3.2. Synthesis
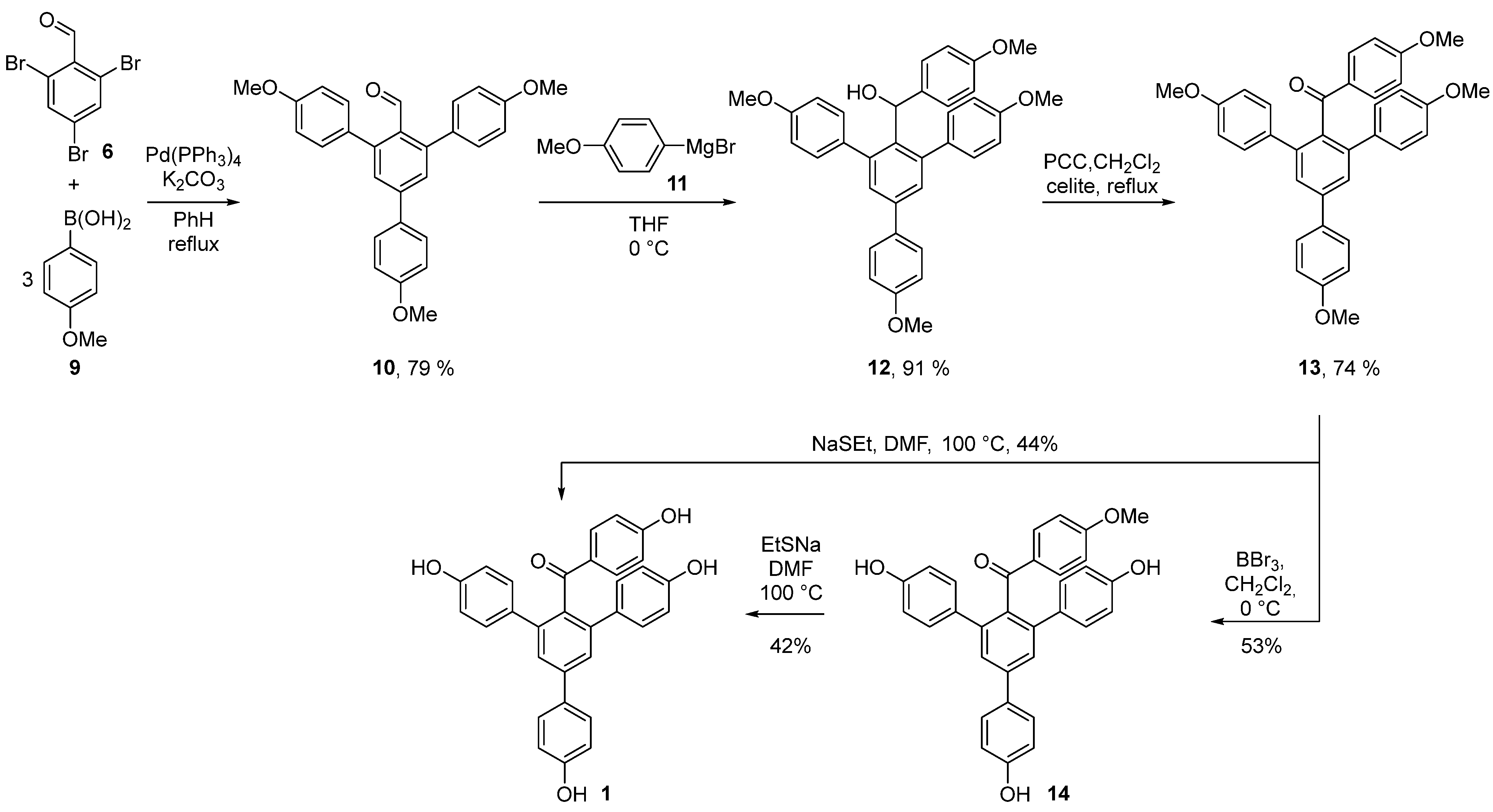
3.2.1. Synthesis of 4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-2′-carbaldehyde (10)
2,4,6-Tribromobenzaldehyde 6 (0.73 mmol, 250 mg), Pd(PPh3)4 (0.037 mmol, 5 mol%, 42 mg), K2CO3 (2.56 mmol, 353 mg), and (4-methoxyphenyl)boronic acid 9 (2.3 mmol, 355 mg) were dissolved in a degassed mixture of benzene and H2O (5:1, 6 mL). The reaction was heated in a closed vial at 90 °C for 16 h. Then, the reaction mixture was concentrated and the product was purified with column chromatography (EA:Hex 1:6 to 1:4). The reaction yielded 574 mg (93%) of product in the form of a yellow glassy oil. Rf = 0.3 [EA:Hex (1:4)]; IR (KBr) 3033, 2999, 2954, 2933, 2906, 2835, 2754, 1693, 1608 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.70–7.59 (m, 2H), 7.54 (s, 2H), 7.41–7.32 (m, 4H), 7.10–6.91 (m, 6H), 3.88 (s, 6H), 3.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.4, 160.2, 159.4 (2C), 145.1 (2C), 143.9, 132.3 (2C), 132.0, 131.3, 131.0 (4C), 128.6 (2C), 128.4 (2C), 114.6 (2C), 113.8 (4C), 55.5 (3C); HRMS (ESI) calculated for C28H25O4 (MS + H+): 425.1747; found 425.1746.
3.2.2. Synthesis of (4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanol (12)
A solution of (4-methoxyphenyl)magnesium bromide 11 (1 M in THF, 0.1 mmol, 0.1 mL) was added in a dropwise manner to a solution of 4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-2′-carbaldehyde 10 (0.071 mmol, 34 mg) in THF (1 mL) at 0 °C. After stirring the reaction in the ice bath for 40 min, it was quenched with saturated NH4Cl solution (3 mL). The product was extracted with EA (3 × 5 mL). Combined organic phases were dried over Na2SO4, then filtered and concentrated. The product was purified by column chromatography (gradient eluent EA:Hex 1:6 to 1:4). The reaction yielded 30 mg (79%) of product in the form of a colorless glassy oil. Rf = 0.2 [EA:Hex (1:4)]; IR (KBr) 3556, 3033, 2999, 2952, 2933, 2906, 2835, 2044, 1888, 1736, 1608 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.62–7.56 (m, 2H), 7.43 (s, 2H), 7.13 (d, J = 7.9 Hz, 4H), 6.99–6.93 (m, 2H), 6.91–6.85 (m, 2H), 6.84–6.78 (m, 4H), 6.73–6.67 (m, 2H), 5.95 (d, J = 9.2 Hz, 1H), 3.84 (s, 3H), 3.81 (s, 6H), 3.77 (s, 3H), 2.14 (d, J = 10.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 159.5, 158.9 (2C), 158.1, 142.8 (2C), 139.1, 138.0 (2C), 134.0 (2C), 132.7, 130.8 (4C), 128.8 (2C), 128.2 (2C), 126.7 (2C), 114.4 (2C), 113.5 (4C), 113.2 (2C), 72.3, 55.5, 55.4 (3C); HRMS (ESI) calculated for C35H31O4: 515.2217; found 515.2219.
3.2.3. Synthesis of (4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone (13)
PCC (0.483 mmol, 104 mg), (4,4′
’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′
’-terphenyl]-4′-yl)(4-methoxyphenyl)methanol
12 (0.4 mmol, 214 mg), and Celite
® (214 mg) were suspended in DCM (15 mL) and refluxed for 24 hours. Then reaction mixture was filtered through a plug of Celite
® and concentrated. On TLC, the conversion was visible only when the plate was stained with Hanessian’s stain (CAM). The product was purified by column chromatography (EA:Hex 1:6 to 1:4). Reaction yielded 156 mg (74%) of product in the form of a colorless glassy oil. R
f = 0.2 [EA:Hex (1:4)]; mp = 180–182 °C (DCM:MeOH), lit. 173–174 °C [
25] (AcOH); IR (KBr) 3033, 3001, 2956, 2933, 2908, 2835, 2048, 1660, 1606, 1597 cm
−1;
1H NMR (400 MHz, CDCl
3) δ 7.67–7.61 (m, 2H), 7.58–7.53 (m, 4H), 7.26–7.22 (m, 4H), 7.03–6.98 (m, 2H), 6.79–6.73 (m, 4H), 6.70–6.64 (m, 2H), 3.87 (s, 3H), 3.76 (s, 3H), 3.74 (s, 6H);
13C NMR (100 MHz, CDCl
3) δ 197.9, 163.2, 159.7, 158.9 (2C), 141.2, 140.9, 136.9 (2C), 133.1 (2C), 132.8, 131.9, 131.7 (2C), 130.4 (4C), 128.4 (2C), 127.2 (2C), 114.5 (2C), 113.7 (4C), 113.4 (2C), 55.5, 55.4, 55.3 (2C); HRMS (ESI) calculated for C
35H
31O
5 (M + H): 531.2166; found 531.2167.
3.2.4. Synthesis of (4,4′’-dihydroxy-5′-(4-hydroxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone (14)
BBr3 solution (1 M in heptane, 0.415 mmol, 0.415 mL) was added into a solution of (4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone (0.094 mmol, 50 mg) in DCM (2 mL) in a dropwise manner at 0 °C. The reaction was stirred at room temperature for 16 h. The reaction mixture was quenched with a solution of NaHSO3 (50%, 5 mL), then extracted with EA (3 × 7 mL). Organic phases were combined, washed with brine (5 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The product was purified by column chromatography (EA:Hex 1:4 to 1:1). The reaction yielded 24 mg (53%) of product in the form of a colorless glassy oil. Rf = 0.2 [EA:Hex (1:1)]; IR (KBr) 3323, 3070, 3033, 3014, 2964, 2935, 2839, 2044, 1894, 1643, 1610, 1591 cm−1; 1H NMR (600 MHz, MeOD-d4) δ 7.59–7.56 (m, 2H), 7.51–7.48 (m, 4H), 7.12–7.08 (m, 4H), 6.92–6.88 (m, 2H), 6.78–6.74 (m, 2H), 6.64–6.60 (m, 4H), 3.77 (s, 3H); 13C NMR (150 MHz, MeOD-d4) δ 200.7, 165.2 (2C), 158.8, 157.9 (2C), 143.1, 142.4 (2C), 137.5, 133.2 (2C), 133.1, 132.7, 132.6, 131.5 (4C), 129.3 (2C), 127.6 (2C), 116.8 (2C), 115.9 (4C), 114.5 (2C), 56.0; HRMS (ESI) calculated for C32H25O5 (M + H): 489.1697; found 489.1697.
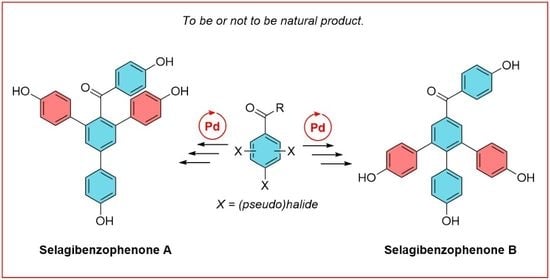
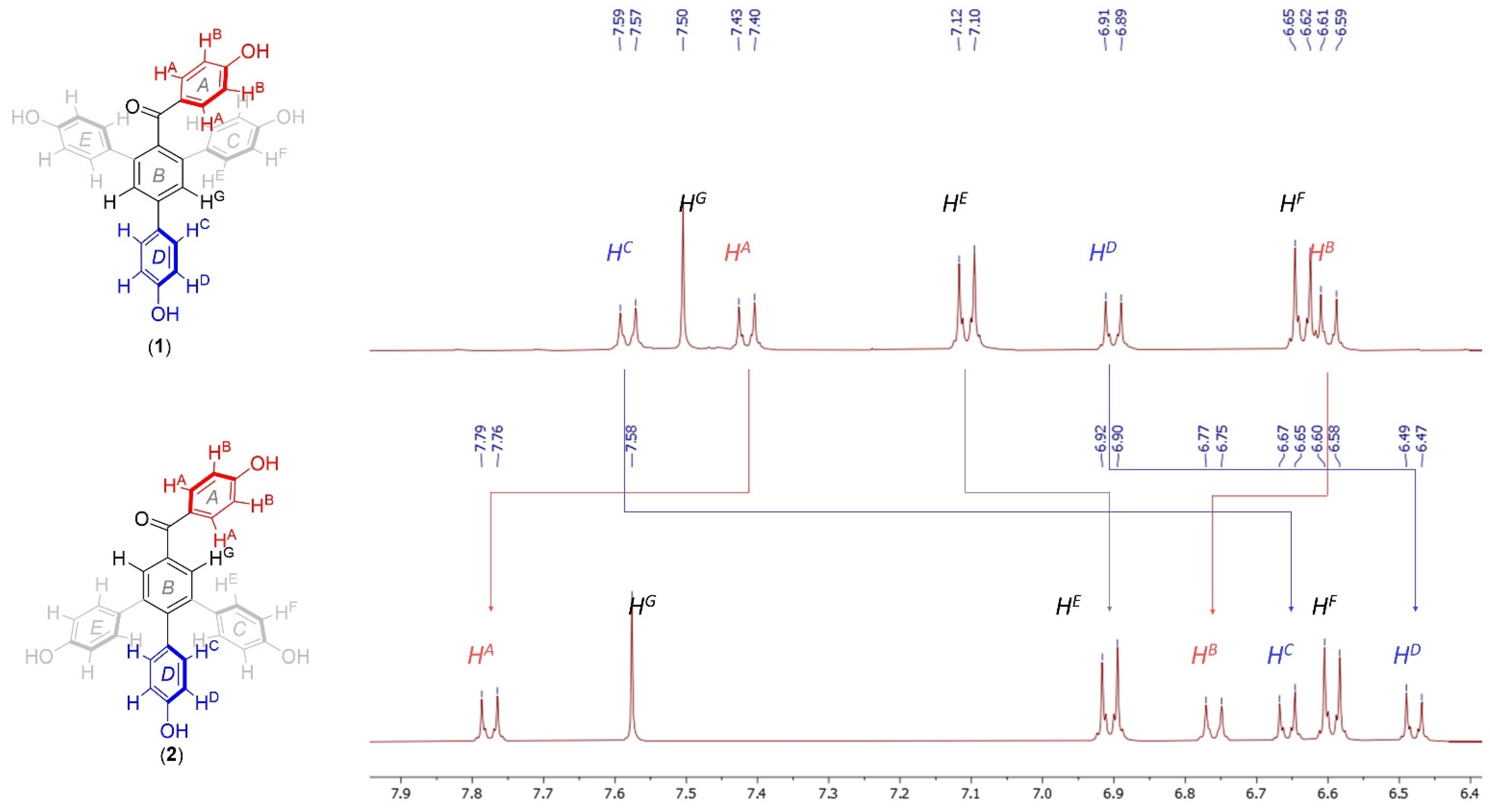
3.2.5. Synthesis of (4,4′’-dihydroxy-5′-(4-hydroxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-hydroxyphenyl)methanone—selagibenzophenone A (1)
Procedure A: A solution of (4,4′’-dihydroxy-5′-(4-hydroxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone 13 (0.01 mmol, 5 mg) and NaSEt (0.24 mmol, 20 mmol) in DMF (0.8 mL, anhydrous) was heated in a vial at 100 °C. After 10 h, the reaction mixture was cooled down and diluted with an aqueous HCl (1 M, 8 mL). The product was extracted with EA (3 × 10 mL). Combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced vacuum. The product was purified by preparative TLC with EA:Hex (1:2 × 3). The reaction yielded 2 mg (42%) of product in the form of an off-white glassy oil.
Procedure B: A solution of (4,4′’-dimethoxy-5′-(4-methoxyphenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone 14 (0.038 mmol, 20 mg) and NaSEt (0.667 mmol, 56 mg) in DMF was loaded into a vial, closed, and heated at 100 °C. After 18 h, the reaction mixture was cooled down and diluted with HCl solution (1 M, 15 mL). The product was extracted with EA (3 × 20 mL). Organic phases were washed with brine (1 × 4 mL), dried over Na2SO4, filtered, and concentrated. The product was purified using column chromatography (EA:Hex, 1:4 to 2:1). The reaction yielded 8 mg (44%) of product in the form of an off-white glassy oil.
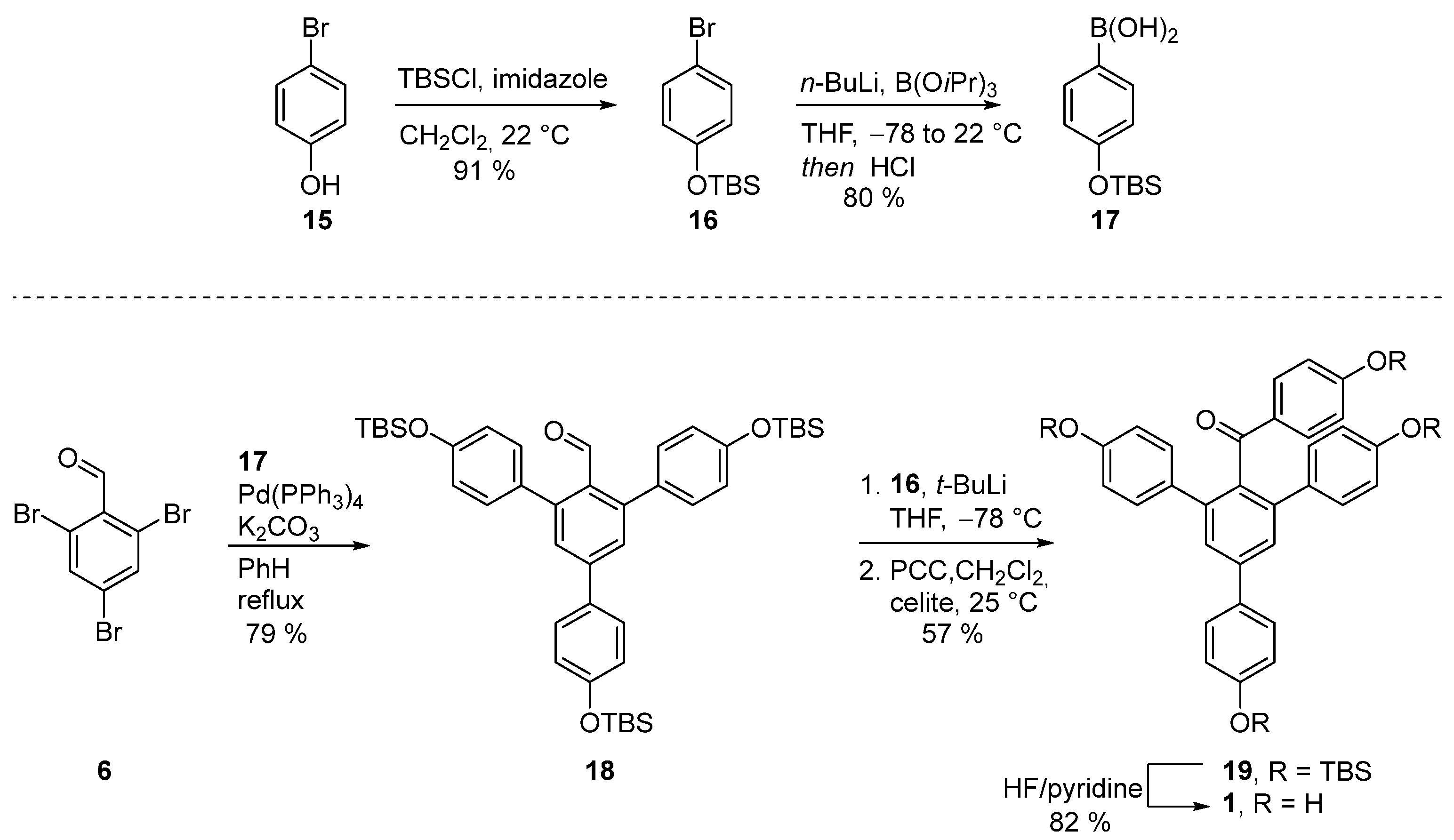
Procedure C: Olah’s reagent (HF·pyridine, 12 mmol, 0.3 mL) was added to a solution of (4,4′
’-bis((tert-butyldimethylsilyl)oxy)-5′-(4-((tert-butyldimethylsilyl)oxy)phenyl)-[1,1′:3′,1′
’-terphenyl]-4′-yl)(4-((tert-butyldimethylsilyl)oxy)phenyl)methanone (
19) (0.02 mmol, 20 mg) in THF (5 mL) in a plastic flask and stirred at 22 °C for 2 h. Then, the reaction mixture was quenched with saturated solution NaHCO
3 (5 mL) and extracted with EA (3 × 10 mL). Combined organic layers were washed with HCl (1M, 1 × 20 mL), dried over Na
2SO
4, filtrated, and concentrated under reduced pressure. The selagibenzophenone A (
1) was purified with column chromatography (DCM:MeOH 20:1). The reaction yielded 8.4 mg (82%) of product in the form of a slightly yellow solid. R
f = 0.1 [1:1 (EA:Hex)];
1H NMR (600 MHz, MeOD-
d4) δ 7.60–7.56 (m, 2H), 7.50 (s, 2H), 7.43–7.39 (m, 2H), 7.12–7.09 (m, 4H), 6.92–6.88 (m, 2H), 6.65–6.62 (m, 4H), 6.61–6.58 (m, 2H);
13C NMR (150 MHz, MeOD-
d4) δ 200.7, 163.8, 158.8, 157.9 (2C), 143.0, 142.4 (2C), 137.6, 133.5 (2C), 133.2 (2C), 132.8, 131.6, 131.5 (4C), 129.3 (2C), 127.6 (2C), 116.8 (2C), 116.0 (2C), 115.8 (4C). The recorded values are in a good agreement with the published data [
2].
3.2.6. Synthesis of (4-bromophenoxy)(tert-butyl)dimethylsilane (16)
Tert-butyl dimethyl silyl chloride (104 mmol, 16 g) and imidazole (104 mmol, 7.5 g) were added to a solution of a 4-bromophenol
15 (87 mmol, 15.5 g) in DCM (80 mL) at 0 °C. After 16 hours of stirring at 22 °C, the reaction mixture was filtered through a short pad of silica gel and washed with hexanes (200 mL). The product was used in the next reaction without additional purification. The reaction yielded 22.5 g (91%) of product in the form of a transparent oil. R
f = 0.4 [Hex];
1H NMR (400 MHz, CDCl
3) δ 7.37–7.34 (m, 2H), 6.77–6.74 (m, 2H), 1.03 (s, 9H), 0.23 (s, 6H).
13C NMR (100 MHz, CDCl
3) δ 155.0, 132.4 (2C), 122.0 (2C), 113.8, 25.8 (3C), 18.3, −4.4 (2C). The recorded values are in good agreement with the published data [
26].
3.2.7. Synthesis of (4-((tert-butyldimethylsilyl)oxy)phenyl)boronic acid (17)
A solution of
n-BuLi (1.6 M in hexane, 3.8 mmol, 2.4 mL) was slowly added to a solution of (4-bromophenoxy)(tert-butyl)dimethylsilane (
16) (3.5 mmol, 1 g) in THF (10 mL) at −78 °C. After 30 min, triisopropyl borate (10.5 mmol, 2.3 mL) was added in a dropwise manner at −78 °C and the reaction was then allowed to warm to 22 °C and stirred for 16 h. The reaction mixture was acidified with HCl (1 M, 3 mL). Then, the organic layer was washed with brine (1 × 5 mL), dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The product was purified with column chromatography (DCM to DCM:MeOH 20:1). The reaction yielded 700 mg (80%) of product in the form of an off-white solid.
1H NMR (400 MHz, CDCl
3) δ 8.13–8.11 (m, 2H), 6.97–6.95 (m, 2H), 1.02 (s, 9H), 0.26 (s, 6H).
13C NMR (100 MHz, CDCl
3) δ 159.9, 137.6 (2C), 123.1, 119.9 (2C), 25.9 (3C), 18.5, −4.2 (2C). The recorded values are in good agreement with the published data [
26].
3.2.8. Synthesis of 4,4′’-bis((tert-butyldimethylsilyl)oxy)-5′-(4-((tert-butyldimethylsilyl)oxy)phenyl)-[1,1′:3′,1′’-terphenyl]-2′-carbaldehyde (18)
2,4,6-Tribromobenzaldehyde 6 (0.29 mmol, 100 mg), Pd(PPh3)4 (0.029 mmol, 10 mol%, 34 mg), K2CO3 (1.02 mmol, 140 mg), and (4-methoxyphenyl)boronic acid 9 (0.92 mmol, 140 mg) were dissolved in a degassed mixture of benzene and H2O (5:1, 2.2 mL). The reaction was heated in a closed vial at 90 °C for 16 h. Then, the reaction mixture was concentrated and the product was purified with column chromatography (EA:Hex 1:20 to 1:4). The reaction yielded 166 mg (79%) of product in the form of a yellow glassy oil. Rf = 0.14 [EA:Hex (1:30)]; IR (KBr) 3033, 2956, 2858, 2540, 1699, 1604, 1508, 1263, 914, 837, 781 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 7.57 (d, J = 8.6 Hz, 2H), 7.52 (s, 2H), 7.30–7.22 (m, 4H), 6.96–6.86 (m, 6H), 1.02 (s, 18H), 1.00 (s, 9H), 0.25 (s, 12H), 0.23 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 193.5, 156.4, 155.6 (2C), 145.1 (2C), 143.8, 132.9 (2C), 132.7, 131.3, 130.9 (4C), 128.6 (2C), 128.4 (2C), 120.7 (2C), 119.8 (4C), 25.8 (9C), 18.4 (3C), −4.2 (6C); HRMS (ESI) calculated for C43H61O4Si3 (MS + H+): 725.3872; found 725.3895.
3.2.9. Synthesis of (4,4′’-bis((tert-butyldimethylsilyl)oxy)-5′-(4-((tert-butyldimethylsilyl)oxy)phenyl)-[1,1′:3′,1′’-terphenyl]-4′-yl)(4-((tert-butyldimethylsilyl)oxy)phenyl)methanone (19)
A solution of t-BuLi (1.7 M in heptane, 0.25 mmol, 0.16 mL) was added in a dropwise manner to a solution of (4-bromophenoxy)(tert-butyl)dimethylsilane (16, 0.25 mmol, 70 mg) in THF (3 mL) at −78 °C. After 20 min, a solution of 4,4′’-bis((tert-butyldimethylsilyl)oxy)-5′-(4-((tert-butyldimethylsilyl)oxy)phenyl)-[1,1′:3′,1′’-terphenyl]-2′-carbaldehyde (18, 0.18 mmol, 130 mg) in THF (3 mL) was added and the reaction was stirred at −78 °C for 1 h. The reaction mixture was warmed up to room temperature, quenched with water (10 mL), and extracted with EA (3 × 20 mL). Combined organic layers were dried over MgSO4, filtered, and concentrated. The resulting crude product was redissolved in DCM (10 mL) and Celite® (200 mg) and PCC (0.4 mmol, 90 mg) was added to it. After 16 h, the reaction mixture was filtered through a pad of Celite®. The product was purified with column chromatography (DCM:Hex, 1:3 to 1:1). The reaction yielded 96 mg (57%) of product in the form of a yellow oil. Rf = 0.2 [DCM:Hex (1:2)]; IR (KBr) 2966, 2929, 2887, 2858, 1666, 1597, 1508, 1263 cm−1; 1H NMR (400 MHz, CD3OD) 7.59–7.57 (m, 2H), 7.55 (s, 2H), 7.44–7.42 (m, 2H), 7.17–7.15 (m, 4H), 6.94–6.92 (m, 2H), 6.68–6.66 (m, 4H), 6.59–6.57 (m, 2H), 1.01 (s, 9H), 0.94 (s, 27H), 0.24 (s, 6H), 0.16 (s, 6H), 0.13 (s, 12H). 13C NMR (100 MHz, CD3OD) δ 197.9, 159.8, 155.9, 155.1 (2C), 141.3, 141.1 (2C), 136.9, 133.8 (2C), 133.5, 132.4, 131.7 (2C), 130.5 (4C), 128.4 (2C), 127.2 (2C), 120.6 (2C), 119.8 (4C), 119.4 (2C), 25.9 (3C), 25.8 (6C), 25.7 (3C), 18.4, 18.3 (3C), −4.2 (2C), −4.3 (2C), −4.3 (4C); HRMS (ESI) calculated for C55H78O5Si4 (MS + H+): 931.4999; found 931.5030.
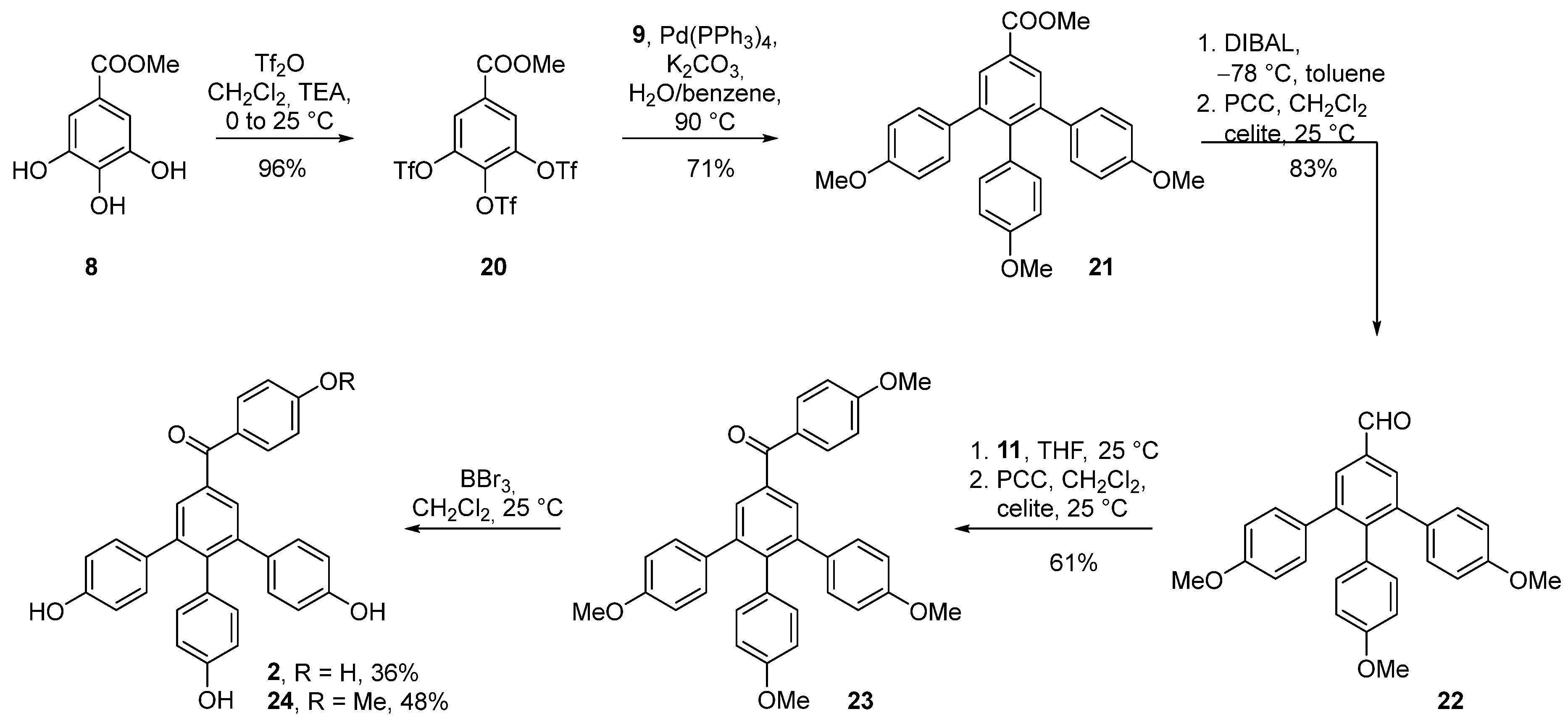
3.2.10. Synthesis of Methyl 3,4,5-tris(((trifluoromethyl)sulfonyl)oxy)benzoate (20)
In a flame-dried flask, methyl gallate (7.6 mmol, 1.5 g) was dissolved in CH2Cl2 (20 mL) under an inert atmosphere. Triethyl amine (24.2 mmol, 3.4 mL) was added, whereupon the suspension dissolved. The mixture was cooled to 0 °C and Tf2O (24.2 mmol, 4.07 mL) was added. The mixture was heated to the room temperature and stirred for 5 minutes. After this, the reaction was quenched via the addition of 5% HCl (20 mL). The organic phase was separated and washed with NaHCO3 (15 mL) and brine (15 mL). The organic phase was dried with MgSO4 and concentrated under reduced pressure. The mixture was used without further purification for the next step. Rf = 0.3 [EA:Hex (1:10)]; mp = 49–51 °C (DCM); IR (KBr) 3116, 2968, 2682, 2355, 1728, 1597, 1435, 1321 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.20 (s, 2H), 4.02 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 162.7, 142.4, 132.1, 125.1, 124.1, 120.8, 120.7, 116.6, 116.4, 112.3, 53.8; HRMS (ESI) calculated for C11H9F9NO11S3 (MS + NH4+): 597.9198; found 597.9192.
3.2.11. Synthesis of Methyl 4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-carboxylate (21)
Aldehyde 20 (2.60 mmol, 1.49 mg), Pd(PPh3)4 (0.26 mmol, 10 mol%, 300 mg), K2CO3 (9.1 mmol, 1.25 mg), and (4-methoxyphenyl)boronic acid 9 (8.20 mmol, 1.24 mg) were dissolved in a degassed mixture of benzene and H2O (5:1, 20 mL). The reaction was heated in a closed vial at 90 °C for 16 h. Then, the reaction mixture was concentrated and the product was purified with column chromatography (EA:Hex 1:99 to 1:10). The reaction yielded 865 mg (71%) of product in the form of a yellow glassy oil. Rf = 0.15 [EA:Hex (1:4)]; mp = 133–136 °C (DCM); IR (KBr) 3066, 3005, 2993, 2935, 2839, 2065, 1716, 1608, 1514, 1435 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 2H), 7.03–6.96 (m, 4H), 6.80–6.68 (m, 6H), 6.61–6.54 (m, 2H), 3.94 (s, 3H), 3.76 (s, 6H), 3.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.2, 158.3 (2C), 158.0, 143.4, 142.2, 133.8, 132.5 (2C), 131.4, 131.0 (4C), 130.4 (2C), 128.8, 113.3 (4C), 113.1 (2C), 55.3, 55.1, 52.3; HRMS (ESI) calculated for C29H27O5 (MS + H+): 455.1853; found 455.1853.
3.2.12. Synthesis of 4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-carbaldehyde (22)
A solution of LiAlH4 (1M in THF, 1.6 mmol, 1.6 mL) was added dropwise into a solution of methyl 4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-carboxylate (21) (0.82 mmol, 400 mg) in THF (10 mL) at 0 °C. After 1 h, the reaction mixture was quenched with Na2SO4 (300 mg), filtered through a pad of Celite®, then the filtrate was concentrated under reduced pressure. The crude mixture was then dissolved in DCM (10 mL). After adding Celite® (400 mg) and PCC (1.6 mmol, 356 mg), the reaction mixture was left to stir for 16 h. Then, it was filtered through a plug of Celite®, the filtrate was concentrated, and the product was purified with column chromatography (EA:Hex 1:5). The reaction yielded 270 mg (72%) of product in the form of a yellow glassy oil. Rf = 0.2 [EA:Hex (1:4)]; IR (KBr) 3032, 2999, 2956, 2933, 2835, 2729, 2536, 2044, 1888, 1695, 1512, 1441cm−1; 1H NMR (400 MHz, CDCl3) δ 10.09 (s, 1H), 7.86 (s, 2H), 7.00–6.98 (m, 4H), 6.74–6.72 (m, 6H) 6.59–6.57 (m, 2H), 3.77 (s, 6H), 3.71 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.3, 158.5 (2C), 158.2, 145.1, 142.9 (2C), 135.2, 133.5 (2C), 132.4 (2C), 131.2, 130.9 (4C), 130.6 (2C), 113.4 (4C), 113.2 (2C), 55.3 (2C), 55.2; HRMS (ESI) calculated for C28H25O4 (MS + H+): 425.1747; found 425.1749.
3.2.13. Synthesis of (4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone) (23)
A solution of (4-methoxyphenyl)magnesium bromide (1M in THF, 1 mL, 1 mmol) was added in a dropwise manner to a solution of 4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-carbaldehyde (22, 0.64 mmol, 270 mg) in THF (10 mL) at 0 °C. After 16 h, the reaction was quenched with a saturated solution of NH4Cl (20 mL) and extracted with EA (3 × 30 mL). The combined organic phases were dried over Na2SO4, filtered, and evaporated. The crude mixture was then dissolved in DCM (20 mL) and Celite® (240 mg) and PCC (0.9 mmol, 196 mg) was added to it. After 16 h, the reaction mixture was filtered over a Celite® pad and the filtrate was concentrated. The product was purified with column chromatography (EA:Hex 1:10 to 4:1). The reaction yielded 141 mg (70%) of product in the form of a slightly yellow solid. Rf = 0.2 [EA:Hex (1:5)]; mp = 77–78 °C (DCM); IR (KBr) 3032, 3001, 2956, 2931, 2908, 2835, 1510, 1250, 1034, 831 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.95–7.93 (m, 2H), 7.75 (s, 2H), 7.01–6.97 (m, 6H), 6.77–6.70 (m, 2H), 6.72–6.70 (m, 4H), 6.60–6.57 (m, 2H), 3.89 (s, 3H), 3.76 (s, 3H), 3.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 198.4, 163.4, 158.3 (2C), 158.1, 142.4, 141.9 (2C), 137.1, 133.9 (2C), 132.7 (2C), 132.6 (2C), 131.5, 131.1 (4C), 130.7 (2C), 130.4, 113.8 (2C), 113.3 (4C), 113.1 (2C), 55.6, 55.3(2C), 55.2; HRMS (ESI) calculated for C35H31O5 (MS + H+): 531.2166; found 531.2161.
3.2.14. Synthesis of (4,4′’-dihydroxy-6′-(4-hydroxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-yl)(4-hydroxyphenyl)methanone—selagibenzophenone B (2)
A solution of a BBr3 (1M in heptane, 0.2 mmol, 0.2 ml) was added in a dropwise manner to a solution of the (4,4′’-dimethoxy-6′-(4-methoxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone (23, 0.026 mmol, 14 mg) in DCM (2 mL) at room temperature. After 16 h, the reaction was quenched with a saturated solution of NH4Cl (5 mL) and extracted with EA (3 × 10 mL). Combined organic phases were dried over Na2SO4, filtered, and evaporated. The product was purified with preparative TLC (DCM:MeOH, 20:1). The reaction yielded 6 mg of the monomethoxy derivative (4,4′’-dihydroxy-6′-(4-hydroxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone (48% yield) and 6 mg of the selagibenzophenone B (2) in the form yellow solids (36% yield). Selagibenzophenone B (2): Rf = 0.2 [DCM:MeOH 20:1]; mp = 250 °C (decomp.); IR (KBr) 3459, 2927, 2792, 1587, 1342 cm−1; 1H NMR (600 MHz, MeOD-d4) δ 7.79–7.55 (m, 2H), 7.58 (s, 2H), 6.93–6.88 (m, 4H), 6.77–6.74 (m, 2H), 6.77–6.74 (m, 2H), 6.61–6.58 (m, 4H), 6.50–6.46 (m, 2H); 13C NMR (150 MHz, MeOD-d4) δ 197.5, 170.5, 157.2 (2C), 156.9, 143.7, 143.4 (2C), 138.8, 134.6 (2C), 134.2, 133.7 (2C), 132.1 (4C), 131.9, 131.0 (2C), 126.5, 118.2 (2C), 115.6 (4C), 115.4 (2C). HRMS (ESI) calculated for C31H23O5 (MS + H+): 475.1540, found 475.1545.
3.2.15. Synthesis of (4,4′’-dihydroxy-6′-(4-hydroxyphenyl)-[1,1′:2′,1′’-terphenyl]-4′-yl)(4-methoxyphenyl)methanone—(monomethoxy-selagibenzophenone B) (24)
Rf = 0.4 [DCM:MeOH 20:1]; mp = 253–254 °C (DCM); IR (KBr) 3302, 3032, 2958, 2841, 1888, 1699, 1595, 1512, 1419, 1342, 1257 cm−1; 1H NMR (400 MHz, CD3OD) δ 7.90–7.88 (m, 2H), 7.62 (s, 2H), 7.07–7.05 (m, 2H), 6.90–6.88 (m, 4H), 6.66–6.64 (m, 2H), 6.60–6.58 (m, 4H), 6.49–6.47 (m, 2H,), 3.89 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 197.5, 165.2, 157.2 (2C), 156.9, 144.4, 143.7 (2C), 137.8, 134.0, 133.7 (3C), 133.7 (2C), 132.0 (4C), 131.7, 131.3 (2C), 131.2, 115.6 (4C), 115.4 (2C), 114.9 (2C), 56.1; HRMS (ESI) calculated for C32H25O5 (MS + H+): 489.1697 found 489.1691.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





