
Following optimization, all iridium NHC amino acid complexes synthesized in this work were screened for ATH of acetophenone using 2.5 mol% of catalyst and 2 equivalents of NaOH in IPA at 25 °C (Table 5). Although the majority of catalysts gave lower conversions at 25 °C than seen in the initial trials at 80 °C, enantioselectivity was improved in all cases. Catalysts featuring the amino acids Aze, Pro, and Pip generated 1-phenylethanol in higher enantiomeric excess than those containing primary amino acids, indicating secondary amino acids are essential to achieve reasonable enantioselectivity in this system. While the l-Aze catalyst gave a notable increase in ee at room temperature, the proline-based catalysts are clearly superior to all other in terms of activity and enantioselectivity. Incorporation of IEt or IiPr NHC ligands in lieu of IMe achieved nearly complete conversion of acetophenone, but at the expense of enantioselectivity. Enantiomeric excess notably decreased as the steric bulk of the NHC N-substituents increased. The high conversions indicate the face of the metal is not completely blocked, but as suggested earlier when evaluating the X-ray crystal structure of the glycine complex, larger N-substituents appear to obstruct the alignment of the incoming substrate, eroding enantioselectivity.
The majority of these catalysts generated the R-enantiomer of 1-phenylethanol in excess. Changing the configuration of a particular amino acid led to an excess of the opposite enantiomer, as demonstrated from the trials employing l- and d-proline. Of all the catalysts screened that incorporated l-amino acids, only the l-Pip variant gave an excess of the S-enantiomer. This phenomenon was also observed for noble metal amino acid ATH catalysts of the form (Cp*R)Ir(aa)Cl previously reported by the Merola group, with those studies finding that the configurations of the alcohol products directly correlated to the crystallographically observed configurations of the chelated amino acid nitrogens [34]. The crystal structure of (Cp*)Ir(l-Pip)Cl had the opposite configuration at the nitrogen (R) than those containing other secondary l-amino acids (S); thus, the opposite product enantiomer was generated when using the l-Pip catalyst. This could justify why the same trend was observed for the iridium NHC amino acid catalysts as well. Unfortunately, suitable single-crystals of the NHC complexes featuring chiral amino acids could not be grown to confirm the absolute configurations, which may be related to the presence of numerous other isomers, as seen in solution.
2.3.6. Optimized Results: Substrate Scope
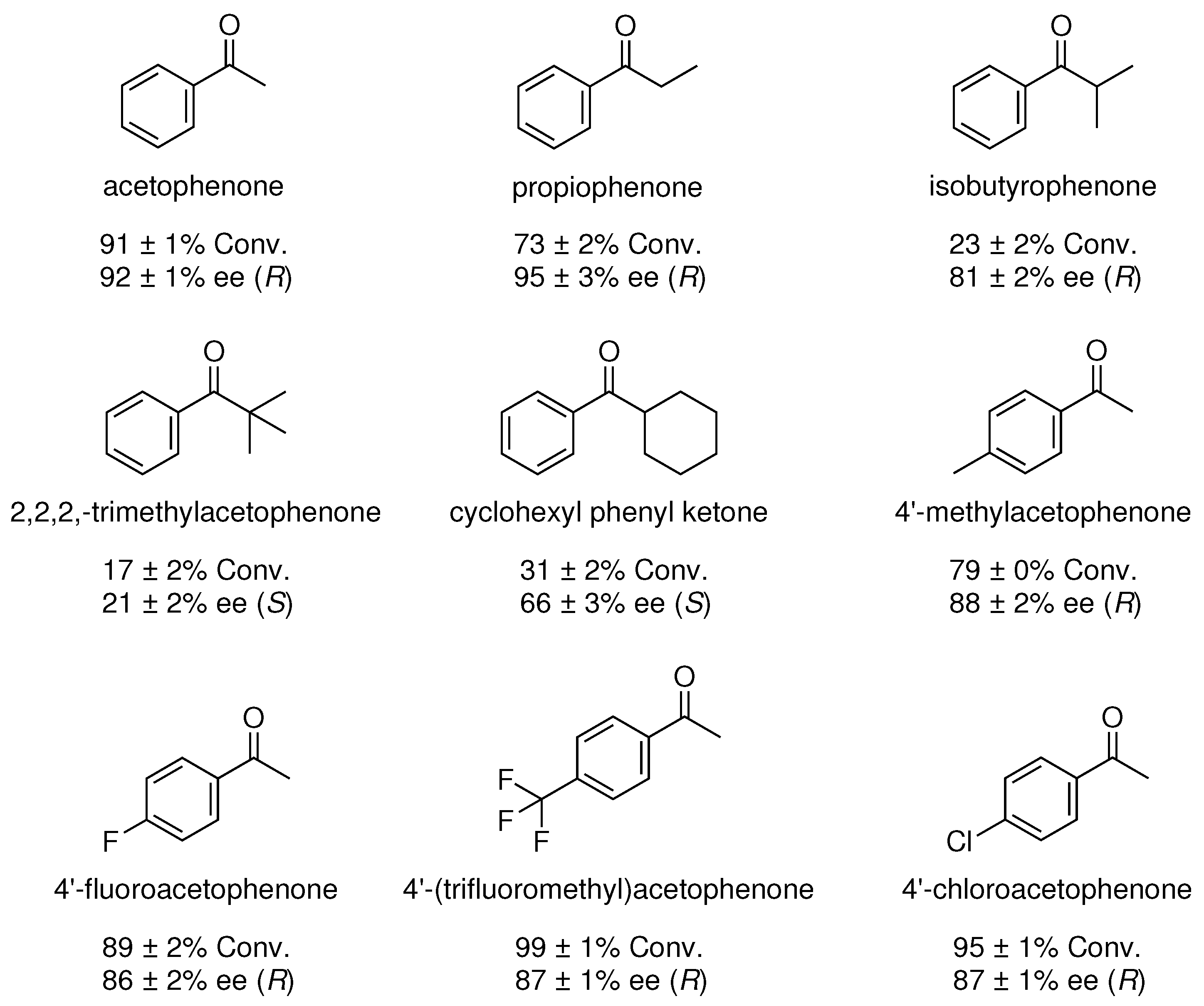
The Ir(IMe)2(l-Pro)(H)(I) catalyst was employed in an expanded substrate scope featuring a variety of acetophenone derivatives. All trials were carried out under the optimized catalytic conditions using 2.5 mol% of catalyst and 2 equivalents of NaOH in IPA at 25 °C. Conversion and enantiomeric excess were quantified by chiral GC, and the results are summarized below in Figure 13. The majority of aryl ketone substrates were reduced in good conversions and enantiomeric excess. The highest ee’s in the study were observed for acetophenone and propiophenone, giving 92% and 95% ee, respectively. Conversion and enantioselectivity were lowered upon screening branched acetophenone derivatives, which may be attributable to unfavorable steric interactions between the isopropyl or tert-butyl groups on the respective substrates and the N-substituents on the NHC ligands. Enantiomeric excess was only marginally lower for acetophenone derivatives featuring CH, F, CF, or Cl substituents on the phenyl ring.
2.3.7. Proposed Catalytic Mechanism
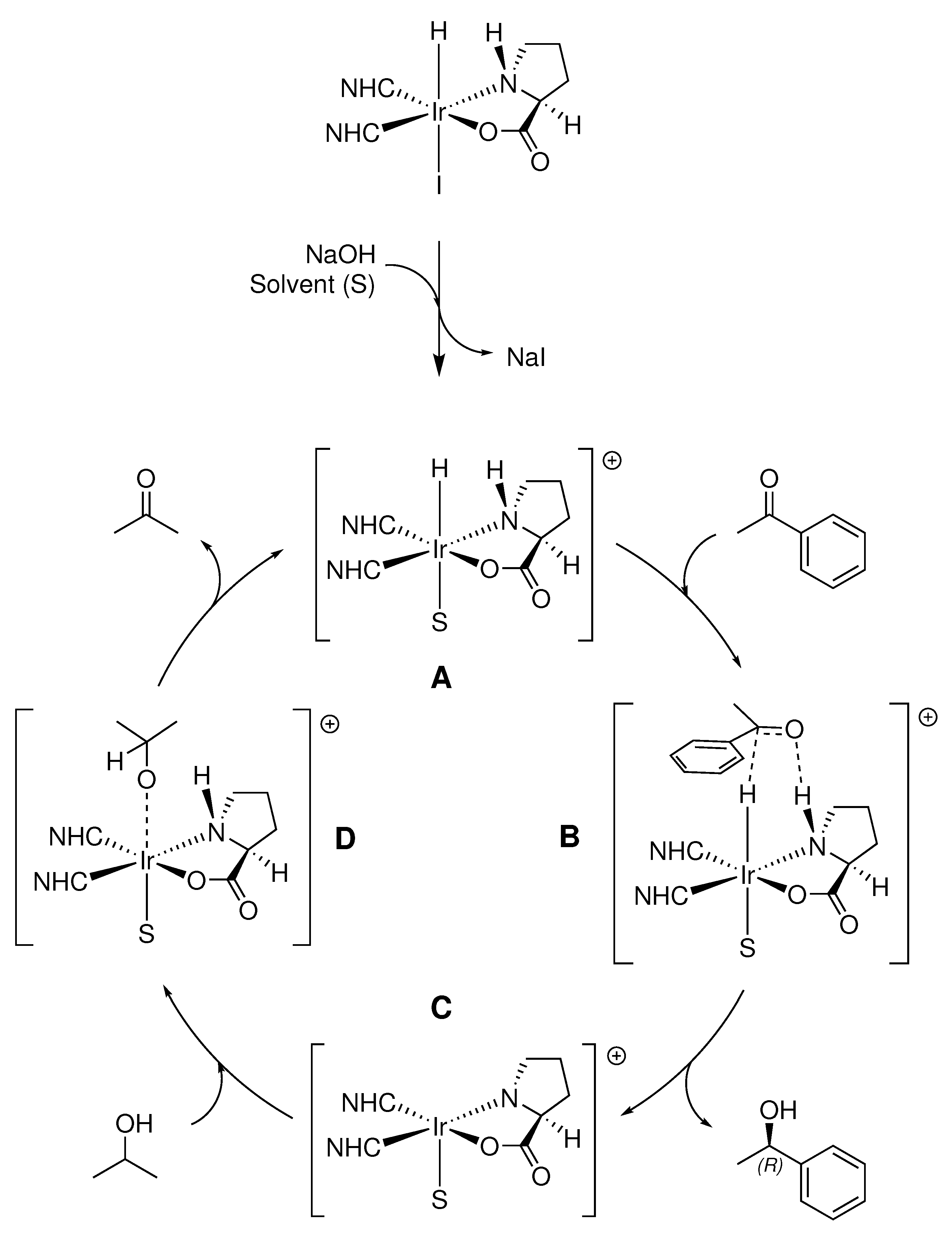
While detailed experimental and computational mechanistic studies are ongoing, it is likely that transfer hydrogenation in this system proceeds through an outer-sphere mechanism in a similar fashion to Noyori’s metal-ligand bifunctional ruthenium diamine ATH catalyst (Scheme 4). Upon addition of base, the active catalytic species is generated by dissociation of the iodide ligand (A). Simultaneous transfer of the proton and hydride from the catalyst to the substrate occurs via a cyclic six-membered transition state (B). Although the active catalyst can then potentially be regenerated by hydrogen transfer through another cyclic six-membered transition state between IPA and the coordinatively unsaturated species (C), the requirement of a strong base to achieve any substrate conversion in this system (as seen in the base optimization studies) suggests the active catalyst is regenerated by coordination of J (D) and subsequent β-hydride elimination of acetone.
The small decrease in enantiomeric excess observed when screening electron-withdrawing acetophenone derivatives suggests that if CH3/π interactions are occurring between the aryl group of the substrate and a methyl group on one of the NHC ligands, they are likely not significant. Therefore, it seems most probable that steric inhibition between the alkyl groups of the substrate and NHC N-substituents dominate the enantiodetermining step, as highlighted by the diminished conversions and ee’s obtained for branched acetophenone derivatives. This is also supported by the catalytic trials featuring bulkier NHC N-substituents (IEt, IiPr), which still favorably generated the same enantiomer of 1-phenylethanol as the IMe-based catalyst, but in notably lower enantiomeric excess.
3. Materials and Methods
Reagent grade solvents and all materials for synthesis, purification, characterization, and catalysis were purchased from commercial sources and used as received unless otherwise stated. Iridium(III) chloride hydrate was purchased from Pressure Chemical (Pittsburgh, PA, USA). 1,3-Diisopropylimidazolium chloride, l-phenylglycine, acetophenone, 4’-chloroacetophenone, and 4’-methylacetophenone were purchased from Sigma Aldrich (St. Louis, MO, USA). l-Alanine, l-pipecolic acid, d-proline, l-proline, l-valine, cyclohexyl phenyl ketone, 4’-fluoroacetophenone, isobutyrophenone, propiophenone, 4’-(trifluoromethyl)acetophenone, and 2,2,2-trimethylacetophenone were purchased from Alfa Aesar (Ward Hill, MA, USA). l-Azetidine-2-carboxylic acid and cis-4-fluoro-l-proline were purchased from Indofine Chemical (Hillsborough, NJ, USA). Glycine was purchased Qiagen Sciences (Germanstown, MD 20874, USA). Deuterated solvents for NMR spectroscopy were obtained from Cambridge Isotope Laboratories (Andover, MA, USA).
H NMR, C NMR, and F NMR spectra were collected on an Agilent U4-DD2 or Agilent MR4 400 MHz spectrometer. C NMR were correspondingly recorded at 101 MHz and F NMR at 376 MHz. High-resolution mass spectra were collected on an Agilent 6220 Accurate Mass TOF LC-MS. X-ray crystallographic data were collected at 100 K on a Rigaku Oxford Diffraction Synergy diffractometer operating with Mo Kα radiation. Crystals were coated in Paratone oil and mounted on a fiber. Data collection and data reduction were performed using Agilent’s CrysAlisPro software [54]. Structure solution was performed using SHELXT [55] and refined using SHELXL [56] via Olex2. The final refinement model involved anisotropic displacement parameters for non-hydrogen atoms and a riding model for all hydrogen atoms. Olex2 was used for molecular graphics generation [57]. Searches of the Cambridge Structural Database (CSD) [58] were carried out using the program Conquest [59], and the results were analyzed with the statistical functions of the program Mercury [60,61]. GC chromatograms were acquired on a Hewlett Packard 5890 equipped with a flame-ionization detector and CP-ChiraSil-Dex CB 25 m × 0.25 mm column for determination of conversion and enantiomeric excess (inlet temp = 240 °C, detector temp = 250 °C). Column temperatures are provided with the respective GC chromatograms in the Supplemental Information.
3.1. Synthesis of Imidazolium Salts and Iridium Complex Precursors
3.2. General Procedure for the Synthesis of Ir(NHC)2(aa)(H)(X) Complexes
A Schlenk flask was charged with the desired iridium COD bis-NHC complex, amino acid, and 15 mL of water. Upon heating the mixture to 100 °C, both reagents fully dissolved, and solution was allowed to react for period of 12 h, over which time the color changed from bright orange to light yellow. The water was removed by rotary evaporation, and the crude product was extracted into dichloromethane. After drying with magnesium sulfate, the mixture was filtered through Celite, and the dichloromethane was removed by rotary evaporation. Following trituration with diethyl ether, the products were isolated on a fritted glass filter as off-white solids, and dried overnight under vacuum.
3.2.1. Synthesis of Ir(IMe)2(Gly)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and glycine (0.0500 g, 0.6666 mmol) were reacted in water to give Ir(IMe)2(Gly)(H)(I) (0.0999 g, 53%). H NMR (400 MHz, DMSO-d6, δ): 7.45 (d, J = 2.0 Hz, 1H, NHC backbone), 7.33 (d, J = 2.0 Hz, 1H, NHC backbone), 7.31 (d, J = 2.0 Hz, 1H, NHC backbone), 7.27 (d, J = 1.9 Hz, 1H, NHC backbone), 5.21 (m, 1H, NH), 4.94 (m, 1H, NH), 4.06 (s, 3H, NHC CH), 3.80 (s, 3H, NHC CH), 3.33–3.25 (m, 1H, αCH), 3.18 (s, 3H, NHC CH), 3.17–3.10 (m, 1H, αCH), 2.89 (s, 3H, NHC CH), −24.33 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 178.8 (C(O)), 149.4 (Ccarbene), 143.5 (Ccarbene), 123.5 (NHC backbone), 123.2 (NHC backbone), 122.8 (NHC backbone), 122.7 (NHC backbone), 47.0 (αC), 38.3 (NHC CH), 38.2 (NHC CH), 36.8 (NHC CH), 35.0 (NHC CH). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 460.1319; Found: 460.1304.
3.2.2. Synthesis of Ir(IMe)2(l-Ala)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-alanine (0.0575 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Ala)(H)(I) (0.1503 g, 78%). Analysis for major isomer (51%): H NMR (400 MHz, DMSO-d6, δ): 7.46 (d, J = 1.9 Hz, 1H, NHC backbone), 7.42 (d, J = 1.9 Hz, 1H, NHC backbone), 7.32 (d, J = 1.9 Hz, 1H, NHC backbone), 7.29 (d, J = 2.0 Hz, 1H, NHC backbone), 5.07 (dd, J = 12.5, 6.1 Hz, 1H, NH), 4.29 (dd, J = 12.7, 9.6 Hz, 1H, NH), 4.11 (s, 3H, NHC CH), 3.76 (s, 3H, NHC CH), 3.19 (s, 3H, NHC CH), 2.98 (s, 3H, NHC CH), 2.55–2.41 (m, 1H, αCH), 1.23 (d, J = 7.0 Hz, 3H, ala CH), −24.00 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 179.9 (C(O)), 149.0 (Ccarbene), 142.1 (Ccarbene), 123.3 (NHC backbone), 123.2 (NHC backbone), 122.8 (NHC backbone), 122.7 (NHC backbone), 53.7 (αC), 38.6 (NHC CH), 38.2 (NHC CH), 36.8 (NHC CH), 35.2 (NHC CH), 20.6 (ala CH). Analysis for minor isomer (49%): H NMR (400 MHz, DMSO-d6, δ): 7.34 (d, J = 2.0 Hz, 1H, NHC backbone), 7.32 (d, J = 2.0 Hz, 1H, NHC backbone), 7.25 (d, J = 1.9 Hz, 1H, NHC backbone), 7.24 (d, J = 1.9 Hz, 1H, NHC backbone), 5.59 (dd, J = 11.6, 5.1 Hz, 1H, NH), 4.52 (t, J = 11.4 Hz, 1H, NH), 4.06 (s, 3H, NHC CH), 3.94 (s, 3H, NHC CH), 3.41 (ddt, J = 13.9, 10.0, 6.9 Hz, 1H, αCH), 3.18 (s, 3H, NHC CH), 2.86 (s, 3H, NHC CH), 1.14 (d, J = 6.9 Hz, 3H, Ala CH), −24.23 (s, 1H, hydride). CNMR (101 MHz, DMSO-d6, δ): 179.0 (C(O)), 149.0 (Ccarbene), 143.6 (Ccarbene), 123.8 (NHC backbone), 123.3 (NHC backbone), 122.9 (NHC backbone), 122.8 (NHC backbone), 53.3 (αC), 38.7 (NHC CH), 38.3 (NHC CH), 36.6 (NHC CH), 34.9 (NHC CH), 20.0 (Ala CH). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 474.1476; Found: 474.1455.
3.2.3. Synthesis of Ir(IMe)2(l-Val)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-valine (0.0756 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Val)(H)(I) (0.1756 g, 87%). Analysis for major isomer (55 %): H NMR (400 MHz, DMSO-d6, δ): 7.44 (d, J = 2.0 Hz, 1H, NHC backbone), 7.36 (d, J = 2.0 Hz, 1H, NHC backbone), 7.28 (d, J = 2.0 Hz, 1H, NHC backbone), 7.26 (d, J = 2.0 Hz, 2H, NHC backbone), 4.78 (dd, J = 12.5, 6.2 Hz, 1H, NH), 4.17 (br d, J = 12.4 Hz, 1H, NH), 4.13 (s, 3H, NHC CH), 3.96 (s, 3H, NHC CH), 3.25 (s, 3H, NHC CH), 3.10 (s, 3H, NHC CH), 2.32 (ddd, J = 11.2, 6.0, 3.1 Hz, 1H, αCH), 2.13 (ddp, J = 9.9, 6.9, 3.5 Hz, 1H, Val CH), 0.93 (d, J = 7.1 Hz, 3H, Val CH), 0.83 (d, J = 6.9 Hz, 3H, Val CH), −23.80 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 178.5 (C(O)), 148.0 (Ccarbene), 141.2 (Ccarbene), 124.0 (NHC backbone), 123.9 (NHC backbone), 123.0 (NHC backbone), 122.9 (NHC backbone), 62.1 (αC), 38.7 (NHC CH), 38.4 (NHC CH), 36.9 (NHC CH), 35.3 (NHC CH), 31.5 (Val CH), 18.4 (Val CH), 16.9 (Val CH). Analysis for minor isomer (45%): H NMR (400 MHz, DMSO-d6, δ): 7.45 (d, J = 2.0 Hz, 1H, NHC backbone), 7.34 (d, J = 2.0 Hz, 1H, NHC backbone), 7.31 (d, J = 2.0 Hz, 1H, NHC backbone), 7.26 (d, J = 1.7 Hz, 2H, NHC backbone), 5.43 (dd, J = 12.0, 6.6 Hz, 1H, NH), 4.09 (s, 3H, NHC CH), 3.89 (s, 3H, NHC CH), 3.23 (s, 3H, NHC CH), 3.16 (ddd, J = 9.0, 6.7, 4.5 Hz, 1H, αCH), 2.95 (s, 3H, NHC CH), 2.04 (dtd, J = 13.9, 6.9, 4.5 Hz, 1H, Val CH), 0.90 (d, J = 6.9 Hz, 3H, Val CH), 0.49 (d, J = 6.9 Hz, 3H, Val CH), −23.84 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 179.0 (C(O)), 149.3 (Ccarbene), 142.1 (Ccarbene), 123.8 (NHC backbone), 123.3 (NHC backbone), 123.1 (NHC backbone), 122.9 (NHC backbone), 61.3 (αC), 38.6 (NHC CH), 37.5 (NHC CH), 36.8 (NHC CH), 35.2 (NHC CH), 31.0 (val CH), 19.3 (Val CH), 16.5 (Val CH). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 502.1789; Found: 502.1777.
3.3. Synthesis of Ir(IMe)2(l-Phg)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-phenylglycine (0.0976 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Phg)(H)(I) (0.1401 g, 66%). Phenyl H NMR signals from both isomers ranged from 7.35–7.13 and overlapped with each other and with the signals for the NHC backbones, while phenyl C NMR signals from both isomers ranged from 128.3–127.1 and overlapped with each other. Analysis for major isomer (55%): H NMR (400 MHz, DMSO-d6, δ): 7.38 (d, J = 2.0 Hz, 1H, NHC backbone), 7.31 (dd, J = 2.2, 0.7 Hz, 2H, NHC backbone), 7.21 (d, J = 1.9 Hz, 1H, NHC backbone), 6.06 (m, 1H, NH), 4.66–4.51 (m, 2H, overlapped αCH and NH), 4.12 (s, 3H, NHC CH), 3.65 (s, 3H, NHC CH), 3.24 (s, 3H, NHC CH), 2.87 (s, 3H, NHC CH), −24.33 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 177.0 (C(O)), 148.4 (Ccarbene), 142.3 (Ccarbene), 140.1 (Ph), 123.3 (NHC backbone), 123.0 (NHC backbone), 122.9 (NHC backbone), 122.8 (NHC backbone), 61.5 (αC), 38.2 (NHC CH), 36.8 (NHC CH), 35.0 (NHC CH). Analysis for minor isomer (45%): H NMR (400 MHz, DMSO-d6, δ): 7.50 (d, J = 2.0 Hz, 1H, NHC backbone), 7.25 (d, J = 1.9 Hz, 2H, NHC backbone), 7.15 (dd, J = 1.9, 0.5 Hz, 1H, NHC backbone), 5.66 (dd, J = 12.3, 6.6 Hz, 1H, NH), 4.78–4.67 (m, 1H, NH), 4.18 (s, 3H, NHC CH), 3.96 (s, 3H, NHC CH), 3.61 (dd, J=10.5, 6.5 Hz, 1H, αCH), 3.33 (s, overlapped with H2O peak, NHC CH), 3.11 (s, 3H, NHC CH), −24.04 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 178.1 (C(O)), 147.8 (Ccarbene), 141.1 (Ccarbene), 141.9 (Ph), 124.0 (NHC backbone), 123.9 (NHC backbone), 123.4 (NHC backbone), 123.2 (NHC backbone), 61.1 (αC), 38.3 (NHC CH), 37.0 (NHC CH), 35.4 (NHC CH). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 536.1632; Found: 536.1636.
3.3.1. Synthesis of Ir(IMe)2(l-Aze)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-azetidine-2-carboxylic acid (0.0653 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Aze)(H)(I) (0.1088 g, 55%). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 486.1476; Found: 486.1476. Due to the presence of many isomers (that could not be separated by column chromatography) and overlapping resonances, the NMR spectra were not assigned for this structure. Additional characterization was not further pursued due to poor catalytic enantioselectivity.
3.3.2. Synthesis of Ir(IMe)2(l-Pro)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-proline (0.0743 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Pro)(H)(I) (0.1861 g, 92%). H NMR (400 MHz, DMSO-d6, δ): 7.42 (d, J = 1.9 Hz, 1H, NHC backbone), 7.35 (d, J = 2.0 Hz, 1H, NHC backbone), 7.31 (d, J = 2.0 Hz, 1H, NHC backbone), 7.25 (d, J = 2.0 Hz, 1H, NHC backbone), 5.97 (dt, J = 10.7, 5.5 Hz, 1H, NH), 4.09 (s, 3H, NHC CH), 3.94 (s, 3H, NHC CH), 3.63 (dd, J = 8.6, 5.5 Hz, 1H, αCH), 3.31 (s, 3H, NHC CH), 3.00 (s, 3H, NHC CH), 2.02 (dq, J = 12.9, 8.0 Hz, 1H, Pro CH), 1.93 (ddd, J = 17.9, 8.9, 4.7 Hz, 1H, Pro CH), 1.77 (dt, J = 13.3, 7.1 Hz, 1H, Pro CH), 1.69 (t, J = 7.5 Hz, 1H, Pro CH), 1.64–1.55 (m, 1H, Pro CH), 1.49 (qd, J = 11.6, 7.2 Hz, 1H, Pro CH), −24.12 (s, 1H, hydride). C NMR (101 MHz, DMSO-d6, δ): 178.9 (C(O)), 150.0 (Ccarbene), 140.2 (Ccarbene), 123.9 (NHC backbone), 123.6 (NHC backbone), 123.3 (NHC backbone), 122.6 (NHC backbone), 60.7 (Pro αCH), 52.4 (Pro CH), 39.8 (NHC CH), 38.3 (NHC CH), 36.8 (NHC CH), 35.4 (NHC CH), 28.9 (Pro CH), 24.1 (Pro CH). HRMS/ESI+ (m/z): Calc. for C15H25O2[193Ir]N5: 500.1632; Found: 500.1624.
3.3.3. Synthesis of Ir(IMe)2(d-Pro)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and d-proline (0.0743 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(d-Pro)(H)(I) (0.1450 g, 72%). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 500.1632; Found: 500.1631. NMR spectra were identical to Ir(IMe)2(l-Pro)(H)(I).
3.3.4. Synthesis of Ir(IMe)2(l-F-Pro)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and cis-4-fluoro-l-proline (0.0859 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-F-Pro)(H)(I) (0.0921 g, 44%). H NMR (400 MHz, DMSO-d6, δ): 7.41 (d, J = 1.9 Hz, 1H, NHC backbone), 7.35 (d, J = 1.9 Hz, 1H, NHC backbone), 7.31 (d, J = 1.9 Hz, 1H, NHC backbone), 7.26 (d, J = 2.0 Hz, 1H, NHC backbone), 6.42 (dt, J = 11.5, 5.4 Hz, 1H, NH), 5.28–4.96 (tm, J = 112.1, 53.9, 1H, FCH), 4.14 (dd, J = 11.4, 5.1 Hz, 1H, αCH), 4.09 (s, 3H, NHC CH), 4.06–3.98 (m, 1H, Pro CH), 3.96 (s, 3H, NHC CH), 3.34 (s, 3H, NHC CH), 3.00 (s, 3H, NHC CH), 2.35–2.15 (m, 2H, proline CH), 1.79 (app dtd, J = 25.4, 13.3, 3.7 1H, Pro CH), −24.19 (s, 1H, hydride). C NMR (400 MHz, DMSO-d6, δ): 149.3 (Ccarbene), 138.9 (Ccarbene), 124.1 (NHC backbone), 123.7 (NHC backbone), 123.4 (NHC backbone), 122.7 (NHC backbone), 93.9 (d, J = 174.8 Hz, Pro CF), 63.4 (Pro αCH), 62.0 (d, J = 18.5 Hz, Pro CH), 38.4 (NHC CH), 37.5 (NHC CH), 36.9 (NHC CH), 35.5 (NHC CH), 35.4 (Pro CH), F NMR (376 MHz, DMSO-d6, δ): −169.18 – −173.96 (m). HRMS/ESI+ (m/z): Calc. for CHFO[193Ir]N: 518.1543; Found: 518.1503.
3.3.5. Synthesis of Ir(IMe)2(l-Pip)(H)(I)
Following the general procedure: [Ir(COD)(IMe)2]I (0.2000 g, 0.3228 mmol) and l-pipecolic acid (0.0834 g, 0.6456 mmol) were reacted in water to give Ir(IMe)2(l-Pip)(H)(I) (0.1359 g, 66%). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 514.1789; Found: 514.1783. Due to the presence of many isomers (that could not be separated by column chromatography) and overlapping resonances, the NMR spectra were not assigned for this structure. Additional characterization was not further pursued due to poor catalytic enantioselectivity.
3.3.6. Synthesis of Ir(IEt)2(l-Pro)(H)(I)
Following the general procedure: [Ir(COD)(IEt)2]I (0.2000 g, 0.2960 mmol) and l-proline (0.0682 g, 0.5920 mmol) were reacted in water to give Ir(IEt)2(l-Pro)(H)(I) (0.1369 g, 68%). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 556.2259; Found: 556.2249. Due to the presence of many isomers (that could not be separated by column chromatography) and overlapping resonances, the NMR spectra were not assigned for this structure. Additional characterization was not further pursued due to poor catalytic enantioselectivity.
3.3.7. Synthesis of Ir(IiPr)2(l-Pro)(H)(Cl)
Following the general procedure: [Ir(COD)(IiPr)2]Cl (0.2000 g, 0.2960 mmol) and l-proline (0.0682 g, 0.5920 mmol) were reacted in water to give Ir(IiPr)2(l-Pro)(H)(Cl) (0.1580 g, 82%). HRMS/ESI+ (m/z): Calc. for CHO[193Ir]N: 612.2885; Found: 612.2887. Due to the presence of many isomers (that could not be separated by column chromatography) and overlapping resonances, the NMR spectra were not assigned for this structure. Additional characterization was not further pursued due to poor catalytic enantioselectivity.
3.4. General Procedure for Asymmetric Transfer Hydrogenation of Ketones
Using the optimized reaction conditions, the catalyst (0.005 mmol) was dissolved in 2 mL of isopropyl alcohol and stirred at 25 °C. Catalysis was initiated by addition of the desired substrate (0.200 mmol) and NaOH (2 equiv.), and maintained at 25 °C for the appropriate period of time (usually 24 h). When initially screening ATH substrates, the products were isolated for characterization via NMR spectroscopy to confirm their structures. Isopropyl alcohol was removed by rotary evaporation and water was added to the reaction vial. The products were then extracted with diethyl ether, and dried over magnesium sulfate. Following filtration and solvent removal under vacuum, the 1H and 13C NMR spectra of all products were acquired, and consistent with previously reported data.
Conversion and enantiomeric excess were determined by chiral GC-FID using a CP-ChiraSil-Dex CB 25 m × 0.25 mm column (inlet temp = 240 °C, detector temp = 250 °C.) For quantification, 1,3,5-trimethoxybenzene was employed as an internal standard. Absolute configurations of the products were assigned by using the appropriate alcohol standard. Column temperatures are provided with the respective GC chromatograms in the Supplemental Information.
4. Conclusions
Novel complexes of the form Ir(NHC)2(aa)(H)(X) were synthesized by oxidative addition of α-amino acids to [Ir(COD)(NHC)2]X precursors and screened for asymmetric transfer hydrogenation of ketones. Following optimization, high enantioselectivity was achieved when employing the Ir(IMe)2(l-Pro)(H)(I) catalyst, which reduces a range of acetophenone derivatives in up to 95% ee. These results are comparable to other well-regarded ATH catalysts previously reported in the literature, and superior to many of those featuring NHC ligands. This work illustrates the design of highly enantioselective noble metal catalysts by incorporation of inexpensive, “off-the-shelf” chiral amino acids in combination with easily accessible N-heterocyclic carbenes; thus, providing an alternative to costly or synthetically challenging chiral ligands. The facile modularity of this catalytic system may also allow for simple tailoring towards additional ATH substrates, such as alkyl ketones, and potentially other enantioselective transformations.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal11060671/s1, Figure S1: Displacement ellipsoid plot of Ir(IMe)(Gly)(H)(I), Table S1: Selected bond lengths and angles from the structure of Ir(IMe)(Gly)(H)(I), Figure S2: NMR spectra of Ir(IMe)(Gly)(H)(I), Figure S3: NMR spectra of the major isomer of Ir(IMe)(l-Ala)(H)(I), Figure S4: NMR spectra of the minor isomer of Ir(IMe)(l-Ala)(H)(I), Figure S5: NMR spectra of the major isomer of Ir(IMe)(l-Val)(H)(I), Figure S6: NMR spectra of the minor isomer of Ir(IMe)(l-Val)(H)(I), Figure S7: NMR spectra of the major isomer of Ir(IMe)(l-Phg)(H)(I), Figure S8: NMR spectra of the minor isomer of Ir(IMe)(l-Phg)(H)(I), Figure S9: NMR spectra of Ir(IMe)(l-Pro)(H)(I), Figure S10: NMR spectra of Ir(IMe)(l-F-Pro)(H)(I), Figure S11: FID trace of 1-phenylethanol, Figure S12: FID trace of 1-phenylpropanol, Figure S13: FID trace of 2-methyl-1-phenylpropanol, Figure S14: FID trace of 2,2-dimethyl-1-phenylpropanol, Figure S15: FID trace of cyclohexyl(phenyl)methanol, Figure S16: FID trace of 1-(p-tolyl)ethanol, Figure S17: FID trace of 1-(4-fluorophenyl)ethanol, Figure S18: FID trace of 1-(4-(trifluoromethyl)phenyl)ethanol, Figure S19: FID trace of 1-(4-chlorophenyl)ethanol. In addition, CSD number 2062242 contains the full supplementary.cif files for this paper. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures (accessed on 23 May 2021).
Author Contributions
Conceptualization, J.S.M.; Data curation, C.M.B.; Funding acquisition, J.S.M.; Investigation, C.M.B.; Methodology, C.M.B.; Supervision, J.S.M.; Writing—original draft, C.M.B.; Writing—review & editing, C.M.B. and J.S.M. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for the Rigaku Oxford Diffraction Synergy diffractometer used in this work was provided by the National Science Foundation (grant No. 1726077).
Acknowledgments
The authors would like to acknowledge Christine M. DuChane, Justin Martinez, and William Owen for providing assistance in synthesis and characterization of the ligands and complexes.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| aa | Amino acid |
| Ala | Alanine |
| ATH | Asymmetric transfer hydrogenation |
| Aze | Azetidine-2-carboxylic acid |
| COD | 1,5-Cyclooctadiene |
| Cp* | Pentamethylcyclopentadienyl ligand |
| DMSO | Dimethyl sulfoxide |
| ee | Enantiomeric excess |
| F-Pro | cis-4-Fluoro-proline |
| FID | Flame ionization detector |
| GC | Gas chromatography |
| Gly | Glycine |
| HRMS/ESI+ | High resolution mass spectrometry/electrospray ionization |
| IiPr | 1,3-Diisopropylimidazol-2-ylidene |
| IEt | 1,3-Diethylimidazol-2-ylidene |
| IMe | 1,3-Dimethylimidazol-2-ylidene |
| IPA | Isopropyl alcohol |
| NHC | N-Heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| Phg | Phenylglycine |
| Pip | Pipecolic acid |
| Pro | Proline |
| TsDPEN | N-Tosyl-1,2-diphenylethylenediamine |
| Val | Valine |
References
- Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 2004, 43, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-Triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2008. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, H.; Noyori, R. The metal-ligand bifunctional catalysis: A theoretical study on the ruthenium(II)-catalyzed hydrogen transfer between alcohols and carbonyl compounds. J. Am. Chem. Soc. 2000, 122, 1466–1478. [Google Scholar] [CrossRef]
- Noyori, R.; Yamakawa, M.; Hashiguchi, S. Metal-ligand bifunctional catalysis: A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001, 66, 7931–7944. [Google Scholar] [CrossRef] [PubMed]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, O.; Crabtree, R.H. Outer sphere hydrogenation catalysis. New J. Chem. 2013, 37, 21–27. [Google Scholar] [CrossRef]
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Milstein, D. Metal-ligand cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef] [PubMed]
- Dub, P.A.; Gordon, J.C. Metal–Ligand bifunctional catalysis: The “accepted” mechanism, the issue of concertedness, and the function of the ligand in catalytic cycles involving hydrogen atoms. ACS Catal. 2017, 7, 6635–6655. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Agbossou-Niedercorn, F.; Michon, C. Bifunctional homogeneous catalysts based on first row transition metals in asymmetric hydrogenation. Coord. Chem. Rev. 2020, 425, 213523. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, X.; Xiao, J. Broader, greener, and more efficient: Recent advances in asymmetric transfer hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef] [PubMed]
- Stefane, B.; Pozgan, F. Asymmetric hydrogenation and transfer hydrogenation of ketones. In Hydrogenation; Karame, I., Ed.; InTechOpen: London, UK, 2012; Chapter 2. [Google Scholar] [CrossRef] [Green Version]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Baráth, E. Hydrogen transfer reactions of carbonyls, alkynes, and alkenes with noble metals in the presence of alcohols/ethers and amines as hydrogen donors. Catalysts 2018, 8, 671. [Google Scholar] [CrossRef] [Green Version]
- Matsunami, A.; Kayaki, Y. Upgrading and expanding the scope of homogeneous transfer hydrogenation. Tetrahedron Lett. 2018, 59, 504–513. [Google Scholar] [CrossRef]
- Seo, C.S.G.; Morris, R.H. Catalytic homogeneous asymmetric hydrogenation: Successes and opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Ohta, T.; Nakahara, S.I.; Shigemura, Y.; Hattori, K.; Furukawa, I. α-Amino acid: An effective ligand for asymmetric catalysis of transfer hydrogenation of ketones. Appl. Organomet. Chem. 2001, 15, 699–709. [Google Scholar] [CrossRef]
- Carmona, D.; Lamata, M.P.; Oro, L.A. Half-sandwich complexes with aminocarboxylate ligands and their use as enantioselective hydrogen transfer catalysts. Eur. J. Inorg. Chem. 2002, 2002, 2239–2251. [Google Scholar] [CrossRef]
- Yim, A.S.; Wills, M. Asymmetric transfer hydrogenation using amino acid derivatives; further studies and a mechanistic proposal. Tetrahedron 2005, 61, 7994–8004. [Google Scholar] [CrossRef]
- Ahlford, K.; Ekström, J.; Zaitsev, A.B.; Ryberg, P.; Eriksson, L.; Adolfsson, H. Asymmetric transfer hydrogenation of ketones catalyzed by amino acid derived rhodium complexes: On the origin of enantioselectivity and enantioswitchability. Chem. Eur. J. 2009, 15, 11197–11209. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, J.; Stodulski, M.; Mlynarski, J. Catalysts based on amino acids for asymmetric reactions in water. Angew. Chem. Int. Ed. 2009, 48, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Nordin, M.; Liao, R.Z.; Ahlford, K.; Adolfsson, H.; Himo, F. Theoretical study of asymmetric transfer hydrogenation of ketones catalyzed by amino acid-derived rhodium complexes. ChemCatChem 2012, 4, 1095–1104. [Google Scholar] [CrossRef]
- Carmona, D.; Lahoz, F.J.; García-Orduña, P.; Oro, L.A.; Lamata, M.P.; Viguri, F. Half-sandwich complexes of osmium(II) with l-α-amino carboxylate ligands as asymmetric transfer hydrogenation catalysts. On the origin of the enantioselectivity. Organometallics 2012, 31, 3333–3345. [Google Scholar] [CrossRef]
- Carmona, D.; Viguri, F.; Pilar Lamata, M.; Ferrer, J.; Bardají, E.; Lahoz, F.J.; García-Orduña, P.; Oro, L.A. Ruthenium amino carboxylate complexes as asymmetric hydrogen transfer catalysts. Dalton Trans. 2012, 41, 10298. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, L.; Abdalghani, I.; Chiellini, F.; Zacchini, S.; Pampaloni, G.; Crucianelli, M.; Marchetti, F. Ruthenium arene complexes with α-aminoacidato ligands: New insights into transfer hydrogenation reactions and cytotoxic behaviour. Eur. J. Inorg. Chem. 2018, 2018, 3041–3057. [Google Scholar] [CrossRef]
- Morris, D.M.; McGeagh, M.; De Peña, D.; Merola, J.S. Extending the range of pentasubstituted cyclopentadienyl compounds: The synthesis of a series of tetramethyl(alkyl or aryl)cyclopentadienes (Cp*R), their iridium complexes and their catalytic activity for asymmetric transfer hydrogenation. Polyhedron 2014, 84, 120–135. [Google Scholar] [CrossRef]
- Morris, D.M. Design and Modification of Half-Sandwich Ir(III), Rh(III), and Ru(II) Amino acid Complexes for Application in Asymmetric Transfer Hydrogenation Reactions. Ph.D. Thesis, Virginia Tech, Blacksburg, VA, USA, 2015. [Google Scholar]
- Herrmann, W.A. N-heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nat. Chem. 2014, 510, 485–496. [Google Scholar] [CrossRef]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Candish, L.; Paul, D.; Glorius, F. N-heterocyclic carbenes in asymmetric hydrogenation. ACS Catal. 2016, 6, 5978–5988. [Google Scholar] [CrossRef]
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Hillier, A.C.; Lee, H.M.; Stevens, E.D.; Nolan, S.P. Cationic iridium complexes bearing imidazol-2-ylidene ligands as transfer hydrogenation catalysts. Organometallics 2001, 20, 4246–4252. [Google Scholar] [CrossRef]
- Albrecht, M.; Crabtree, R.H.; Mata, J.; Peris, E. Chelating bis-carbene rhodium(III) complexes in transfer hydrogenation of ketones and imines. Chem. Commun. 2002, 2, 32–33. [Google Scholar] [CrossRef]
- Poyatos, M.; Mata, J.A.; Falomir, E.; Crabtree, R.H.; Peris, E. New ruthenium(II) cnc-pincer bis(carbene) complexes: Synthesis and catalytic activity. Organometallics 2003, 22, 1110–1114. [Google Scholar] [CrossRef]
- Cross, W.B.; Daly, C.G.; Boutadla, Y.; Singh, K. Variable coordination of amine functionalised N-heterocyclic carbene ligands to Ru, Rh and Ir: C–H and N–H activation and catalytic transfer hydrogenation. Dalton Trans. 2011, 40, 9722. [Google Scholar] [CrossRef]
- Seo, H.; Kim, B.Y.; Lee, J.H.; Park, H.J.; Son, S.U.; Chung, Y.K. Synthesis of chiral ferrocenyl imidazolium salts and their rhodium(I) and iridium(I) complexes. Organometallics 2003, 22, 4783–4791. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Baskakov, D.; Herdtweck, E.; Hoffmann, S.D.; Bunlaksananusorn, T.; Rampf, F.; Rodefeld, L. Chiral N-heterocyclic carbene ligands derived from 2,2′-bipiperidine and partially reduced biisoquinoline: Rhodium and iridium complexes in asymmetric catalysis. Organometallics 2006, 25, 2449–2456. [Google Scholar] [CrossRef]
- Dyson, G.; Frison, J.C.; Whitwood, A.C.; Douthwaite, R.E. Synthesis of rhodium(I) and iridium(I) complexes of chiral N-heterocyclic carbenes and their application to asymmetric transfer hydrogenation. Dalton Trans. 2009, 7141. [Google Scholar] [CrossRef]
- Diez, C.; Nagel, U. Chiral iridium(I) bis(NHC) complexes as catalysts for asymmetric transfer hydrogenation. Appl. Organomet. Chem. 2010, 24, 509–516. [Google Scholar] [CrossRef]
- Chiyojima, H.; Sakaguchi, S. Iridium complex bearing a chiral hydroxy-amide functionalized N-heterocyclic carbene: A catalyst precursor for asymmetric transfer hydrogenation. Tetrahedron Lett. 2011, 52, 6788–6791. [Google Scholar] [CrossRef]
- Sabater, S.; Baya, M.; Mata, J.A. Highly active cp*Ir catalyst at low temperatures bearing an N-heterocyclic carbene ligand and a chelated primary benzylamine in transfer hydrogenation. Organometallics 2014, 33, 6830–6839. [Google Scholar] [CrossRef]
- Ramasamy, B.; Kumar Gangwar, M.; Ghosh, P. Chiral oxazolidine-fused N-heterocyclic carbene complexes of rhodium and iridium and their utility in the asymmetric transfer hydrogenation of ketones. Eur. J. Inorg. Chem. 2017, 2017, 3253–3268. [Google Scholar] [CrossRef]
- Iglesias, M.; Oro, L.A. A leap forward in iridium–NHC catalysis: New horizons and mechanistic insights. Chem. Soc. Rev. 2018, 47, 2772–2808. [Google Scholar] [CrossRef]
- Ramasamy, B.; Gangwar, M.K.; Ghosh, P. Asymmetric transfer hydrogenation of α,β-unsaturated carbonyl compounds to saturated alcohols as catalyzed by iridium complexes of tricyclic bioxazoline-fused imidazole-derived N-heterocyclic carbene ligands. ChemistrySelect 2019, 4, 357–365. [Google Scholar] [CrossRef]
- Yoshida, K.; Kamimura, T.; Kuwabara, H.; Yanagisawa, A. Chiral bicyclic NHC/Ir complexes for catalytic asymmetric transfer hydrogenation of ketones. Chem. Commun. 2015, 51, 15442–15445. [Google Scholar] [CrossRef]
- CrysAlisPRO; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2018.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Gardner, S.; Kawamoto, T.; Curran, D.P. Synthesis of 1,3-Dialkylimidazol-2-ylidene Boranes from 1,3-Dialkylimidazolium Iodides and Sodium Borohydride. J. Org. Chem. 2015, 80, 9794–9797. [Google Scholar] [CrossRef]
- Huang, S.; Qi, X.; Liu, T.; Wang, K.; Zhang, W.; Li, J.; Zhang, Q. Towards Safer Rocket Fuels: Hypergolic Imidazolylidene-Borane Compounds as Replacements for Hydrazine Derivatives. Chem. A Eur. J. 2016, 22, 10187–10193. [Google Scholar] [CrossRef] [PubMed]
- Baghurst, D.R.; Michael, D.; Mingos, P.; Watson, M.J. Application of microwave dielectric loss heating effects for the rapid and convenient synthesis of organometallic compounds. J. Organomet. Chem. 1989, 368, C43–C45. [Google Scholar] [CrossRef]
- Bernier, C.M.; DuChane, C.M.; Merola, J.S. Crystal structures of (η4 -cycloocta-1,5-diene)bis(1,3-dimethylimidazol-2-ylidene)iridium(I) iodide and (η4 -cycloocta-1,5-diene)bis(1,3-diethylimidazol-2-ylidene)iridium(I) iodide. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Scheme 1.
Transition metal catalyzed enantioselective reduction of ketones to chiral alcohols.

Figure 1.
(A) Structure of Noyori’s ruthenium arene TsDPEN active catalyst for ATH of ketones. (B) Noyori’s catalyst operates through a concerted outer-sphere mechanism where the substrate does not directly bind to the metal center. For aryl ketone substrates, the transition state is stabilized by CH/π interactions between the catalyst and ketone.
Figure 1.
(A) Structure of Noyori’s ruthenium arene TsDPEN active catalyst for ATH of ketones. (B) Noyori’s catalyst operates through a concerted outer-sphere mechanism where the substrate does not directly bind to the metal center. For aryl ketone substrates, the transition state is stabilized by CH/π interactions between the catalyst and ketone.

Figure 2.
A few examples of previously reported iridium NHC catalysts and their optimal ee’s for asymmetric transfer hydrogenation of various ketone substrates.
Figure 2.
A few examples of previously reported iridium NHC catalysts and their optimal ee’s for asymmetric transfer hydrogenation of various ketone substrates.

Figure 3.
Highly enantioselective iridium NHC catalyst for ATH of ketones designed by Yoshida.

Figure 4.
Structure and modularity of iridium NHC amino acid complexes reported in this article for asymmetric transfer hydrogenation of ketones, highlighting features that have potential for specific tailoring. These complexes incorporate inexpensive, easily accessible ligands, and conveniently contain a hydride on the metal center prior to initiating the catalysis.
Figure 4.
Structure and modularity of iridium NHC amino acid complexes reported in this article for asymmetric transfer hydrogenation of ketones, highlighting features that have potential for specific tailoring. These complexes incorporate inexpensive, easily accessible ligands, and conveniently contain a hydride on the metal center prior to initiating the catalysis.

Scheme 2.
Synthesis of novel iridium NHC amino acid complexes by oxidative addition of α-amino acids, where aa = chelated amino acid and X = halide.
Scheme 2.
Synthesis of novel iridium NHC amino acid complexes by oxidative addition of α-amino acids, where aa = chelated amino acid and X = halide.

Figure 5.
Displacement ellipsoid plot (50% probability) of Ir(IMe)2(Gly)(H)(I) (CSD 2062242). The complex crystallized in the triclinic space group P. Hydrogen atoms are omitted for clarity.
Figure 5.
Displacement ellipsoid plot (50% probability) of Ir(IMe)2(Gly)(H)(I) (CSD 2062242). The complex crystallized in the triclinic space group P. Hydrogen atoms are omitted for clarity.

Figure 6.
Upon oxidative addition, two metal enantiomers are formed when glycine is used (top-left). Employing a homochiral primary amino acid gives rise to a pair of metal diastereomers (top-right), while incorporation of a secondary amino acid such as l-proline gives rise to four possible metal diastereomers (bottom).
Figure 6.
Upon oxidative addition, two metal enantiomers are formed when glycine is used (top-left). Employing a homochiral primary amino acid gives rise to a pair of metal diastereomers (top-right), while incorporation of a secondary amino acid such as l-proline gives rise to four possible metal diastereomers (bottom).

Figure 7.
1H NMR spectra (400 MHz, DMSO-d6) showing the hydride resonances for complexes of the form Ir(IMe)2(aa)(H)(I), where aa = glycine, l-alanine, l-valine, and l-phenylglycine.
Figure 7.
1H NMR spectra (400 MHz, DMSO-d6) showing the hydride resonances for complexes of the form Ir(IMe)2(aa)(H)(I), where aa = glycine, l-alanine, l-valine, and l-phenylglycine.

Scheme 3.
Reduction of acetophenone catalyzed by Ir(NHC)2(aa)(H)(X) complexes.

Figure 8.
1H NMR spectra (400 MHz, IPA, DMSO-d6 insert) showing the hydride resonances of Ir(IMe)2(l-Pro)(H)(I) before (top) and after (bottom) the addition of NaOH.
Figure 8.
1H NMR spectra (400 MHz, IPA, DMSO-d6 insert) showing the hydride resonances of Ir(IMe)2(l-Pro)(H)(I) before (top) and after (bottom) the addition of NaOH.

Figure 9.
Conversion of acetophenone over time at various temperatures. All conversions appeared to level off after a period of 8 h.
Figure 9.
Conversion of acetophenone over time at various temperatures. All conversions appeared to level off after a period of 8 h.

Figure 10.
Enantiomeric excess of 1-phenylethanol over time at various temperatures. Enantioselectivity degraded over time at temperatures above 25 °C.
Figure 10.
Enantiomeric excess of 1-phenylethanol over time at various temperatures. Enantioselectivity degraded over time at temperatures above 25 °C.

Figure 11.
Effect of varying the amount of NaOH on the conversion of acetophenone. Little to no conversion of acetophenone was observed when employing weaker bases.
Figure 11.
Effect of varying the amount of NaOH on the conversion of acetophenone. Little to no conversion of acetophenone was observed when employing weaker bases.

Figure 12.
Effect of varying the catalyst loading on the conversion of acetophenone.

Figure 13.
Substrate scope for ATH of ketones catalyzed by Ir(IMe)2(l-Pro)(H)(I). All reactions were carried out using the optimized catalytic conditions.
Figure 13.
Substrate scope for ATH of ketones catalyzed by Ir(IMe)2(l-Pro)(H)(I). All reactions were carried out using the optimized catalytic conditions.

Scheme 4.
Proposed catalytic cycle for ATH of aryl ketones in this system, where S is representative of the solvent (IPA). Steps: A. solvent complexed cationic resting state of catalyst; B. positioning of substrate with hydride attacking the carbonyl carbon and N-H bonding to carbonyl oxygen; C. elimination of reduced product; D. coordination of isopropanol followed by reductive elimination to regenerate metal hydride (A). Metal-ligand bifunctional catalysts of this type typically operate through an outer-sphere mechanism, where the substrate does not directly bind to the metal center.
Scheme 4.
Proposed catalytic cycle for ATH of aryl ketones in this system, where S is representative of the solvent (IPA). Steps: A. solvent complexed cationic resting state of catalyst; B. positioning of substrate with hydride attacking the carbonyl carbon and N-H bonding to carbonyl oxygen; C. elimination of reduced product; D. coordination of isopropanol followed by reductive elimination to regenerate metal hydride (A). Metal-ligand bifunctional catalysts of this type typically operate through an outer-sphere mechanism, where the substrate does not directly bind to the metal center.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
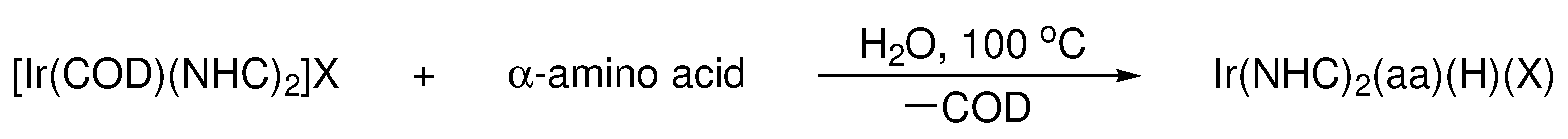{kind=link}
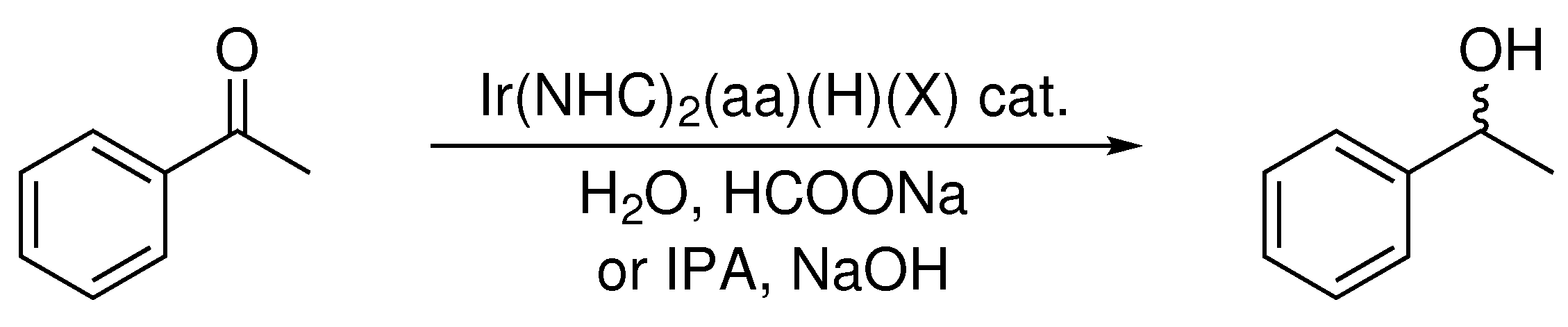{kind=link}
{kind=link}
Table 1.
Scope and synthetic yields of iridium NHC amino acid complexes featured in this work.
| NHC | Amino Acid | Yield (%) |
|---|---|---|
| IMe a | glycine | 53 |
| l-alanine | 78 | |
| l-valine | 87 | |
| l-phenylglycine | 66 | |
| l-azetidine-2-carboxylic acid | 55 | |
| l-proline | 92 | |
| d-proline | 72 | |
| cis-4-fluoro-l-proline | 44 | |
| l-pipecolic acid | 66 | |
| IEt a | l-proline | 68 |
| IiPr b | l-proline | 82 |
a X = I−; b X = Cl−.
Table 2.
Selected bond lengths and angles from the crystal structure of Ir(IMe)(Gly)(H)(I).
| Atoms | Bond Lengths (Å) |
| Ir1–C1 | 1.986 |
| Ir1–C4 | 2.004 |
| Ir1–O1 | 2.231 |
| Ir1–N5 | 2.157 |
| Ir1–I1 | 2.738 |
| Atoms | Bond Angles (°) |
| C1–Ir1–C4 | 89.39 |
| C1–Ir1–O1 | 91.49 |
| C1–Ir1–N5 | 90.53 |
| C4–Ir1–O1 | 101.52 |
| C4–Ir1–I1 | 93.28 |
| O1–Ir1–I1 | 91.60 |
| N5–Ir1–I1 | 86.85 |
Table 3.
Initial catalytic results for the reduction of acetophenone. All trials were carried out using 2.5 mol% of catalyst at 80 °C over a period of 24 h.
Table 3.
Initial catalytic results for the reduction of acetophenone. All trials were carried out using 2.5 mol% of catalyst at 80 °C over a period of 24 h.
| Solvent | NHC | Amino Acid | Conv. (%) d |
|---|---|---|---|
| H2O a | IMe c | glycine | 44 |
| l-alanine | 50 | ||
| l-proline | 59 | ||
| IPA b | IMe c | glycine | 76 |
| l-alanine | 86 | ||
| l-proline | 98 |
a 500 equiv. HCOONa; b 2 equiv. NaOH (equiv. relative to catalyst); c X = I−; d Conversion was quantified via 1 H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard.
Table 4.
Initial catalytic results for the ATH of acetophenone. All trials were carried out using 2.5 mol% of catalyst and 2 equiv. of NaOH in IPA at 80 °C over a period of 24 h.
Table 4.
Initial catalytic results for the ATH of acetophenone. All trials were carried out using 2.5 mol% of catalyst and 2 equiv. of NaOH in IPA at 80 °C over a period of 24 h.
| NHC | Amino Acid | Conv. (%) b | ee (%) b | Conf. |
|---|---|---|---|---|
| IMe a | glycine | 75 | 0 | – |
| l-alanine | 81 | 3 | R | |
| l-valine | 74 | 5 | R | |
| l-phenylglycine | 77 | 6 | R | |
| l-azetidine-2-carboxylic acid | 83 | 17 | R | |
| l-proline | 98 | 68 | R | |
| d-proline | 95 | 68 | S | |
| l-pipecolic acid | 82 | 4 | S |
a X = I−; b Conversion and enantiomeric excess were quantified via chiral GC-FID using 1,3,5-trimethoxybenzene as an internal standard.
Table 5.
Catalyst scope for ATH of acetophenone using the optimized catalytic conditions. Error values are representative of the absolute mean residual from three independent trials.
Table 5.
Catalyst scope for ATH of acetophenone using the optimized catalytic conditions. Error values are representative of the absolute mean residual from three independent trials.
| NHC | Amino Acid | Conv. (%) | ee (%) | Conf. |
|---|---|---|---|---|
| IMe a | glycine | 46 ± 2 | 0 | – |
| l-alanine | 52 ± 1 | 9 ± 1 | R | |
| l-valine | 45 ± 3 | 6 ± 1 | R | |
| l-phenylglycine | 49 ± 2 | 8 ± 1 | R | |
| l-azetidine-2-carboxylic acid | 56 ± 1 | 67 ± 2 | R | |
| l-proline | 91 ± 1 | 92 ± 1 | R | |
| d-proline | 92 ± 2 | 92 ± 1 | S | |
| cis-4-fluoro-l-proline | 44 ± 2 | 89 ± 2 | R | |
| l-pipecolic acid | 55 ± 3 | 17 ± 1 | S | |
| IEt a | l-proline | 98 ± 1 | 55 ± 2 | R |
| IiPr b | l-proline | 98 ± 1 | 19 ± 1 | R |
a X = I−; b X = Cl−.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bernier, C.M.; Merola, J.S. Design of Iridium N-Heterocyclic Carbene Amino Acid Catalysts for Asymmetric Transfer Hydrogenation of Aryl Ketones. Catalysts 2021, 11, 671. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11060671
AMA Style
Bernier CM, Merola JS. Design of Iridium N-Heterocyclic Carbene Amino Acid Catalysts for Asymmetric Transfer Hydrogenation of Aryl Ketones. Catalysts. 2021; 11(6):671. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11060671
Chicago/Turabian StyleBernier, Chad M., and Joseph S. Merola. 2021. "Design of Iridium N-Heterocyclic Carbene Amino Acid Catalysts for Asymmetric Transfer Hydrogenation of Aryl Ketones" Catalysts 11, no. 6: 671. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11060671
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.