
Epoxidation of Terpenes


1
Department of Chemical Engineering, Université Laval, Québec, QC G1V 0A6, Canada
2
Department of Chemistry, The University of Helsinki, 00560 Helsinki, Finland
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(7), 847; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070847
Submission received: 15 June 2021
/
Revised: 1 July 2021
/
Accepted: 2 July 2021
/
Published: 14 July 2021
(This article belongs to the Special Issue Catalytic Epoxidation Reaction)
Abstract
:Terpene epoxides are considered as potential primary intermediates in the synthesis of numerous green polymers including epoxy resins, polycarbonates, nonisocyanate polyurethanes and even some polyamides. In this chapter we describe recent efforts from our group to develop catalytic and noncatalytic processes for terpene epoxidation using a variety of oxidizing agents and process intensification methods. Most experimental tests deal with limonene epoxidation with applicability to some other terpenes also demonstrated.
1. Introduction
In the recent quest for green substituents to carbonaceous monomers of fossil origin, terpenes occupy a special position [1,2,3]. Monoterpenes are naturally occurring olefinic hydrocarbons comprising ten carbon atoms. Their chemical properties are therefore similar to those of significant petroleum derived monomers systematically used today in the production of high tonnage polymers [4].
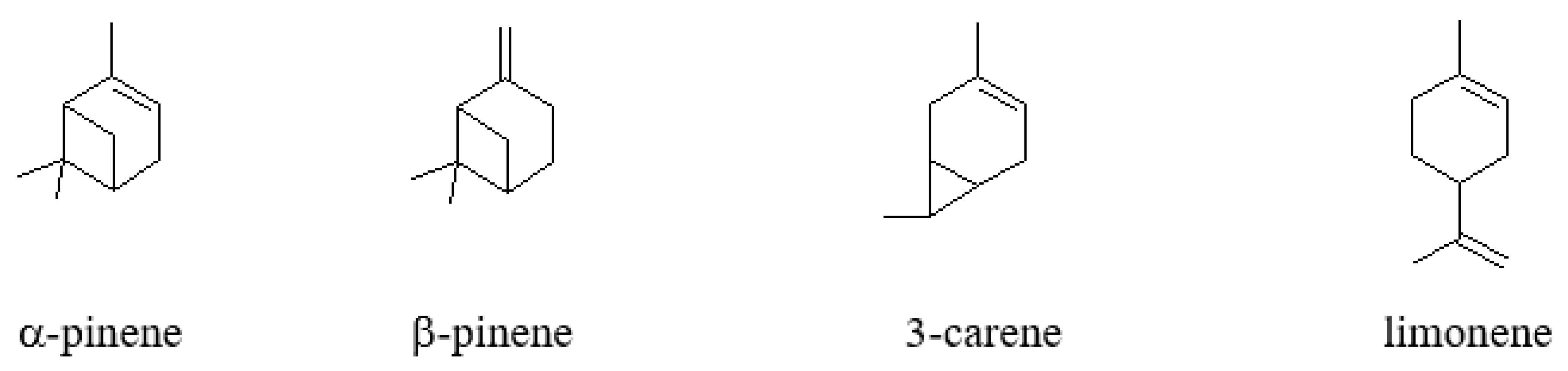
Monoterpenes are dimers of isoprene (2-methyl-1,3-butadiene) biosynthesized by numerous plants at a global scale estimated to around 1000 Mt/year. Isoprene being volatile is released by biomass at a global scale of about 600 Mt/year. Only a few of these monoterpenes are produced commercially at much more modest scale. These are α- and β-pinene, 3-carene, and limonene (as illustrated in Scheme 1).
All four are present in coniferous tree resins. The two pinenes are produced mostly from turpentine by the pulp and paper industry at 300 kt/year (in 2015), whereas limonene is recovered by steam extraction from citrus fruit peels, a waste product of the orange juice industry (90 kt/year) [5].
Notably, the US Company Amyris developed a process to produce farnesene from wood hydrolysates through a proprietary biochemical process. Farnesene is designated as a sesquiterpene, which means a 15-carbon olefin.
Squalene, a 30-carbon triterpene originally extracted from shark liver, is now mostly produced from vegetable sources including olive oil and sugar cane [6].
The biosynthesis of terpenes is well-described in literature [7]. Recent work succeeded in reproducing it in vitro [8], but this approach is still too complex and not liable to any commercial application. Similarly, the in vitro biosynthesis of isoprene was also achieved [9,10,11]. A hybrid process involving glucose fermentation to mesaconic acid, hydrodeoxygenation to 3-methylTHF, and dehydration to isoprene seems closer to an industrial green isoprene production [12,13]. To our knowledge, however, a process for dimerization of isoprene is still to be proposed.
Epoxidation results from the reaction of an alkene with an oxidizing agent. In industrial epoxide production, the choice of the oxidant is determining feasibility, efficiency, and therefore, profitability of the epoxidation process.
Actually, a variety of such oxidants were used industrially. The conventional chlorohydrin process in which alkenes (e.g., propylene) react with chlorine in the presence of sodium hydroxide tends to be replaced by more environmentally friendly ones owing to its large waste production [14,15].
Peracids such as m-CPBA (meta-chloroperbenzoic acid) were also used occasionally. Their oxidizing power depends on the leaving group capacity of the corresponding carboxylate [16]. They leave a stoichiometric amount of the corresponding acid as waste product. They do not require using a catalyst but the acidity makes selective epoxidation challenging [17].
DMDO (dimethyl dioxirane) is a relatively new, highly reactive oxidant generated by reaction of potassium peroxomonosulfate (KHSO5) with acetone. A stabilized form (2KHSO5 + KHSO4 + K2SO4) is commercially available from Dupont Chemicals under the brand name of Oxone. The aqueous solution of Oxone should be maintained at pH between 8 and 9 to avoid its decomposition and acid hydrolysis of the epoxide. The hydrodynamics of injecting this solution in the reaction medium is also a challenge for large-scale application.
Alkylhydroperoxides and hydrogen peroxide are commonly employed using a heterogeneous catalyst [18]. In the large-scale production of propylene oxide, the Halcon process uses ter-butylhydroperoxide (TBHP), the Halcon–Arco process uses ethylbenzene hydroperoxide, whereas cumene hydroperoxide is the oxidant in the Sumitomo process [19]. All these processes generate corresponding alcohols as a stoichioemetric byproduct.
Hydrogen peroxide is especially attractive since its only byproduct is water, and its weight ratio of active oxygen (47%) is the highest after molecular oxygen. Owing to its high rate of decomposition, however, it must be used in large excess, which upsets the large weight ratio asset [20]. The radicals formed during the decomposition process convert the olefins to alcohol or ketones, decreasing epoxide selectivity. For safety reasons, H2O2 is only available commercially in aqueous solutions (30 wt%), while water is a real inhibitor of catalysis by metals and greatly slows down the reaction [21].The epoxide selectivity is often poor in the presence of water or protic solvents due to the ring opening secondary reaction leading to the formation of glycol compounds [22,23].
Oxygen is by far the most desirable oxidant; it has the highest active oxygen content, the obviously lowest cost, and produces essentially no waste product. It is directly employed in ethylene epoxidation, the largest epoxidation process (26 Mt global production in 2018) which uses Ag catalysts. With any other alkene, the selectivity of epoxide formation is not sufficient. Recent developments led to transition metal-based catalysts with relatively high activity and epoxide selectivity [20]. The search for metal catalysts effective in activating molecular oxygen is therefore a current goal of industrial research.
In this text we intend to review the work performed in our laboratory over the last five years, exploring the use of various oxidants in terpenes epoxidation. Our work was primarily targeting limonene epoxidation. As shown in Scheme 1, limonene has two double bonds of different substitution numbers. The 1,2 double bond is trisubstituted and is therefore more easily epoxidized than the 8,9 one, which is only disubstituted, making the study of limonene epoxidation more informative.
Our contributions will be classified according to the oxidizing agent used for epoxidation.
2. Hydroperoxides
Hydroperoxides are generally far superior to H2O2. Hydrogen peroxide dissolution is limited to polar solvents, whereas hydroperoxides are also soluble in hydrocarbons [21,24,25]. Chiker et al. demonstrated that tert-butyl hydroperoxide (TBHP) is far more selective towards epoxides than H2O2 when supported titanium is used as the catalysts [26]. In dealing with epoxidation of limonene using t-butyl hydroperoxide as the oxidizing agent, we took advantage in the longstanding experience of our laboratory for Ti/SiO2 oxidation catalysts.
Titanium on silica was initially proposed by Sheldon in the 1970’s [21,24] and firstly used by Shell to produce propylene oxide in liquid phase. It was also implemented by the Enichen scientists in designing titanium silicalites (TS-1), a work which provided inspiration for numerous oxidation catalysts [25,26,27,28]. In alkene epoxidation over TS-1 the oxidizing agent is, however, limited to H2O2, owing to diffusional restriction in silicalite microchannels for larger ones such as TBHP [25]. Similar restrictions obviously apply to bulky reactants and products with kinetic diameter larger than 0.6 nm. In all Ti/SiO2 oxidation catalysts, the active site is a coordination unsaturated Ti substituted in the silica lattice. In TS-1 for example the substitutional limit is 1.9% of the T atoms. Thus, to lift these two kinds of limitations, Berubé et al. initially studied the incorporation of titanium in mesostructued SBA-15 silica by coprecipitation; they concluded that this direct method was not the most suitable technique [29,30]. For the epoxidation reaction of cyclohexene by TBHP, Berubé et al. then developed a liquid phase soft-templating titanium postgrafting impregnation of SBA-15 using in situ generated acetylacetonate as the stabilizing chelate [31,32]. This synthetic method allows a high content of low coordination titanium on the surface of silica, reaching up to 13.8% of T atoms [31,32]. Up to 5%, isolated four coordinated Ti atoms were obtained, whereas intermediate 4–6 coordinations Ti were formed at 5–13.8% content. Over this latter range the rate of cyclohexene epoxidation by aqueous TBHP stood essentially constant but the catalyst became stable and reusable.
In a more recent contribution, we studied coordination unsaturated titanium catalysts supported on SBA-16 mesoporous silica using anhydrous TBHP as the oxidizing agent for the epoxidation of limonene. The postgrafting impregnation of titanium method developed by Berube et al. was first modified by using soluble acetylacetonate dissolved in toluene. This approach was simpler and yielded the same catalytic activity as the in situ synthesized acetylacetonate titanium [33]. The reaction conditions including temperature, TBHP concentration, and solvent polarity were optimized. The conversion of limonene reached 80% with a 1,2-limonene oxide selectivity of 79%. The only observed secondary product was 8.9-limonene oxide. When SBA-16 was used, the mass transfer was enhanced compared to SBA-15, leading to a higher conversion of limonene [33]. For a similar Ti/Si atomic ratio, the conversion was only 38% when SBA-15 was used, compared to 80% for that of SBA-16. An increase of the 1,2-limonene epoxide trans-isomers was observed as the Ti/Si atomic ratio was increased to 13.4%.
Attempts at increasing conversion to limonene dioxide (LDO) over Ti/SBA-16 using TBHP met with very little success [33,34]. Interestingly, a similar result was obtained in the recent work by Cunningham et al. [35]. When trying to epoxide limonene, over a Venturella phase transfer catalyst (VPTC; PW4O24(PTC)3) using H2O2 as the oxidizing agent, a high yield of 1,2-limonene epoxide was obtained with 30% aqueous H2O2 solution, whereas a 50% H2O2 solution was needed to reach partial conversion to LDO.
3. Dimethyl Dioxirane (DMDO)
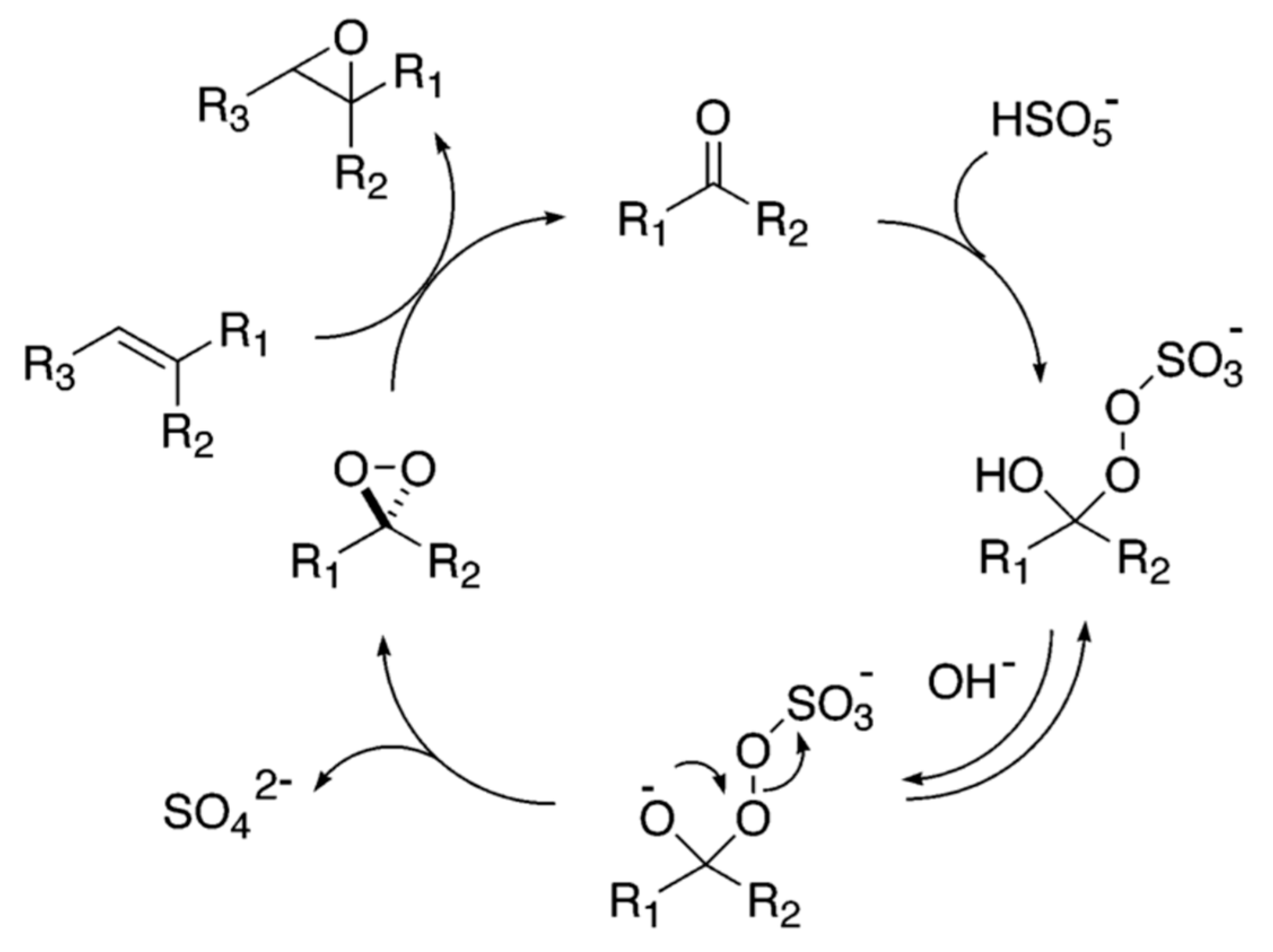
DMDO, sometimes designated as Murray’s reagent, is usually generated in situ from potassium peroxomonosulfate (in oxone) and a ketone (typically acetone) [36]. In this case the reaction medium is multiphasic with an organic phase containing the organic substrate (limonene) and acetone, and an aqueous solution of oxone with sodium bicarbonate as a buffer. DMDO is formed at the interface (as illustrated in Scheme 2).
Epoxidation can also be performed using externally generated DMDO. The asymmetric epoxidation of alkenes is known as the Shi epoxidation reaction [38].
DMDO and other cyclic peroxides are known to be especially efficient in transferring oxygen to a wide variety of alkenes while being rather mild towards oxidizing the substrate and reaction products [39]. DMDO is active in both electron poor and electron-rich alkenes in very high yield [40].
3.1. Epoxidation of Limonene in Semi-Batch Reactor
The biphasic nature of the reaction medium is not the most demanding aspect in terms of reactor design. The aqueous solutions of oxone are acidic (pH ≅ 1–2) and would lead to hydrolysis of any epoxide formed at their contact. Thus, the aqueous phase must be buffered at pH of 7–9, which is accomplished by introducing sodium bicarbonate. Too high basic pH would lead to decomposition of potassium monoperoxysulfate [36,41]. This very strict pH control is to be performed not only on the instantaneous average pH value of the aqueous phase, but also on local values at any reaction time. This imposes constraints on the feed flow rate of oxone to the reactor. The results shown in Table 1 were obtained in a round bottom flask, initially loaded with 10 mmol limonene dissolved in 30 mL acetone and 4 g sodium bicarbonate. Adjusted numbers of moles of oxone were dissolved in 50 mL deionized water, and this solution was fed to the reactor at the flow rate of 1 mL/min. This dropwise feeding allows the solution droplet to be rapidly dispersed in the aqueous phase, thus avoiding over concentration of oxone after a few minutes of reaction time.
The results in Table 1 were obtained after two hours of reaction at room temperature. They clearly indicate that a complete conversion of limonene to LDO is possible under these conditions. No products of allylic oxidation, nor any trace of 8,9-limonene epoxide are observed. The over stoichiometric consumption of oxone is likely related to some slow oxone decomposition.
3.2. Epoxidation under Ultrasound
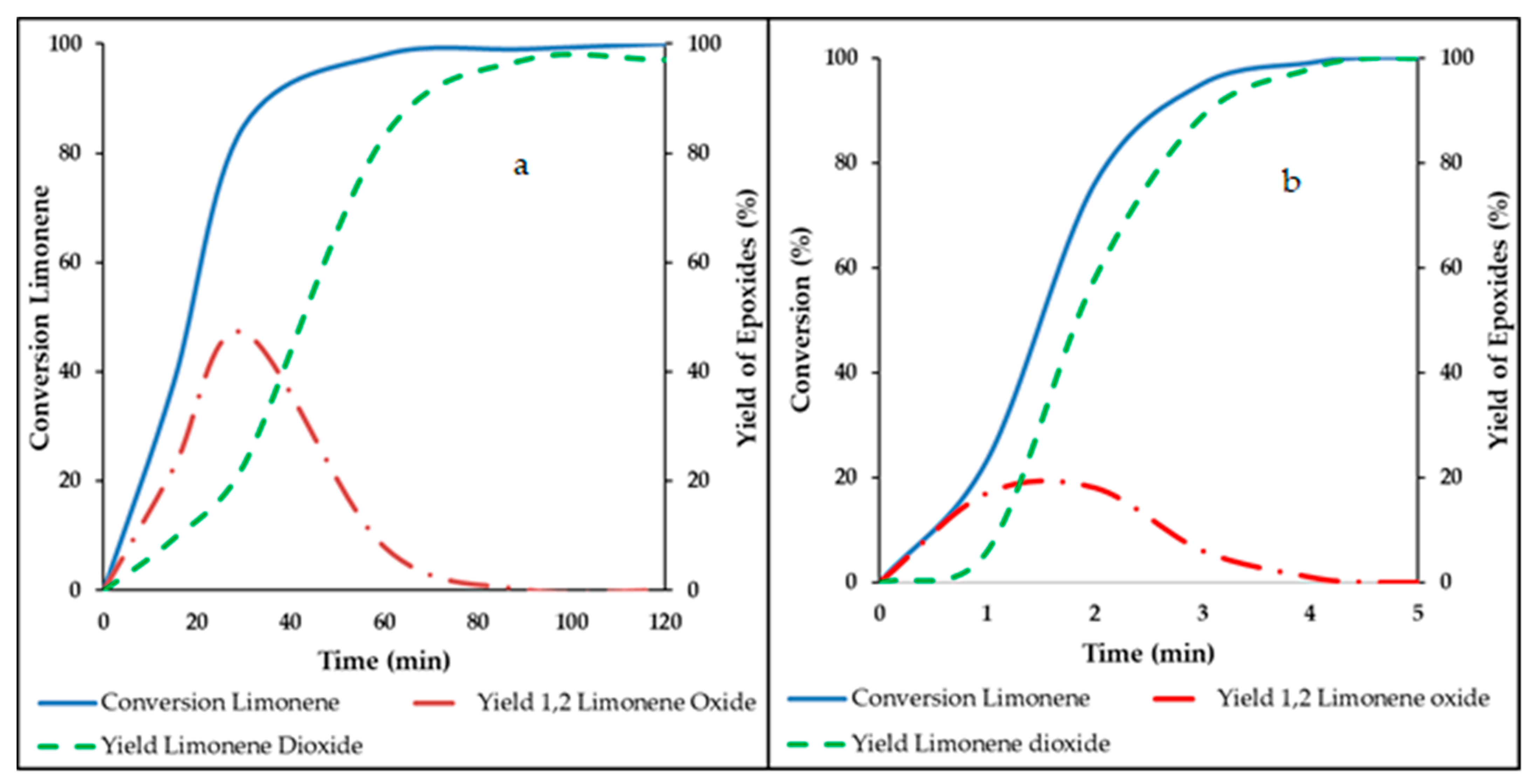
Obviously, the epoxidation process described in Scheme 2 is occurring in a zone localized close to the interface between the aqueous and organic phases. This makes the process rate directly proportional to the surface area of this interface. One of the beneficial effects of applying ultrasound to a reaction medium is precisely an efficient and rapid dispersion in a biphasic system. Thus, a large rate increase of limonene epoxidation by in situ generated DMDO upon applying ultrasound may be expected.
The results shown in Figure 1a,b were obtained in similar conditions. In Figure 1a, 10 mmol limonene was dissolved in 30 mL acetone with 4 g sodium bicarbonate. 50 mL of a 0.52 M oxone solution was added at a flowrate of 1 mL min−1. In Figure 1b, 10 mmol limonene was dissolved in 40 mL of acetone with 4 g sodium bicarbonate. 60 mL of a 0.52 M oxone solution was added at a flowrate of 20 mL min−1, under ultrasound (power 50 W, frequency 26 kHz). a and b tests were run at room temperature.
Comparing a and b highlights the very significant increase in epoxidation rate brought about by ultrasonication. Similar rate enhancements were demonstrated with some other terpenes including the two pinenes, carvone, carveol, and farnesol [42].
3.3. Heterogeneous Polydioxiranes
In a yet unpublished preliminary work, we tested, for limonene epoxidation, a method proposed by Kazemnejadi et al. for epoxidation of alkenes. In this method, a polydioxirane (PDOX) was generated by reaction of polysalicylaldehyde with oxone [43]. After epoxidation, PDOX is back converted to the polyaldehyde, which can be recovered by filtration. In [43], the converted PDOX was reused for eight cycles without significant loss of activity.
Formerly, heterogeneous solid catalysts such as α-fluorotropinone supported on SiO2 and Merrified resin was studied for stereoselective epoxidation of alkenes by oxone [44]. For this purpose, trifluoromethylketone supported on silica, as a solid dioxirane support, was a good catalyst for epoxidation by dioxirane generating alkene epoxides in the presence of oxone [45,46]. Our preliminary results indicate that solid PDOX generated in the presence of oxone can also be employed in the case of terpenes.
3.4. In Microemulsions
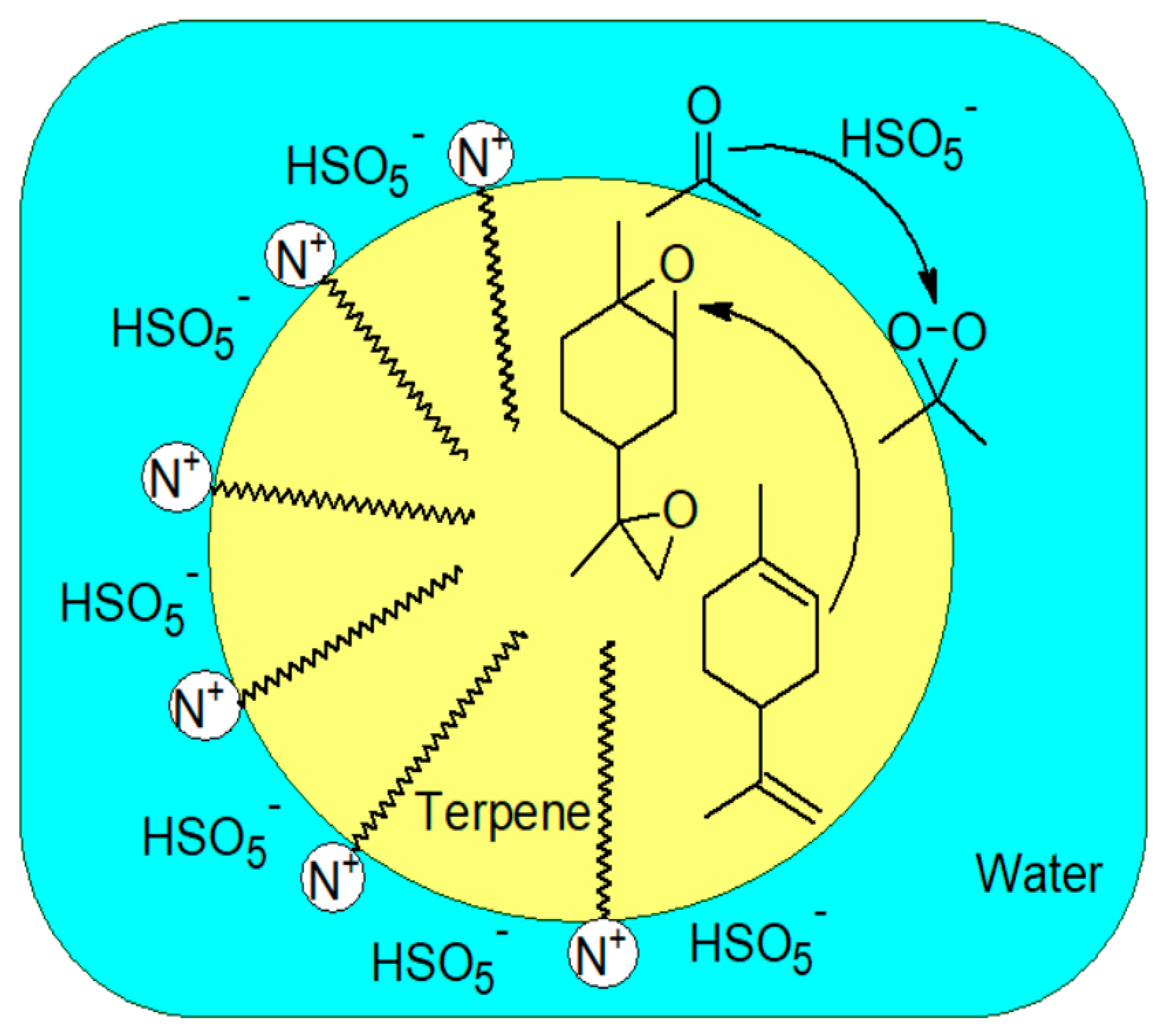
Another way besides ultrasonication to generate uniform dispersion of an organic phase into an aqueous oxone solution is the production of a microemulsion. Introducing minute amounts of an appropriate surfactant, a stable dispersion of organic phase droplets with diameters in the 5 to 50 nm range can be generated [47]. We established that cetyltrimethylammonium hydrogen sulfate (CTHAS) is especially effective in microemulsion epoxidation of terpenes by in situ generated DMDO. Figure 2 shows the respective positions of the quaternary ammonium ions of CTHAS in the terpene droplet and the HSO5− ions attracted to the interface by electrostatic interaction. This favors the local formation of DMDO and consequently a rapid terpene epoxidation.
The data reported in Table 2 illustrate this increase in limonene epoxidation rate, since that introduction of small amounts of CTHAS in the reaction system results systematically in a significant increase in epoxide yield and active oxygen utilization. In these tests, acetone as the only organic solvent is used in small quantities. Also, almost 100% yield LDO can be reached under realistic conditions. No allylic oxidation products or 8,9-LO was observed.
The results shown in Table 3 also indicate that CTHAS in an appropriate surfactant for epoxidation of pinenes by DMDO.
4. Aerobic Epoxidation
Among the various oxidizing agents proposed for alkene epoxidation, molecular oxygen (dioxygen) is by far the greenest and less expensive one [49]. Using molecular oxygen is however limited by a spin forbidden process which makes it unreactive towards organic molecules at low temperature or suffer from lack of selectivity [50].
Works in our group aimed at developing heterogeneous catalysts for the two most relevant liquid phase aerobic epoxidation processes, as applied to limonene epoxidation.
4.1. Aerobic Epoxidation Using an Aldehyde as Sacrificial Reductant (Mukaiyama Reaction)
A solution of cobalt acetylacetonate (Co(acac)2) was used to synthesize Co/SBA-16 by postgrafting impregnation method. The resultant catalyst was employed in the epoxidation of limonene with oxygen in the presence of isobutyraldehyde and ethylacetate as a green solvent and was found to be active under very mild conditions [51]. The nature of the metal affects the epoxide selectivity of the catalyst. Co+2 has a high ability to catalyze chain branching and promote the chain initiation. Also, the activation of dioxygen and/or aldehyde via coordination on the metal center were found the reason for higher activity of Co+2 compounds. Therefore, it could justify the alkene epoxidation by O2 in the presence of an aldehyde. In the case of limonene, Co2+ was also confirmed more active than other ions including Fe2+, Ni2+, and Mn2+ [51]. Interaction of coordinating ligands such as acetylacetone (acac) with surface hydroxyls contributes in low coordination oxidic species grafting on the surface of the support and provides high dispersion of the active Co sites.
The effect of cobalt loading on catalytic performances of Co/SBA-16 is shown in Table 4. Very low activity was observed in the absence of catalyst. The selectivities of both 1,2-limonene oxide and limonene dioxide increased with Co/Si ratio up to 4.5%, and further enhancing the cobalt loading resulted in a negative impact on epoxides selectivity.
The decrease in catalytic activity at higher cobalt loading (4.5 to 9.7%), is associated to the formation of octahedrally coordinated Co3+ in the form of Co3O4, which leads to reducing the number of accessible active sites. The low coordination isolated cobalt (II) species, present on the surface of mesoporous SBA-16, are the highly active and selective catalytic sites in the epoxidation reaction. On the other hand, those additional cobalt species that are grafted on the materials are not catalytically selective for epoxidation reaction.
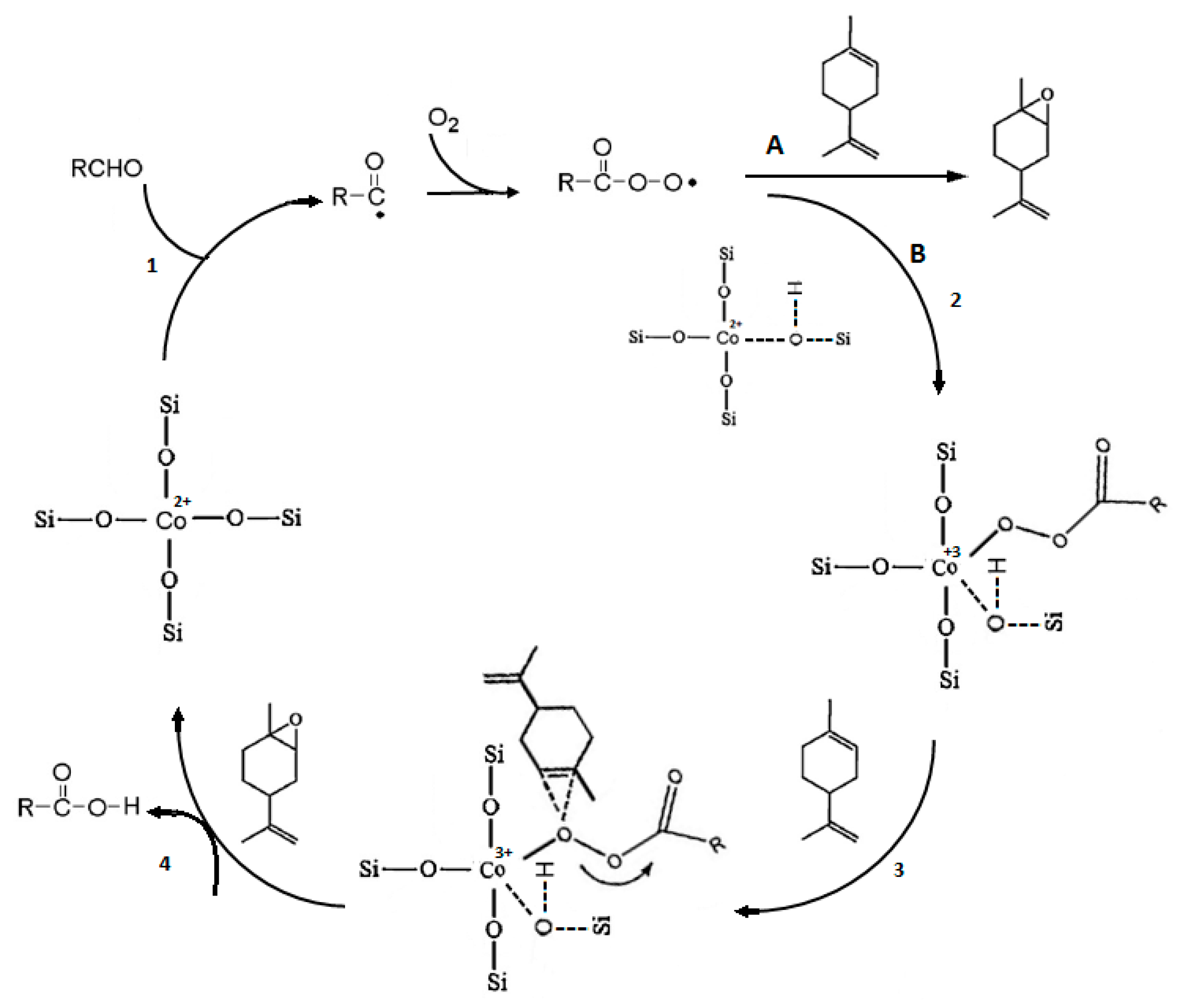
A radical mechanism as exhibited in Scheme 3 was proposed for aerobic epoxidation of limonene in the presence of an aldehyde catalyzed by SBA-16 supported Co2+. The acylperoxy radical is the main epoxidizing agent, generated in the reaction of isobutyraldehyde (RCHO) with molecular oxygen and initiated over cobalt active species. This species could react either directly with limonene through pathway A to produce epoxide or with isolated Co2+ to generate Co-peroxy species (pathway B) and then transfer oxygen by the reaction of Co-peroxy with limonene through a concerted mechanism.
In another study, a novel “pH-adjusting” method was used to graft cobalt in suitable state onto mesoporous silica SBA-16 to be employed for aerobic epoxidation of limonene [52]. Using the optimum reaction conditions established in [51], the catalytic performances of Co/SBA-16 prepared by pH adjusting method at different cobalt loadings were investigated and reported in Table 5. Co/SBA-16 (1.1%) showed the highest optimized epoxide yield (85%), which has a lower cobalt loading compared to the optimal Co/SBA-16 (4.5%) synthesized by postgrafting of Co(acac)2 (as illustrated in Table 4). The higher density of silanol groups on the surface of the catalyst in the pH adjustment method, resulting from no calcination of the support SBA-16 before introduction of the cobalt precursor, accessible to the Co2+ ions, contributes to higher density of isolated tetrahedral Co2+ species. A reverse trend at epoxide yield was observed when cobalt loading increased (as illustrated in Table 5). This suggests some parallel oxidation catalyzed by the higher coordination Co2+ ions, generated in the samples at higher cobalt loading.
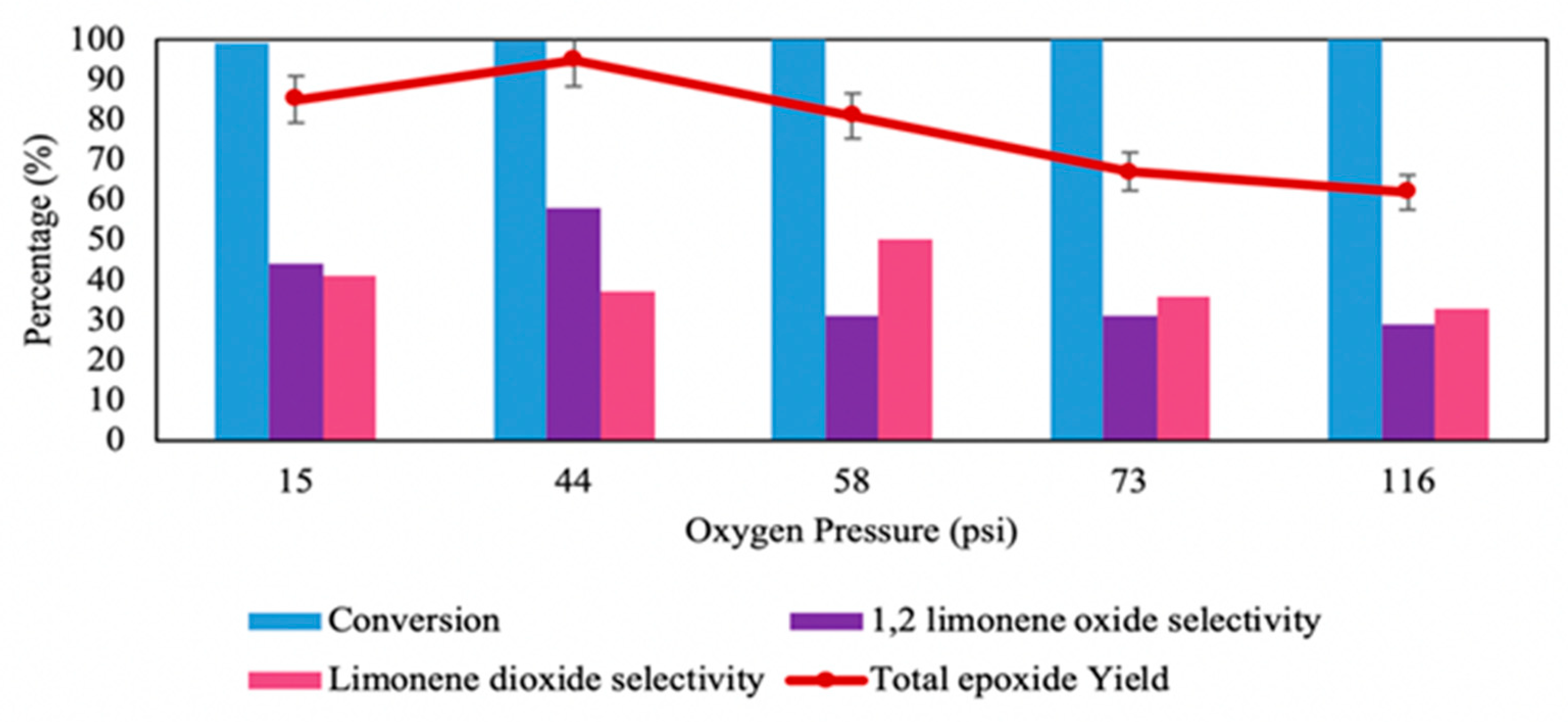
As illustrated in Figure 3, the raise in oxygen pressure from 15 to 44 psi increased the formation of limonene epoxides to the highest combined yield of 95%, which then decreased with further elevation of oxygen pressure up to 58 psi. This can be explained by possible secondary oxidation or ring opening of epoxide at higher pressure.
An example of time evolution of the reaction medium during limonene epoxidation in the presence of isobutyraldehyde and oxygen at 44 psi oxygen pressure with Co/SBA-16 (1.1%) as catalyst is shown in Figure 4.
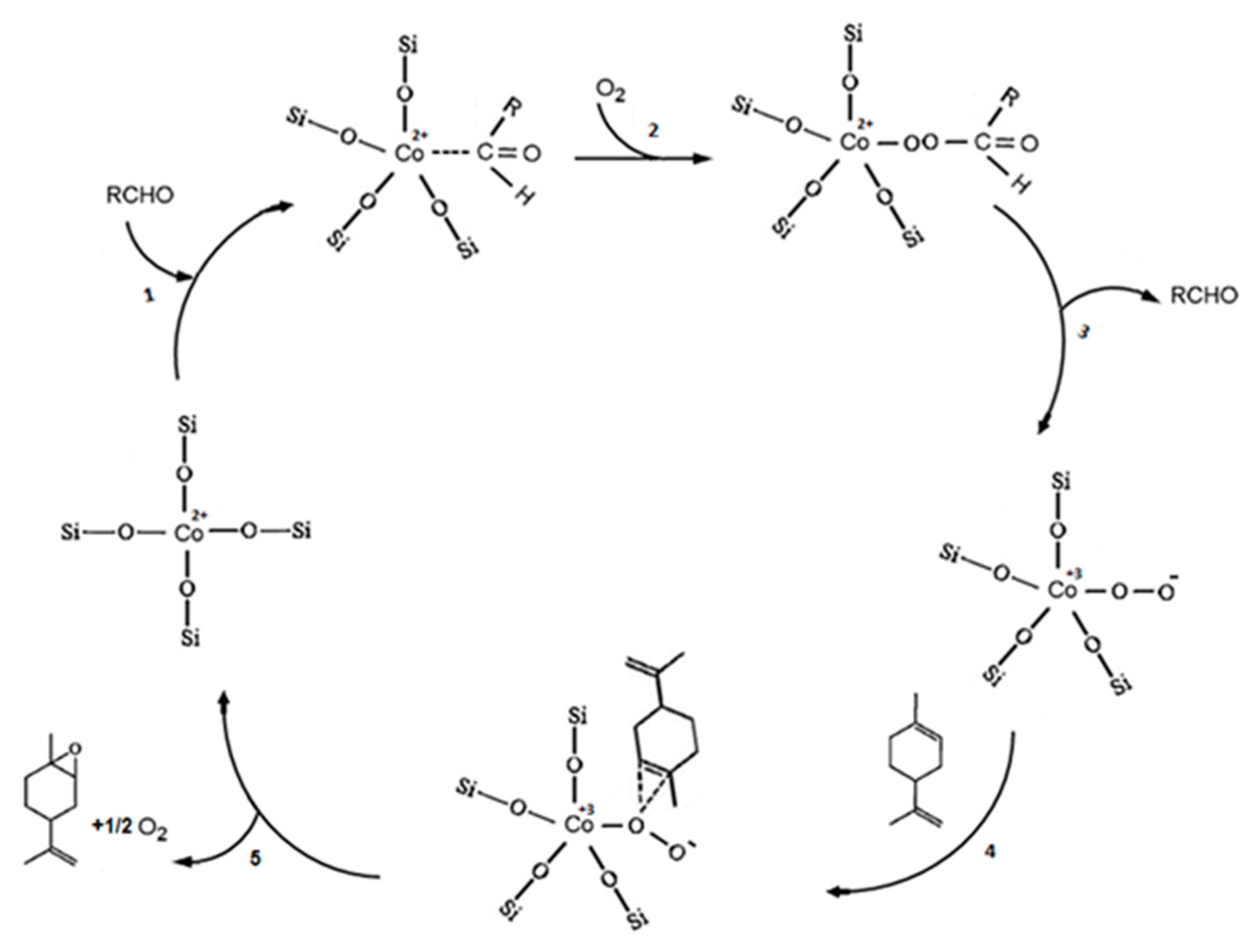
Diepoxide is generated by secondary epoxidation of monoepoxides. 58% selectivity of 1,2 limonene and 37% limonene dioxide were achieved for the total use of limonene at 250 min of the reaction. Different conclusions were made about the mechanism of aerobic epoxidation of limonene. This was done through investigating the initial reaction rates using the optimized catalyst. Since the initial rates of limonene epoxidation (Re) and isobutyraldehyde oxidation (Ra) offered a systematic difference, mechanism pathway B in Scheme 3, which suggests Re = Ra, could be excluded. Therefore, the other reaction pathway C which uses the peroxy intermediate Co3+OO−, was considered as proposed in Scheme 4. However, the strong dependence of Re on the aldehyde concentration leads to assumption that RCHO is not consumed in the process but is nevertheless involved in the formation of this intermediate.
The observed unusual effect of a strong decrease in initial epoxidation rate at increasing limonene concentration imposes that the reaction be only important at very low limonene concentration. This, of course, precludes large scale application of this process.
4.2. Solvent-Free and Initiator-Free Epoxidation
Direct aerobic epoxidation of limonene without using a solvent, sacrificial reductant or radical initiator was investigated using highly dispersed Ru supported on activated carbon (AC) fabricated using various immobilization strategies [53]. Ruthenium/activated carbon was found an active catalyst for the environmentally friendly epoxidation of limonene using molecular oxygen as the only oxidant under very mild condition without any solvent and initiator. The selectivity of the catalyst toward epoxidation was found to be greatly influenced by the preparation method of the catalyst. The catalytic activity of variety Ru supported catalysts using different ACs with various morphologies and different preparation methods for the aerobic epoxidation of limonene under solvent-free conditions and without any radical initiator is presented in Table 6.
Despite higher conversion obtained over the catalyst prepared by the sol immobilization (SI) method (40.4%), the one prepared by cation exchange Ru/G60(CE) showed the highest selectivity and yield toward epoxidation products. The results indicated that despite the formation of small Ru nanoparticles on carbon materials in sample prepared by the SI method (particle size of 2.5 nm and dispersion of 53%), it suffered from high selectivity to the allylic oxidation products. All catalysts synthesized by incipient wetness impregnation (IWI) yielded lower epoxide selectivity.
The catalyst synthesized using a cation exchange method showed a good compromise between activity and selectivity to the epoxides. A higher oxygen functional groups surface density generated by preoxidation of high surface area G60 support, led to AC supported ruthenium catalysts with uniform and isolated Ru nanoparticles.
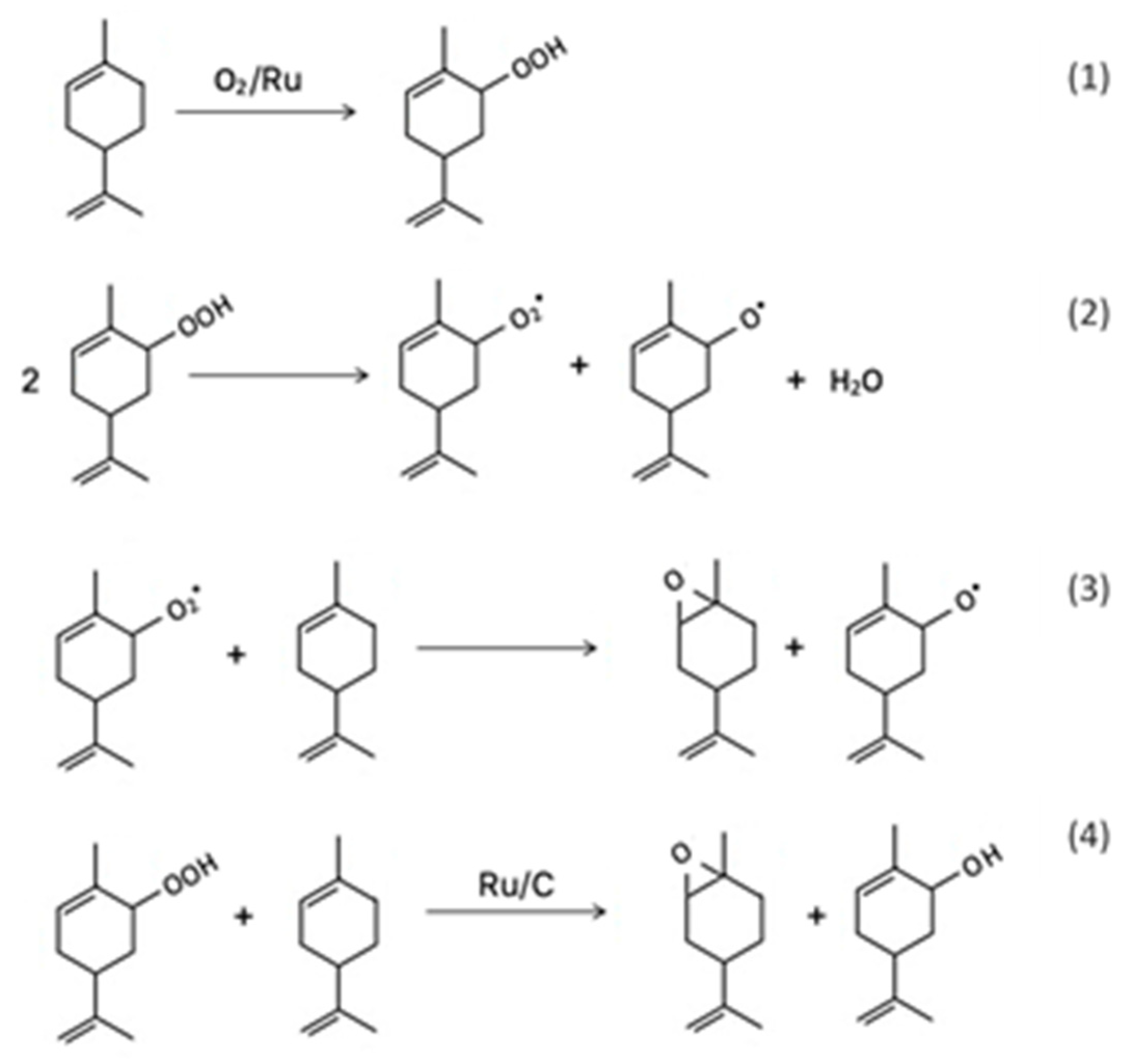
Both epoxidation and allylic oxidation products may occur through oxidation of olefins with dioxygen. The allylic oxidation to epoxide products ratio are determined by the competition between the abstraction of the allylic hydrogen and the addition of the alkyl peroxy radical to the double bond. In catalytic epoxidation of limonene with molecular oxygen generally, electrophilic attack on the double bond of limonene results in the formation of 1,2- and 8,9- epoxy limonene as the primary epoxidation products and 1,2,8,9- diepoxy limonene as the secondary epoxidation product while allylic H-abstraction results in carveol and carvone formation. The main oxygenated byproducts could be generated by the two reaction pathways shown in Scheme 5.
One corresponds to the formation of metal–alkene adducts by chemisorption on the catalyst surface and activation of limonene and oxygen using ruthenium with appropriate catalyst design. The other pathway is the limonene autoxidation via a free radical chain mechanism in which the formation of limonene hydroperoxide by activation of allylic C-H bonds of limonene leads to decomposition of limonene hydroperoxide and formation of radicals capable of reacting with oxygen and yielding variously oxidized products, such as epoxides and allylic products. However, the results in this study suggests the former pathway is favored over the latter due to the higher ratio of epoxide to allylic oxidation products.
The performance shown in Table 6 for the original catalyst Ru/G60(CE) is the best reported epoxide yield for aerobic epoxidation of limonene. This being obtained in absence of any solvent or initiator makes this process close to large scale applicability.
5. Importance and Perspective of Stereoselective Epoxidation
Limonene is a chiral molecule with the (R)-enantiomer, often designated as D-limonene ((+)-limonene), being the most abundant in orange peel essential oils. As shown in Scheme 6, each of the two limonene enantiomers gives rise to two isomers with different configurations of the epoxide bridges in limonene diepoxide (LDO). These various isomers show different reactivities in reactions of interest for further polymerizations. In the above cited ref [35], Cunningham et al. studied the epoxidation of a wide range of terpenes having trisubstituted alkene bonds with a tungsten-based polyoxometalate catalyst using aqueous H2O2 as the oxidant and without solvent. Their work indicates that in all cases, two diastereomers (i.e., a racemic mixture of cis- and trans- epoxides of each terpene) are formed. In the example of (R)-(+)-limonene, epoxidation resulted in the formation of an almost 1:1 mixture of cis- and trans- 1,2-epoxide of limonene. Aerobic epoxidation of limonene over cobalt substituted mesoporous SBA-16, was studied in [51]. Epoxidation with molecular oxygen resulted in the formation of two cis- and trans- diastereomers of limonene dioxide (LDO). Epoxidized samples from this work were quantified in [54]. The products consisted of 60:40 cis:trans limonene dioxide with the exact distribution shown in Scheme 6.
These various terpene isomers (cis- and trans-) are not equally reactive in polymerization reactions. Poly(limonene trans-R-carbonate) and poly(limonene trans-S-carbonate) were synthesized from a cis/trans mixture of (R)- and (S)-limonene oxide, respectively, at room temperature using a zinc b-diminate complex [55,56,57,58]. The catalyst readily allows polymerizing the trans- diastereomers of the epoxide, leaving the cis- diastereomers unreacted, which resulted in a highly regio, diastereoiso, and enantiomerically pure polymer. Similar results were observed in recent studies using tetrabutylammonium halides (TBAX) as catalysts; the formation of polycarbonates and cyclic carbonates from a commercial mixture of (40:60, cis:trans LDO) exhibits a significantly higher conversion of the trans- isomer than the cis- isomer [58,59]. Very recently, Schutz et al. compared the reactivity of trans- and cis- limonene dioxide (LDO) by reacting them separately to form epoxy resins upon crosslinking with branched polyethyleneimine (BPEI) [54]. The results from Fourier transform infrared spectroscopy, DFT calculations, and kinetics of cross-linking studied by differential scanning calorimetry confirmed that the two (trans-LDO) are really functional, while the other two (cis-LDO) possess an epoxide group that is significantly less reactive. In the same vein, it was reported that the use of pure trans-limonene carbonate to form nonisocyanate polyurethanes (NIPU) significantly improved such properties as glass transition temperature, stiffness, and strength [60]. However, the low reactivity of diastereomeric cis- compared to that of trans- is related to its high energy of activation due to the “boat-like” structure of its transition state [61,62].
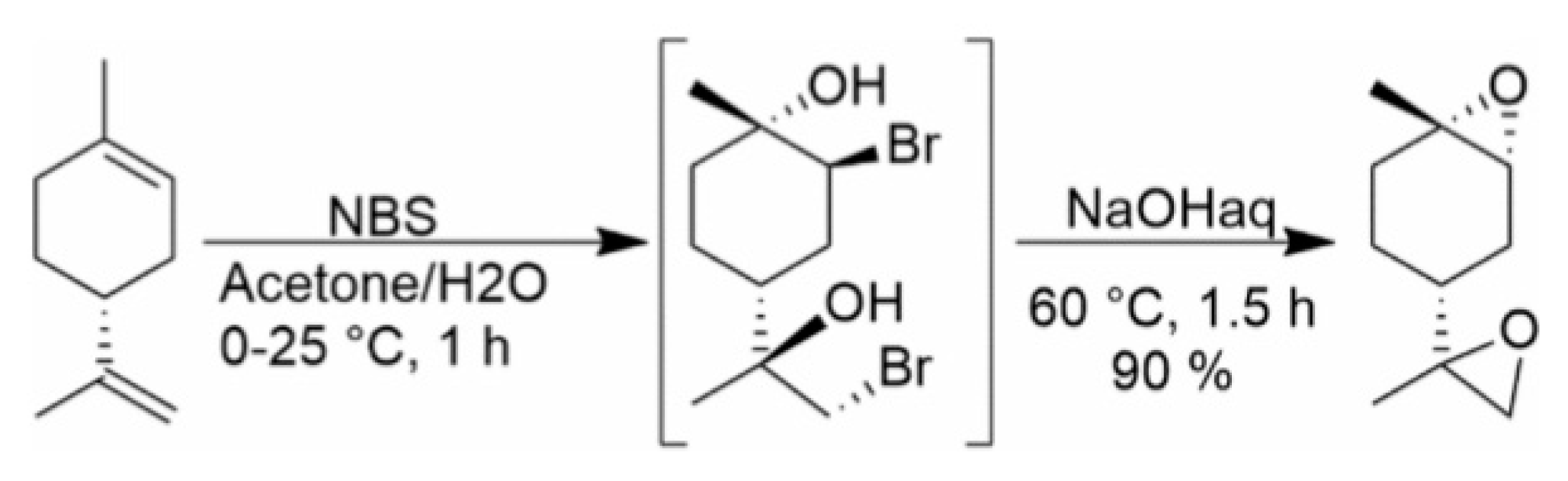
Evidently, the development of a strereoselective epoxidation technique to further synthesize the trans- configuration of terpenes is a necessity. Hauenstein et al. developed a large-scale stereoselective synthesis technique for the trans-isomer 1,2-oxide of limonene (>1 kg/batch) and suggested easy transfer to pilot plant scale [58]. The synthesis involves regio- and stereo-selective endocyclic bromohydrin of limonene in aqueous acetone with N-bromosuccinimide (NBS) as the bromine source to form a bromohydrate intermediate. This intermediate is readily converted to the corresponding limonene trans-1,2-oxide in the presence of aqueous sodium hydroxide. The byproducts thus formed contain more than 85% of the limonene trans-1,2-oxide. Very recently, this same method of stereoselective synthesis was employed to synthesize limonene trans-dioxide. (as illustrated in Scheme 7) [63].
The technique consists in a first step to form a di-bromohydrate of the endocyclic and exocyclic double bonds of limonene to form the di-bromohydrate intermediate of limonene. The latter reacts with an aqueous sodium hydroxide solution to form limonene trans-dioxide. The authors investigated such parameters as reaction temperature, limonene: N-bromosuccinimide molar ratio, and reaction time to promote a very high yield of limonene trans-dioxide. A 97% yield of limonene trans-dioxide was obtained using a 1:2 ratio of limonene: N-bromosuccinimide at 60 °C after 5 min reaction time.
Other techniques for the stereoselective epoxidation of chiral alkenes in the presence of a dioxyrane were investigated by several authors. The stereoselective epoxidation of chiral olefins over Jacobsen’s catalyst immobilized on Al-MCM-41 and NH2-Si-MCM-41 in the presence of in situ generated DMDO as oxygen source was studied [64]. The experimental results indicate that selectivities to cis- and trans- epoxides can be obtained up to 86 and 72%, respectively. The developed catalyst did not undergo any significant change in its chemical structure upon reaction. Therefore, the reusability of the catalyst could be achieved when it was immobilized by chemical bonding of the salt ligand. In the same vein, a chiral dioxirane was developed for the highly stereoselective epoxidation of cis-olefins [65]. The authors were able to produce cis-epoxides of a wide variety of olefins with selectivity of 77–95%. Other similar studies demonstrated that in situ generated dioxiranes from chiral ketones are highly stereoselective for the asymmetric epoxidation of trans-olefins and trisubstituted olefins (See Refs. 4–6 in [41]).
Indeed, the above cited technologies for alkene stereoselective epoxidation need to be tested for setereoselective epoxidation of terpenes and, in particular, for the especially crucial formation of trans-limonene diepoxides.
Enantioselective Separation
Steiner et al. developed an efficient separation process for a 1:1 mixture of cis and trans 1,2-limonene oxide using nucleophilic amines [4]. To isolate cis 1,2-limonene oxide, pyrrolidine and piperidine were used and reacted with only trans 1,2-limonene oxide by ring opening reaction. The unchanged cis 1,2-limonene oxide was isolated by liquid–liquid extraction and distillation. The recovery yield reached up to 88%. To obtain pure trans 1,2-limonene oxide, less nucleophilic amines such as triazole and pyrazole were used. The recovery of trans 1,2-limonene oxide using the same isolation procedure as for cis 1,2-limonene oxide, yielded up to 80% recovery. In both cases the isolated isomer had a purity higher than 98% [61]. Nucleophilic amines were not explored for the isolation of pure trans limonene dioxide. This approach may however be a potential alternative to stereoselective epoxidation.
6. Conclusions
This minireview of our recent terpenes epoxidation works is a necessarily shortened presentation of some of the most significant results. The interested reader may like to access to our cited references. Even more detailed presentations may be found in the already published theses [66,67,68] and in the ones in preparation.
This kind of report is an opportunity to illustrate the philosophy that underlies our work. Firstly, we recognize that the chemical and parachemical industries make significant contributions to the wellbeing of mankind. For several reasons, this is not always acknowledged in the general public. This is not to say that mistakes were never made in these sectors and that significant changes are not necessary. Actually, these industries are increasingly conscious of the need to adapt to a changing world [69]. Our activities are attempts to help this coming adaptation. Epoxidation is an example of a process, currently used on a large scale to valorize compounds of fossil origin, that needs to be studied for application with natural products such as terpenes. In doing so, we considered several possible options, looking systematically at the technical feasibility and possibility of scaling up of the various processes, in addition to a scientific search for fundamental understanding. We believe both exercises should be conducted simultaneously. Note that at this stage, a strict economic assessment of profitability would be premature and probably counterproductive [69].
Funding
This series of works were funded through several grants from CRSNG and CRIBIQ with financial support of Soprema.
Data Availability Statement
There is no supporting information to this minireview. Please refer to the cited references.
Acknowledgments
We wish to thank Soprema (Drummondville, QC, Canada) for its continued support and interest in this work, as well as Consortium de Recherche et d’Innovation en Biotechnologie Industrielle du Québec (CRIBIQ), and Centre de Recherche en Sciences Naturelles et en Génie du Canada (CRSNG) for joint financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nguyen, H.T.H.; Qi, P.; Rostagno, M.; Feteha, A.; Miller, S.A. The quest for high glass transition temperature bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Kristufek, S.L.; Wacker, K.T.; Tsao, Y.-Y.T.; Su, L.; Wooley, K.L. Monomer design strategies to create natural product-based polymer materials. Nat. Prod. Rep. 2017, 34, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Hernández, N.; Williams, R.C.; Cochran, E.W. The battle for the “green” polymer. Different approaches for biopolymer synthesis: Bioadvantaged vs. bioreplacement. Org. Biomol. Chem. 2014, 12, 2834–2849. [Google Scholar] [CrossRef]
- Harman-Ware, A.E. Conversion of terpenes to chemicals and related products. In Chemical Catalysts for Biomass Upgrading; Crocker, M., Santillan-Jimenez, E., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp. 529–568. [Google Scholar]
- Ciriminna, R.; Lomeli-Rodriguez, M.; Carà, P.D.; Lopez-Sanchez, J.A.; Pagliaro, M. Limonene: A versatile chemical of the bioe-conomy. Chem. Commun. 2014, 50, 15288–15296. [Google Scholar] [CrossRef]
- Pandarus, V.; Ciriminna, R.; Béland, F.; Pagliaro, M.; Kaliaguine, S. Solvent-Free Chemoselective Hydrogenation of Squalene to Squalane. ACS Omega 2017, 2, 3989–3996. [Google Scholar] [CrossRef] [Green Version]
- Lange, B.M. Biosynthesis and Biotechnology of High-Value p-Menthane Monoterpenes, Including Menthol, Carvone, and Limonene. Biotechnol. Isoprenoids 2015, 148, 319–353. [Google Scholar] [CrossRef]
- Korman, T.P.; Opgenorth, P.H.; Bowie, J.U. A synthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat. Commun. 2017, 8, 15526. [Google Scholar] [CrossRef]
- Korman, T.P.; Sahachartsiri, B.; Li, D.; Vinokur, J.M.; Eisenberg, D.; Bowie, J.U. A synthetic biochemistry system for the in vitro production of isoprene from glycolysis intermediates. Protein Sci. 2014, 23, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, R.; Yi, X.; Fang, T.; Yang, J.; Bae, H.-J. Isoprene Production on Enzymatic Hydrolysate of Peanut Hull Using Different Pretreatment Methods. BioMed Res. Int. 2016, 2016, 4342892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Liu, H.; Zou, H.; Chen, N.; Shi, M.; Xie, C.; Zhao, G.; Xian, M. Enzymatic process optimization for the in vitro production of isoprene from mevalonate. Microb. Cell Factories 2017, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, L.D.; Dauenhauer, P.J. Hybrid routes to biofuels. Nat. Cell Biol. 2007, 447, 914–915. [Google Scholar] [CrossRef]
- Lundberg, D.J.; Lundberg, D.J.; Zhang, K.; Dauenhauer, P.J. Process Design and Economic Analysis of Renewable Isoprene from Biomass via Mesaconic Acid. ACS Sustain. Chem. Eng. 2019, 7, 5576–5586. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Q.; Wu, H.; Meng, G.; Tang, Q.; Wang, Y. Iron-based heterogeneous catalysts for epoxidation of alkenes using molecular oxygen. Catal. Commun. 2004, 5, 665–669. [Google Scholar] [CrossRef]
- Jiang, J.; Li, R.R.; Wang, H.; Zheng, Y.; Chen, H.; Ma, J. Highly Active/Selective Heterogeneous Catalyst Co/TS-1 for Epoxi-dation of Styrene by Molecular Oxygen: Effects of Catalyst Preparation Conditions and Reaction Conditions on the Reaction. Catal. Lett. 2008, 120, 221–228. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S.G. Organic Chemistry; Oxford University Press: Oxford, UK; New York, NY, USA, 2012. [Google Scholar]
- Cunningham, W. Catalytic Conversion of Terpene Feedstocks into Value-Added Chemicals and Commodity Chemicals. Ph.D. Thesis, University of Bath, Bath, UK, 2018. [Google Scholar]
- Yadav, G.D.; Pujari, A.A. Epoxidation of Styrene to Styrene Oxide: Synergism of Heteropoly Acid and Phase-Transfer Catalyst under Ishii−Venturello Mechanism. Org. Process. Res. Dev. 2000, 4, 88–93. [Google Scholar] [CrossRef]
- Brégeault, J.-M. Transition-metal complexes for liquid-phase catalytic oxidation: Some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 3289–3302. [Google Scholar] [CrossRef]
- Adolfsson, H. Transition Metal Catalyzed Epoxidation of Alkenes. In Modern Oxidation Methods; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Sheldon, R. Synthetic and mechanistic aspects of metal-catalysed epoxidations with hydroperoxides. J. Mol. Catal. 1980, 7, 107–126. [Google Scholar] [CrossRef]
- Rao, A.S. Addition Reactions with Formation of Carbon–Oxygen Bonds: (i) General Methods of Epoxidation. Oxidation 1991, 357–387. [Google Scholar] [CrossRef]
- Arends, I.; Sheldon, R. Activities and stabilities of heterogeneous catalysts in selective liquid phase oxidations: Recent developments. Appl. Catal. A Gen. 2001, 212, 175–187. [Google Scholar] [CrossRef]
- Sheldon, R. Metal-catalyzed epoxidation of olefins with organic hydroperoxides I. A comparison of various metal catalysts. J. Catal. 1973, 31, 427–437. [Google Scholar] [CrossRef]
- Corma, A.; Esteve, P.; Martinez, A.; Valencia, S. Oxidation of Olefins with Hydrogen Peroxide and tert-Butyl Hydroper-oxide on Ti-Beta Catalyst. J. Catal. 1995, 152, 18–24. [Google Scholar] [CrossRef]
- Chiker, F.; Launay, F.; Nogier, J.P.; Bonardet, J.L. Green and selective epoxidation of alkenes catalysed by new TiO2–SiO2SBA mesoporous solids. Green Chem. 2003, 5, 318–322. [Google Scholar] [CrossRef]
- Kapoor, M.P.; On, D.T.; Gallot, J.E.; Kaliaguine, S. Hydrolytic cleavage of epoxy ring during the epoxidation of al-kenes over boron modified titanium silicalites of MFI topology. Catal. Lett. 1997, 43, 127–131. [Google Scholar] [CrossRef]
- Gallot, J.E.; Kaliaguine, S. Oxidation of hydrocarbons by hydrogen peroxide over Ti catalysts: Kinetics and mechanistic studies. Can. J. Chem. Eng. 1998, 76, 833–852. [Google Scholar] [CrossRef]
- Bérubé, F.; Kleitz, F.; Kaliaguine, S. Surface properties and epoxidation catalytic activity of Ti-SBA15 prepared by direct synthesis. J. Mater. Sci. 2009, 44, 6727–6735. [Google Scholar] [CrossRef]
- Bérubé, F.; Kleitz, F.; Kaliaguine, S. A Comprehensive Study of Titanium-Substituted SBA-15 Mesoporous Materials Prepared by Direct Synthesis. J. Phys. Chem. C 2008, 112, 14403–14411. [Google Scholar] [CrossRef]
- Bérubé, F.; Nohair, B.; Kleitz, F.; Kaliaguine, S. Controlled Postgrafting of Titanium Chelates for Improved Synthesis of Ti-SBA-15 Epoxidation Catalysts. Chem. Mater. 2010, 22, 1988–2000. [Google Scholar] [CrossRef]
- Bérubé, F.; Khadhraoui, A.; Janicke, M.T.; Kleitz, F.; Kaliaguine, S. Optimizing Silica Synthesis for the Preparation of Mesoporous Ti-SBA-15 Epoxidation Catalysts. Ind. Eng. Chem. Res. 2010, 49, 6977–6985. [Google Scholar] [CrossRef]
- Charbonneau, L.; Kaliaguine, S. Epoxidation of limonene over low coordination Ti in Ti-SBA-16. Appl. Catal. A Gen. 2017, 533, 1–8. [Google Scholar] [CrossRef]
- Charbonneau, L.; Foster, X.; Zhao, D.; Kaliaguine, S. Catalyst-Free Epoxidation of Limonene to Limonene Dioxide. ACS Sustain. Chem. Eng. 2018, 6, 5115–5121. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, W.B.; Tibbetts, J.D.; Hutchby, M.; Maltby, K.A.; Davidson, M.G.; Hintermair, U.; Plucinski, P.; Bull, S.D. Sustaina-ble catalytic protocols for the solvent free epoxidation and anti-dihydroxylation of the alkene bonds of biorenewable terpene feedstocks using H2O2 as oxidant. Green Chem. 2020, 22, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.W. Chemistry of dioxiranes. 12. Dioxiranes. Chem. Rev. 1989, 89, 1187–1201. [Google Scholar] [CrossRef]
- Shi, Y. Organocatalytic Asymmetric Epoxidation of Olefins by Chiral Ketones. ACC Chem. Res. 2004, 37, 488–496. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.-X.; Shi, Y. An Efficient Asymmetric Epoxidation Method for trans-Olefins Mediated by a Fructose-Derived Ketone. J. Am. Chem. Soc. 1996, 118, 9806–9807. [Google Scholar] [CrossRef]
- Asouti, A.; Hadjiarapoglou, L.P. Substrate-Induced Diastereoselectivity in the Dimethyldioxirane Epoxidation of Simple Al-kenes and Dienes. Synlett 2001, 2001, 1847–1850. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Edwards, J.O.; Pater, R.H.; Curclf, R.; Di Furia, F. On The Formation and Reactivity of Dioxirane Intermediates in the Reaction of Peroxoanions with Organic Substrates. Photochem. Photobiol. 1979, 30, 63–70. [Google Scholar] [CrossRef]
- Charbonneau, L.; Foster, X.; Kaliaguine, S. Ultrasonic and Catalyst-Free Epoxidation of Limonene and Other Terpenes Using Dimethyl Dioxirane in Semibatch Conditions. ACS Sustain. Chem. Eng. 2018, 6, 12224–12231. [Google Scholar] [CrossRef]
- Kazemnejadi, M.; Shakeri, A.; Nikookar, M.; Shademani, R.; Mohammadi, M. Selective and metal-free epoxidation of terminal alkenes by heterogeneous polydioxirane in mild conditions. R. Soc. Open Sci. 2018, 5, 171541. [Google Scholar] [CrossRef] [Green Version]
- Sartori, G.; Armstrong, A.; Maggi, R.; Mazzacani, A.; Sartorio, R.; Bigi, F.; Dominguez-Fernandez, B. α-Fluorotropinone Immobilized on Silica: A New Stereoselective Heterogeneous Catalyst for Epoxidation of Alkenes with Oxone. J. Org. Chem. 2003, 68, 3232–3237. [Google Scholar] [CrossRef]
- D’Accolti, L.; Annese, C.; De Riccardis, A.; De Giglio, E.; Cafagna, D.; Fanelli, F.; Fusco, C. Dioxirane-Mediated Heterogeneous Epoxidations with Potassium Caroate: A Solid Catalyst Bearing Anchored Ketone Moieties. Eur. J. Org. Chem. 2012, 2012, 4616–4621. [Google Scholar] [CrossRef]
- Song, C.E.; Lim, J.S.; Kim, S.C.; Lee, K.-J.; Chi, D.Y. Immobilisation of ketone catalyst: A method to prevent ketone catalyst from decomposing during dioxirane-mediated epoxidation of alkenes. Chem. Commun. 2000, 24, 2415–2416. [Google Scholar] [CrossRef]
- Menger, F.M. Groups of Organic Molecules That Operate Collectively. Angew. Chem. Int. Ed. 1991, 30, 1086–1099. [Google Scholar] [CrossRef]
- Mahamat Ahmat, Y.; Kaliaguine, S. Epoxidation of limonene and pinenes by dimethyldioxirane in microemulsions. Catal. Today. submitted.
- Zhong, W.; Liu, M.; Dai, J.; Yang, J.; Mao, L.; Yin, D. Synergistic hollow CoMo oxide dual catalysis for tandem oxygen transfer: Preferred aerobic epoxidation of cyclohexene to 1,2-epoxycyclohexane. Appl. Catal. B Environ. 2018, 225, 180–196. [Google Scholar] [CrossRef]
- Acharyya, S.S.; Ghosh, S.; Sharma, S.K.; Bal, R. Cetyl alcohol mediated fabrication of forest of Ag/Mn3O4 nanowhiskers catalyst for the selective oxidation of styrene with molecular oxygen. RSC Adv. 2015, 5, 89879–89887. [Google Scholar] [CrossRef]
- Madadi, S.; Charbonneau, L.; Bergeron, J.Y.; Kaliaguine, S. Aerobic epoxidation of limonene using cobalt substituted mesoporous SBA-16 Part 1: Optimization via Response Surface Methodology (RSM). Appl. Catal. B Environ. 2020, 260, 118049. [Google Scholar] [CrossRef]
- Madadi, S.; Bergeron, J.-Y.; Kaliaguine, S. Kinetic investigation of aerobic epoxidation of limonene over cobalt substituted mesoporous SBA-16. Catal. Sci. Technol. 2020, 11, 594–611. [Google Scholar] [CrossRef]
- Madadi, S.; Kaliaguine, S. Activated carbon supported ruthenium as a catalyst for solvent- and initiator-free aerobic epoxidation of limonene. ACS Sustain. Chem. Eng. submitted.
- Schutz, L.; Kazemi, F.; Mackenzie, E.; Bergeron, J.Y.; Gagnon, E.; Claverie, J.P. Trans-limonene dioxide, a promising bio-based epoxy monomer. J. Polym. Sci. 2021, 59, 321–328. [Google Scholar] [CrossRef]
- Byrne, C.M.; Allen, S.D.; Lobkovsky, A.E.B.; Coates, G.W. Alternating Copolymerization of Limonene Oxide and Carbon Dioxide. J. Am. Chem. Soc. 2004, 126, 11404–11405. [Google Scholar] [CrossRef] [Green Version]
- Auriemma, F.; De Rosa, C.; Di Caprio, M.R.; Di Girolamo, R.; Ellis, W.C.; Coates, G.W. Stereocomplexed Poly(Limonene Carbonate): A Unique Example of the Cocrystallization of Amorphous Enantiomeric Polymers. Angew. Chem. 2014, 127, 1231–1234. [Google Scholar] [CrossRef]
- Parrino, F.; Fidalgo, A.; Palmisano, L.; Ilharco, L.M.; Pagliaro, M.; Ciriminna, R. Polymers of Limonene Oxide and Carbon Dioxide: Polycarbonates of the Solar Economy. ACS Omega 2018, 3, 4884–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauenstein, O.; Reiter, M.; Agarwal, S.; Rieger, B.; Greiner, A. Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency. Green Chem. 2016, 18, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Fernández, A.M.L.; Resul, M.G.; Harvey, A. Highly selective, sustainable synthesis of limonene cyclic carbonate from bio-based limonene oxide and CO2: A kinetic study. J. CO2 Util. 2019, 29, 126–133. [Google Scholar] [CrossRef]
- Schimpf, V.; Ritter, B.S.; Weis, P.; Parison, K.; Mülhaupt, R. High Purity Limonene Dicarbonate as Versatile Building Block for Sustainable Non-Isocyanate Polyhydroxyurethane Thermosets and Thermoplastics. Macromolecules 2017, 50, 944–955. [Google Scholar] [CrossRef]
- Steiner, D.; Ivison, L.; Goralski, C.T.; Appell, R.B.; Gojkovic, J.R.; Singaram, B. A facile and efficient method for the kinetic separation of commercially available cis- and trans-limonene epoxide. Tetrahedron Asymmetry 2002, 13, 2359–2363. [Google Scholar] [CrossRef]
- Morikawa, H.; Minamoto, M.; Gorou, Y.; Yamaguchi, J.-I.; Morinaga, H.; Motokucho, S. Two Diastereomers of d-Limonene-Derived Cyclic Carbonates from d-Limonene Oxide and Carbon Dioxide with a Tetrabutylammonium Chloride Catalyst. Bull. Chem. Soc. Jpn. 2018, 91, 92–94. [Google Scholar] [CrossRef]
- Rehman, A.; Russell, E.; Saleem, F.; Javed, F.; Ahmad, S.; Eze, V.C.; Harvey, A. Synthesis of trans-limonene bis-epoxide by stereoselective epoxidation of (R)-(+)-limonene. J. Environ. Chem. Eng. 2021, 9, 104680. [Google Scholar] [CrossRef]
- Cubillos, J.; Hölderich, W. Jacobsen s catalyst anchored on Al-MCM-41 and NH2 group modified Si-MCM-41 as heterogeneous enantioselective epoxidation catalyst using in situ generated dimethyldioxirane as oxidant. Rev. Fac. Ing. Univ. Antioq. 2007, 41, 31–47. [Google Scholar]
- Tian, H.; She, X.; Shu, L.; Yu, H.; Shi, Y. Highly enantioselective epoxidation of cis-olefins by chiral dioxirane. J. Am. Chem. Soc. 2000, 122, 11551–11552. [Google Scholar] [CrossRef]
- Bérubé, F. Catalyseurs D’époxydation de Type Ti/SBA-15. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2015. [Google Scholar]
- Charbonneau, L. Époxydation du Limonene. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2018. [Google Scholar]
- Madadi, S. Aerobic Epoxidation of Terpenes. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2021. [Google Scholar]
- Jérôme, F. Preface. In Industrial Green Chemistry; Kaliaguine, S., Dubois, J.-L., Eds.; De Gruyter: Berlin, Germany, 2021. [Google Scholar]
Scheme 1.
Most important monoterpenes.

Scheme 2.
Mechanism of dioxirane formation from oxone and ketones [37].
Scheme 2.
Mechanism of dioxirane formation from oxone and ketones [37].

Figure 1.
Comparison of limonene epoxidation results: (a) in absence of ultrasound; (b) under ultrasound. Reprinted with permission from Ref. [42]. Copyright 2018 American Chemical Society.
Figure 1.
Comparison of limonene epoxidation results: (a) in absence of ultrasound; (b) under ultrasound. Reprinted with permission from Ref. [42]. Copyright 2018 American Chemical Society.

Figure 2.
Dispersion of reagents and epoxidation reaction within the micelle.

Scheme 3.
Proposed pathway for aerobic epoxidation of limonene, reproduced with permission from Ref. [51]. Copyright Elsevier, 2020.
Scheme 3.
Proposed pathway for aerobic epoxidation of limonene, reproduced with permission from Ref. [51]. Copyright Elsevier, 2020.

Figure 3.
Epoxidation of limonene using Co/SBA16 (Co/Si = 1.1%) catalyst at different oxygen pressures (see Table 2 for reaction conditions). Reproduced from Ref. [52] with permission from Royal Society of Chemistry.

Figure 4.
Limonene conversion (1), 1,2-limonene oxide (2), 8,9-limonene oxide (3) and limonene dioxide (4) selectivity versus time. Reproduced from Ref. [52] with permission from Royal Society of Chemistry.
Figure 4.
Limonene conversion (1), 1,2-limonene oxide (2), 8,9-limonene oxide (3) and limonene dioxide (4) selectivity versus time. Reproduced from Ref. [52] with permission from Royal Society of Chemistry.

Scheme 4.
Proposed pathway for aerobic epoxidation of limonene. Reproduced from Ref. [52] with permission from Royal Society of Chemistry.
Scheme 4.
Proposed pathway for aerobic epoxidation of limonene. Reproduced from Ref. [52] with permission from Royal Society of Chemistry.

Scheme 5.
Proposed reaction pathway in solvent- and initiator-free aerobic epoxidation of limonene [53].
Scheme 5.
Proposed reaction pathway in solvent- and initiator-free aerobic epoxidation of limonene [53].

Scheme 6.
Different isomers of limonene dioxidesynthesized from pure D-limonene [54].
Scheme 6.
Different isomers of limonene dioxidesynthesized from pure D-limonene [54].

Scheme 7.
Synthesis of trans-LBE via stereoselective di-bromohydration of both double bonds of (R)-(+)-limonene [58].
Scheme 7.
Synthesis of trans-LBE via stereoselective di-bromohydration of both double bonds of (R)-(+)-limonene [58].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Effect of total amount of oxone fed to reactor at constant flowrate [34].
Table 1.
Effect of total amount of oxone fed to reactor at constant flowrate [34].
| Oxone®/Limonene Ratio | pH of Oxone® Solution | Conversion a (%) | 1,2-LO Yield a (%) | Limonene Dioxide Yield a (%) | Oxone Conversion to Epoxide (%) |
|---|---|---|---|---|---|
| 0.65 | 1.78 | 41 | 16 | 22 | 92.6 |
| 1.30 | 1.55 | 65 | 22 | 38 | 75 |
| 2.00 | 1.43 | 98 | 23 | 75 | 86 |
| 2.60 | 1.30 | 100 | 0 | 100 | 77 |
10 mmol of limonene, 30 mL of acetone, 4 g of sodium bicarbonate, various amounts of Oxone® dissolve in 50 mL of water was added to the flask at constant flowrate of 1 mL min−1, room temperature, total reaction time 2 h; a: Limonene conversion and yields were determined by GC-FID after purification.
Table 2.
Epoxidation of limonene in presence and absence of CTAHS [48].
Table 2.
Epoxidation of limonene in presence and absence of CTAHS [48].
| CTAHS (g) | Vacetone Vwater | noxone (mmol) | Conversion (%) | Yield (%) | Carbon Balance (%) | Oxygen Balance (%) | ||
|---|---|---|---|---|---|---|---|---|
| 1.2 LO | LDO | 1.2 LO + LDO | ||||||
| 0 | 1/11 | 12.4 | 71 | 16 | 56 | 72 | 100 | 33 |
| 0.05 | 100 | 0 | 99 | 99 | 99 | 51 | ||
| 0 | 1/7 | 8.6 | 61 | 25 | 36 | 61 | 100 | 35 |
| 0.05 | 100 | 9 | 89 | 98 | 98 | 67 | ||
| 0 | 1/5 | 6.2 | 55 | 27 | 24 | 51 | 92 | 37 |
| 0.05 | 85 | 26 | 58 | 84 | 98 | 71 | ||
| 0 | 1/3 | 3.7 | 45 | 36 | 10 | 46 | 100 | 46 |
| 0.05 | 52 | 33 | 20 | 53 | 100 | 61 | ||
Reaction conditions: 6.2 mmol of limonene diluted in 3 mL acetone; water and sodium bicarbonate are varied proportionally to noxone keeping oxone initial concentration to 0.37 mol/L and bicarbonate/oxone close to 4; room temperature; 90 min reaction time; oxone feeding protocol described in [48].
Table 3.
Effect of CTAHS on terpene epoxidation a.
| Terpene | Reaction Time (min) | CTAHS (g) | Active O/Terpene b | Vacetone/Vwater | Yield (%) c |
|---|---|---|---|---|---|
 | 30 | 0 | 3.2 | 1/9 | 44 |
| 0.05 | 3.2 | 1/9 | 100 | ||
 | 30 | 0 | 1.8 | 1/5 | 54 |
| 0.05 | 1.8 | 1/5 | 98 | ||
 | 45 | 0 | 2 | 1/6 | 47 |
| 0.05 | 2 | 1/6 | 97 |
a Reaction conditions: 6.2 mmol terpene diluted in 3 mL acetone; oxone initial concentration 0.37 mol/L; bicarbonate/oxone 4; room temperature. b active O /terpene = 2 noxone/terpene c yields of LDO, α-pinene oxide, and β-pinene oxide.
Table 4.
Catalytic performances of limonene epoxidation using different cobalt loadings a, reproduced with permission from Ref. [51]. Copyright Elsevier, 2020.
Table 4.
Catalytic performances of limonene epoxidation using different cobalt loadings a, reproduced with permission from Ref. [51]. Copyright Elsevier, 2020.
| Catalyst | Co/Si (%) | Conversion (%) | 1,2-LO Selectivity (%) | LDO Selectivity (%) |
|---|---|---|---|---|
| None | - | 42 | 15 | - |
| Co/SBA16 | 1.1 | 99 | 45 | 25 |
| Co/SBA16 | 2.7 | 99 | 49 | 30 |
| Co/SBA16 | 4.5 | 99 | 50 | 33 |
| Co/SBA16 | 6.9 | 99 | 48 | 31 |
| Co/SBA16 | 9.7 | 99 | 35 | 27 |
a Reaction conditions: temperature 28 °C; oxygen flow rate 10 mL/min; catalyst mass 0.17 g; limonene 3.6 mmol; IBA 12 mmol; 200 min reaction time; solvent ethylacetate (1,2 LO: 1,2-limonene oxide; LDO: limonene dioxide).
Table 5.
Catalytic performances of aerobic epoxidation of limonene using Co/SBA16 catalyst, Reproduced from Ref. [52] with permission from the Royal Society of Chemistry.
Table 5.
Catalytic performances of aerobic epoxidation of limonene using Co/SBA16 catalyst, Reproduced from Ref. [52] with permission from the Royal Society of Chemistry.
| Catalyst | Co/Si (%) | Conversion (%) | 1,2-LO Selectivity (%) | LDO Selectivity (%) | Epoxide Yield (%) |
|---|---|---|---|---|---|
| Co/SBA16 | 1.1 | 99 | 44 | 41 | 85 |
| Co/SBA16 | 3 | 99 | 42 | 31 | 72 |
| Co/SBA16 | 5.2 | 99 | 42 | 28 | 69 |
| Co/SBA16 | 7.5 | 99 | 41 | 25 | 65 |
| Co/SBA16 | 8.8 | 99 | 41 | 25 | 65 |
Reaction conditions: temperature 28 °C; limonene 3.6 mmol; catalyst mass 0.17 g; IBA 12 mmol; oxygen flow rate 10 mL/min; 200 min reaction time; solvent ethylacetate (1,2 LO: 1,2-limonene oxide; LDO: limonene dioxide).
Table 6.
Aerobic epoxidation of limonene over supported ruthenium catalysts on activated carbon prepared by different methods [53].
Table 6.
Aerobic epoxidation of limonene over supported ruthenium catalysts on activated carbon prepared by different methods [53].
| Catalysts | Conversion (%) | 1,2-LO (%) | 8,9-LO (%) | LDO (%) | Carvone (%) | Carveol (%) | Others (%) |
|---|---|---|---|---|---|---|---|
| Ru/AC G60 (IWI) | 28 | 21 | 6.5 | 14.6 | 16.7 | 8 | 33.2 |
| Ru/AC MRX (IWI) | 22 | 21 | 6.7 | 13.5 | 17 | 9.1 | 32.7 |
| Ru/AC RB4C (IWI) | 18 | 18 | 6 | 10 | 15 | 7.5 | 43.5 |
| Ru/AC RX3 (IWI) | 21 | 8.7 | 2.6 | 7.9 | 10 | 8 | 62.8 |
| Ru/Graphite (IWI) | 15 | 16 | 4 | 8.5 | 18 | 9.5 | 44 |
| Ru/AC G60 (CE) | 35 | 28 | 10 | 19 | 19 | 6.5 | 17.5 |
| Ru/AC G60 (SI) | 40.4 | 18.5 | 5.5 | 12.8 | 20.7 | 12.9 | 39.6 |
Reaction conditions: limonene 5 mL; catalyst mass 150 mg; temperature 80 °C; oxygen pressure 3 bar; time 6 h ((1,2-LO: 1,2-limonene oxide; 8,9-LO: 8,9-limonene oxide; LDO: limonene dioxide).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mahamat Ahmat, Y.; Madadi, S.; Charbonneau, L.; Kaliaguine, S. Epoxidation of Terpenes. Catalysts 2021, 11, 847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070847
AMA Style
Mahamat Ahmat Y, Madadi S, Charbonneau L, Kaliaguine S. Epoxidation of Terpenes. Catalysts. 2021; 11(7):847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070847
Chicago/Turabian StyleMahamat Ahmat, Yacoub, Sara Madadi, Luc Charbonneau, and Serge Kaliaguine. 2021. "Epoxidation of Terpenes" Catalysts 11, no. 7: 847. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070847
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.