
Selective C–C Bond Cleavage in Diols and Lignin Models: High-Throughput Screening of Metal Oxide-Anchored Vanadium in Mesoporous Silica

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. General Characteristics of V-(Ti/Al/Zr/Ce)-MS Catalysts
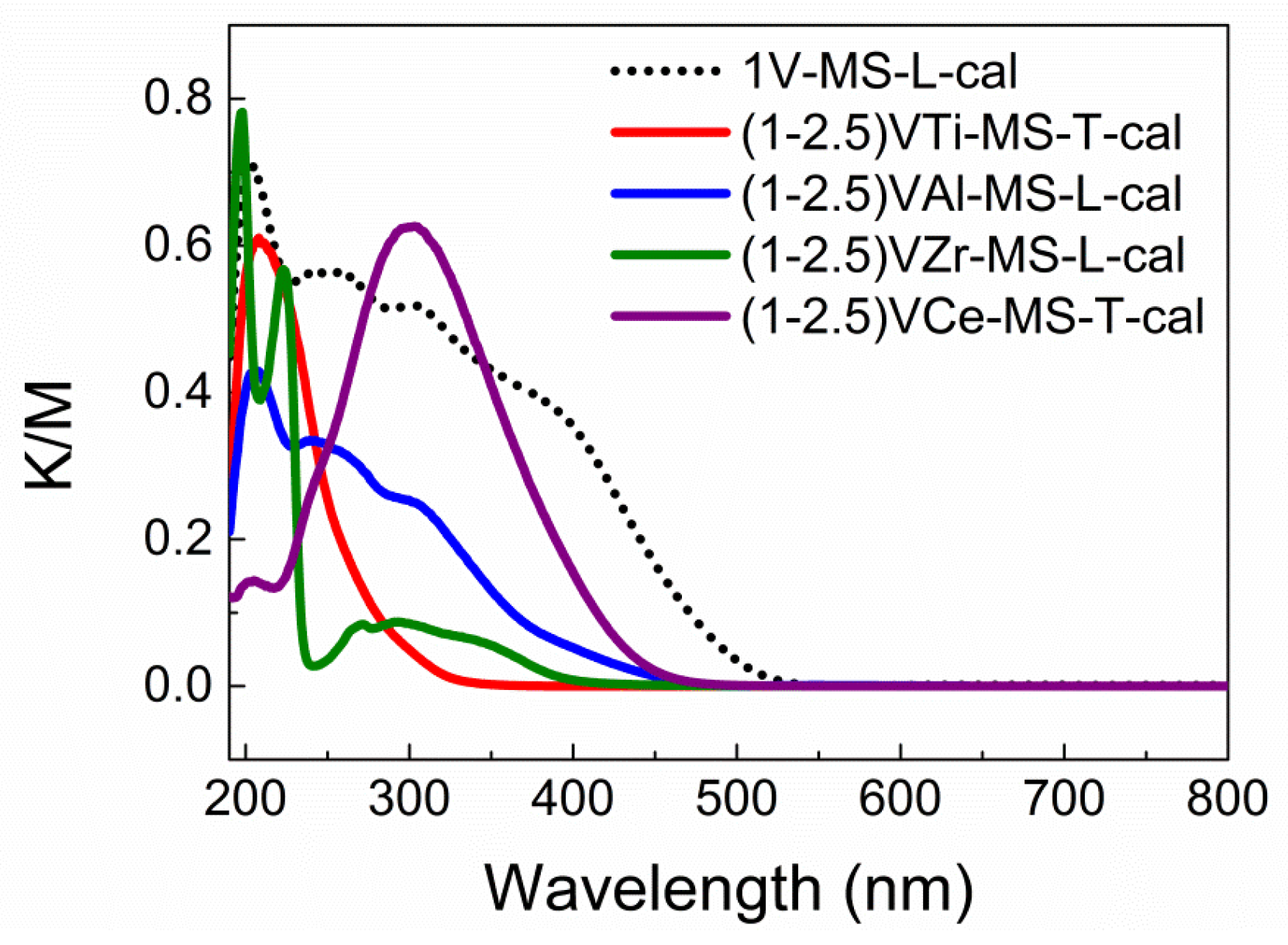
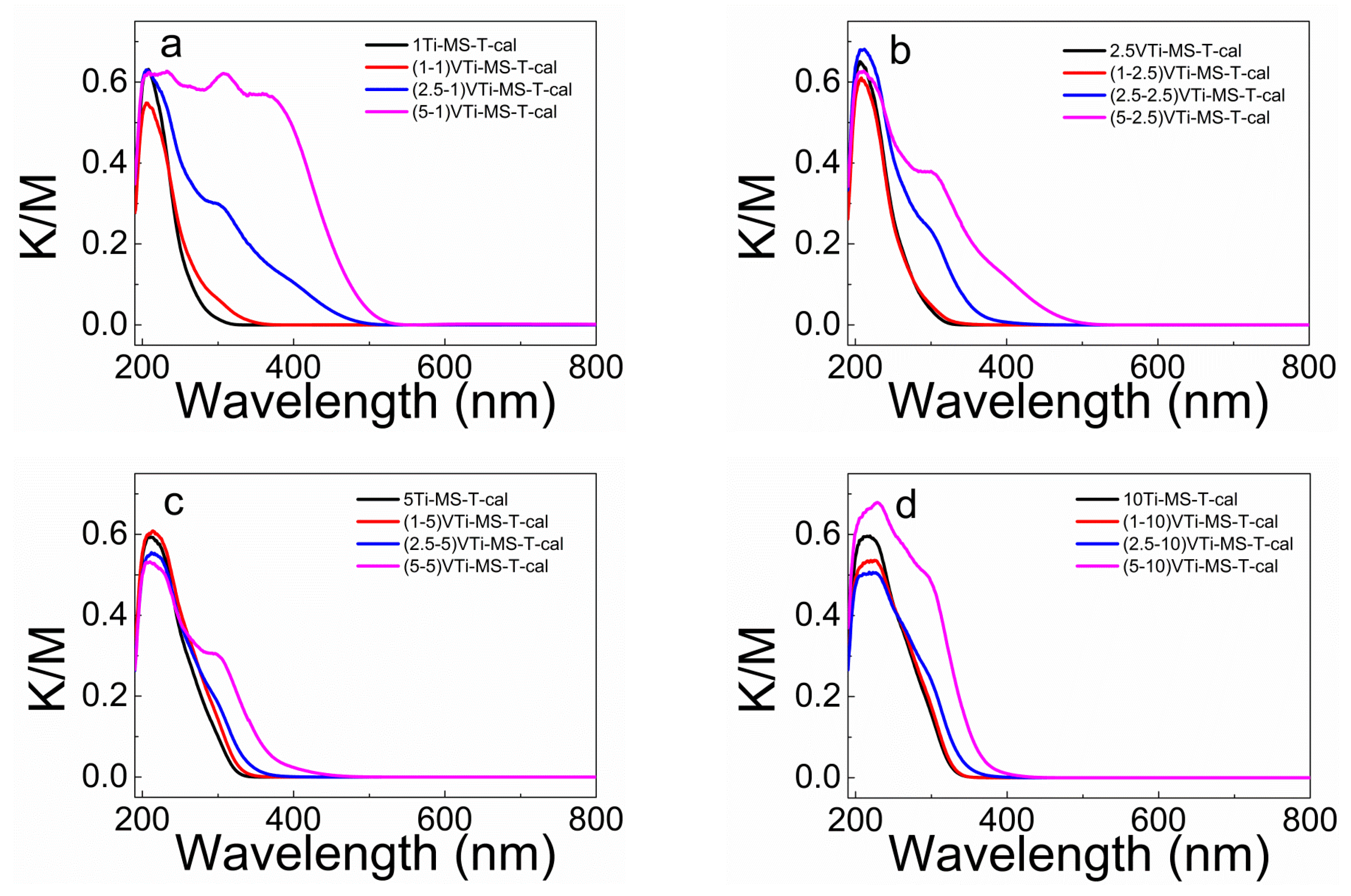
2.3. Effect of Anchoring on the Vanadium LMCT Band Blue-Shift
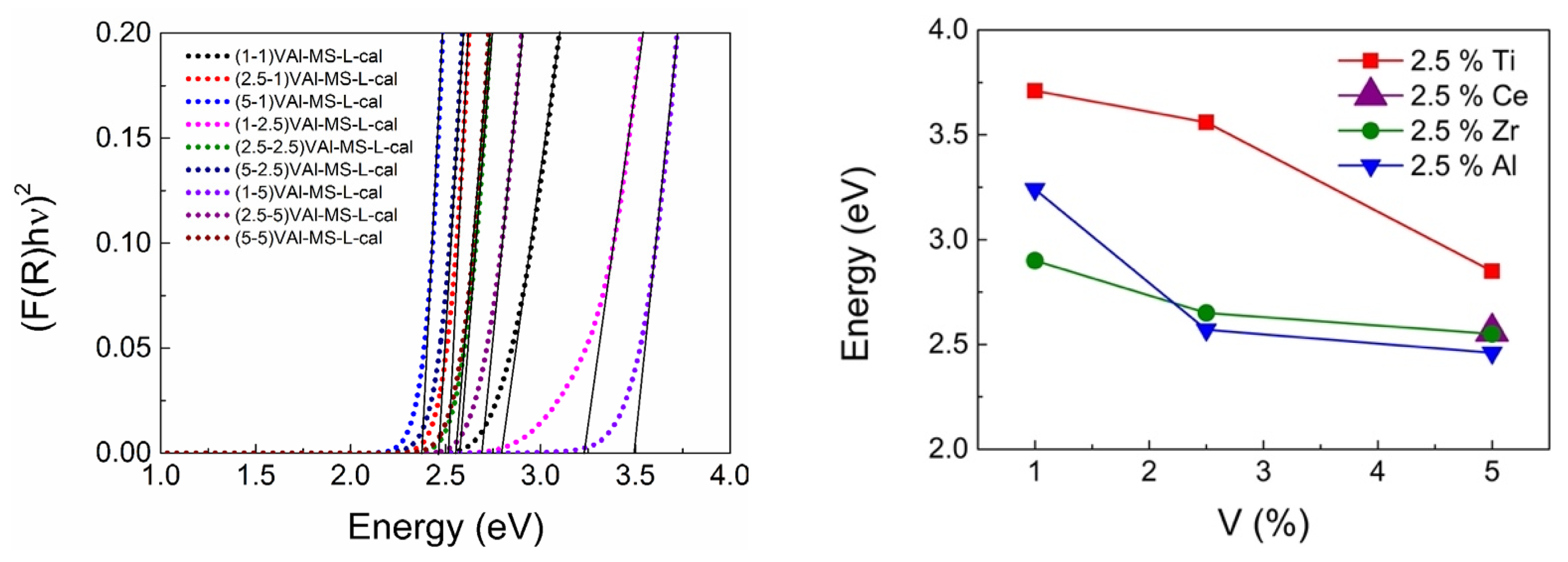
2.4. Measuring the Optical Gap to Quantify the LMCT Band Blue-Shift
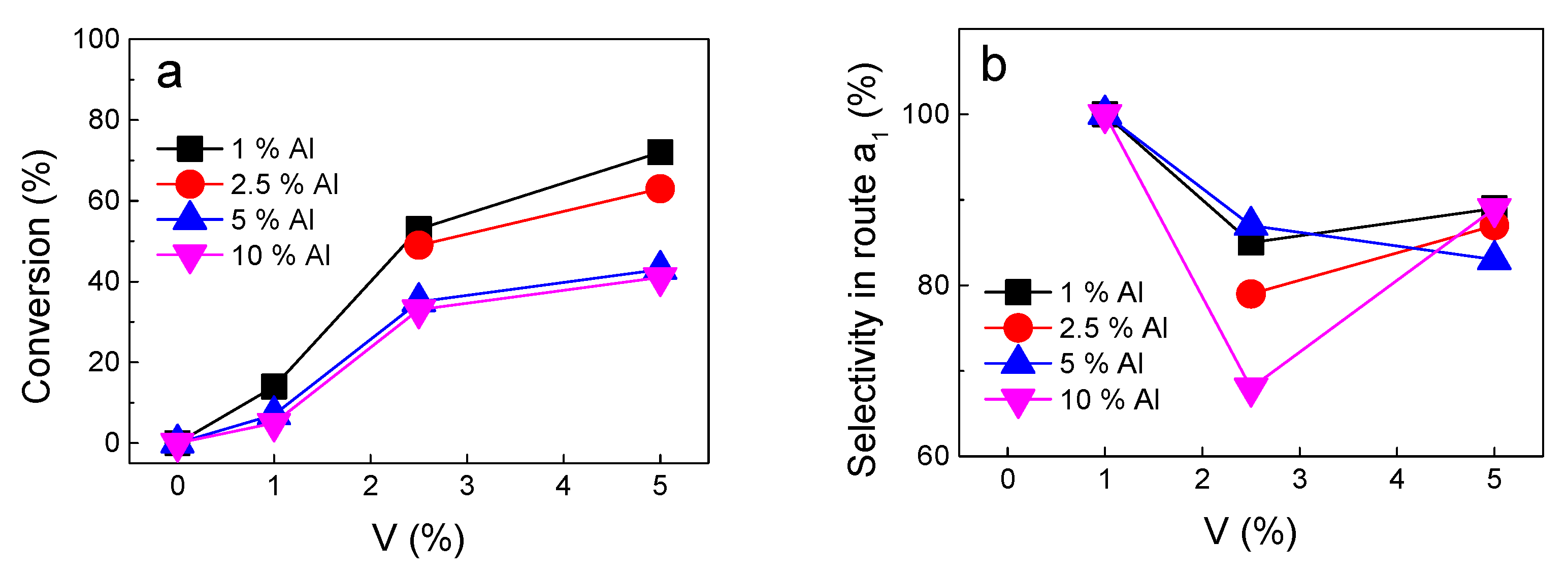
2.5. Catalyst Testing
2.6. Product Analysis and General Selectivity Trends
2.7. High-Throughput Catalytic Tests Using the Lignin Model
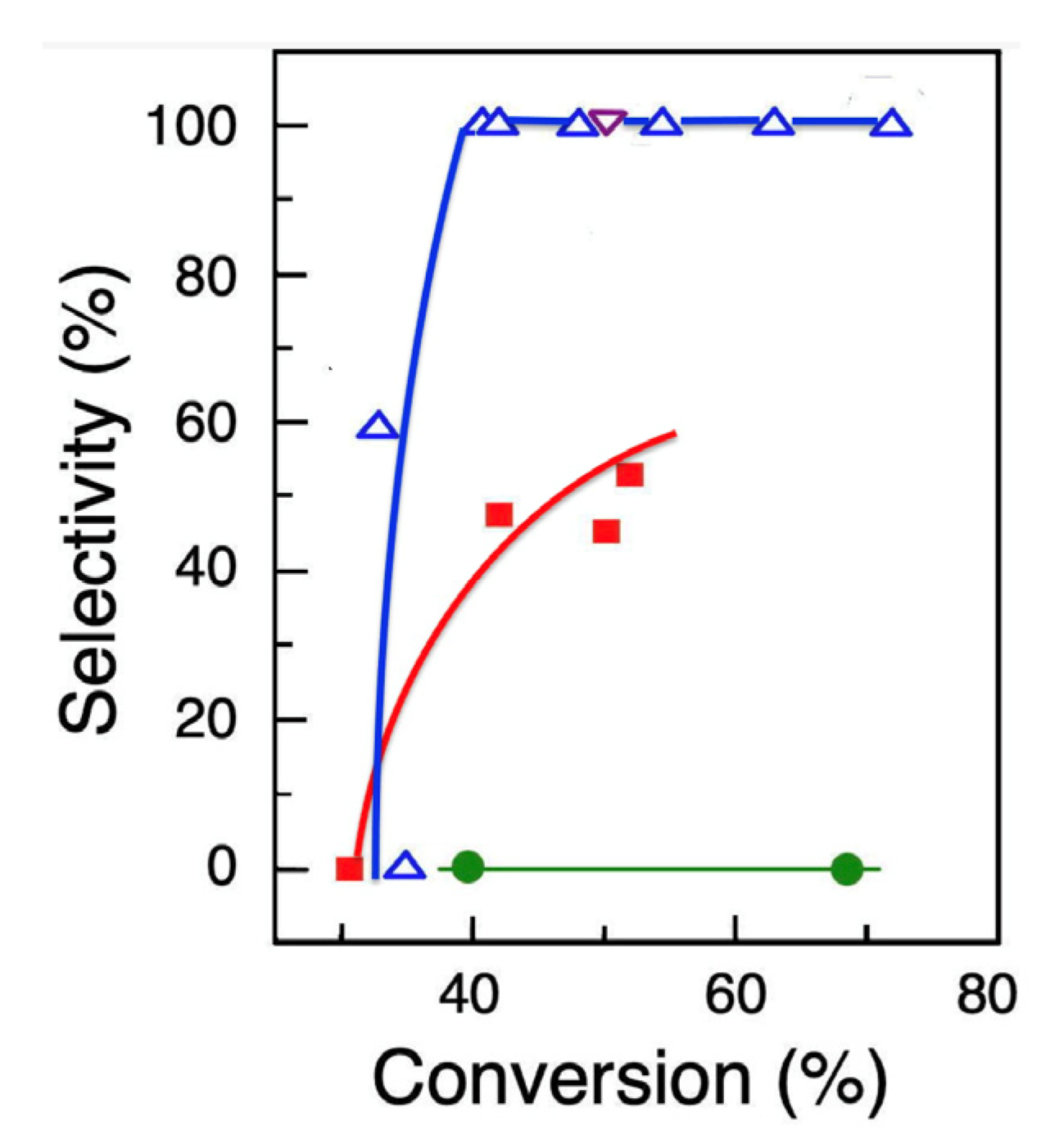
2.8. Reactivity of Substrate 2: Testing Diol Internal C–C Bond Cleavage
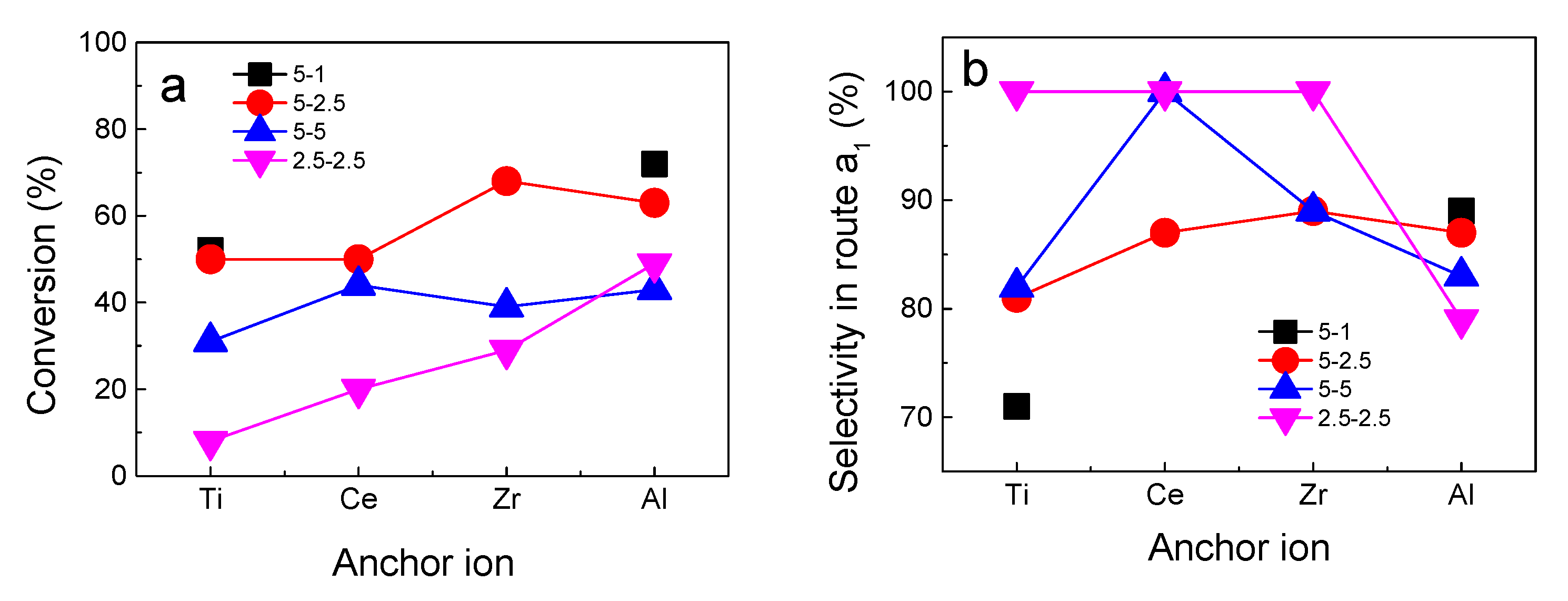
2.9. Activity, Selectivity, and Vanadium Site Nuclearity
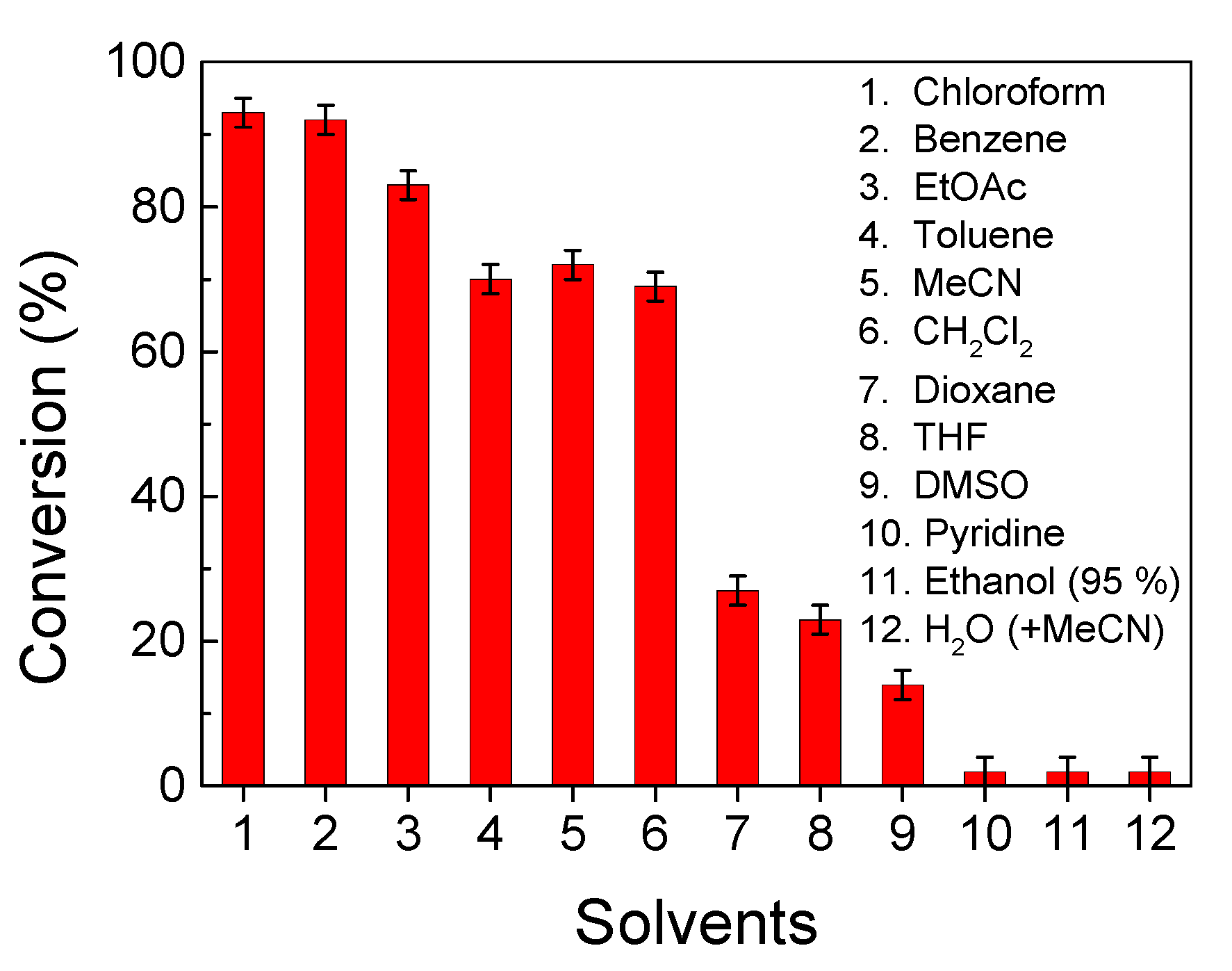
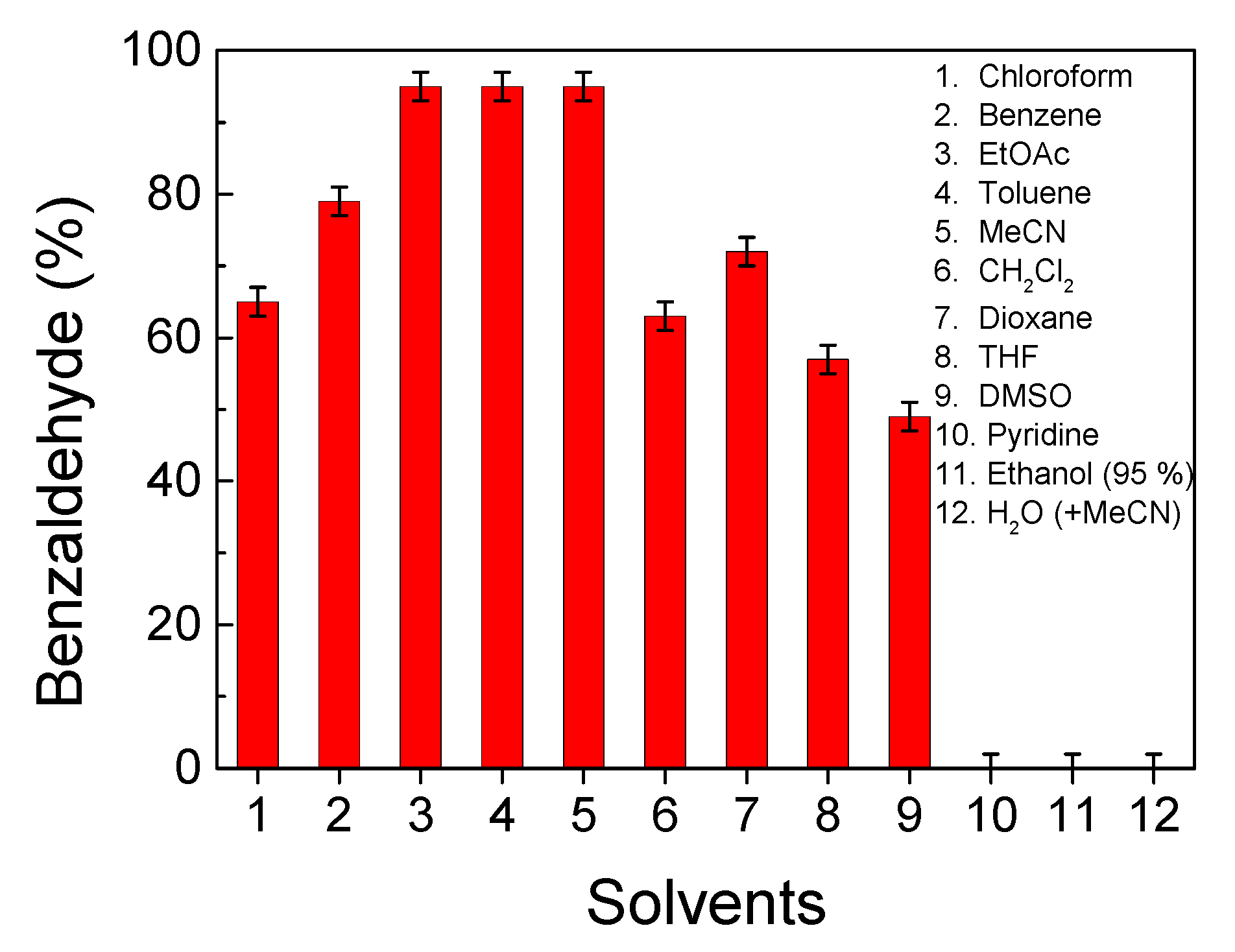
2.10. Solvent Screening for Catalyst Comparison and Vanadium Leaching Test
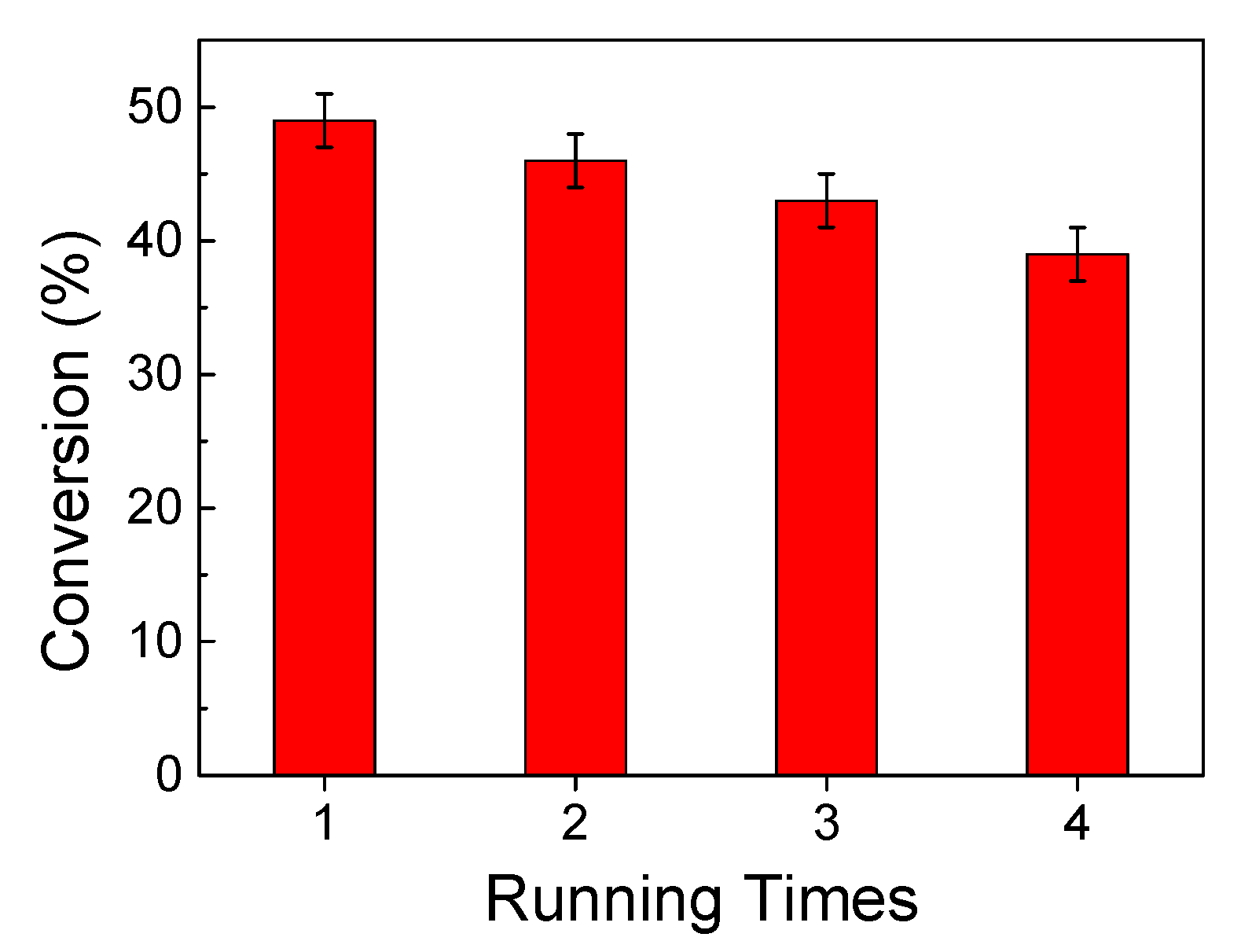
2.11. Leaching Test and Recyclability
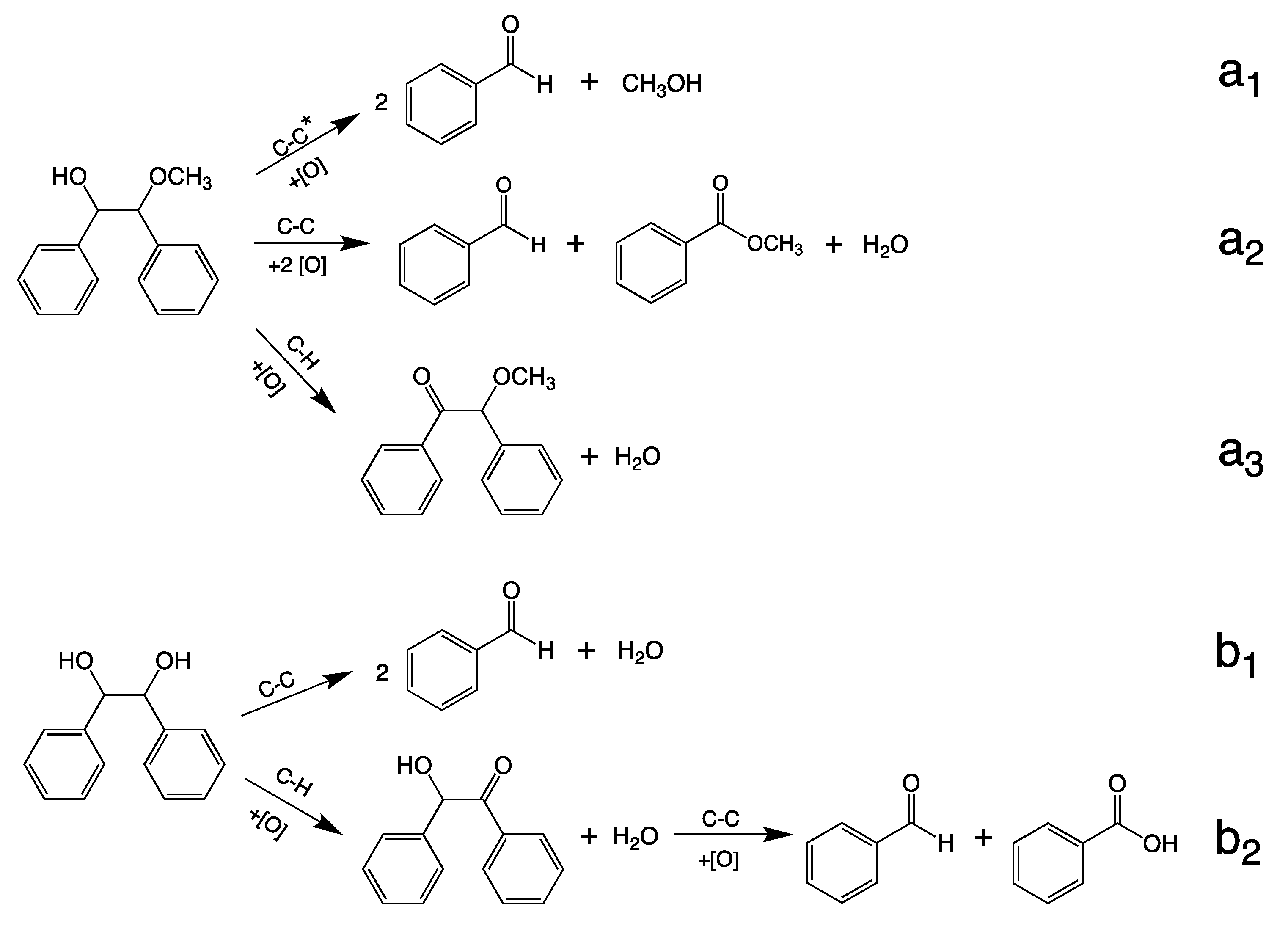
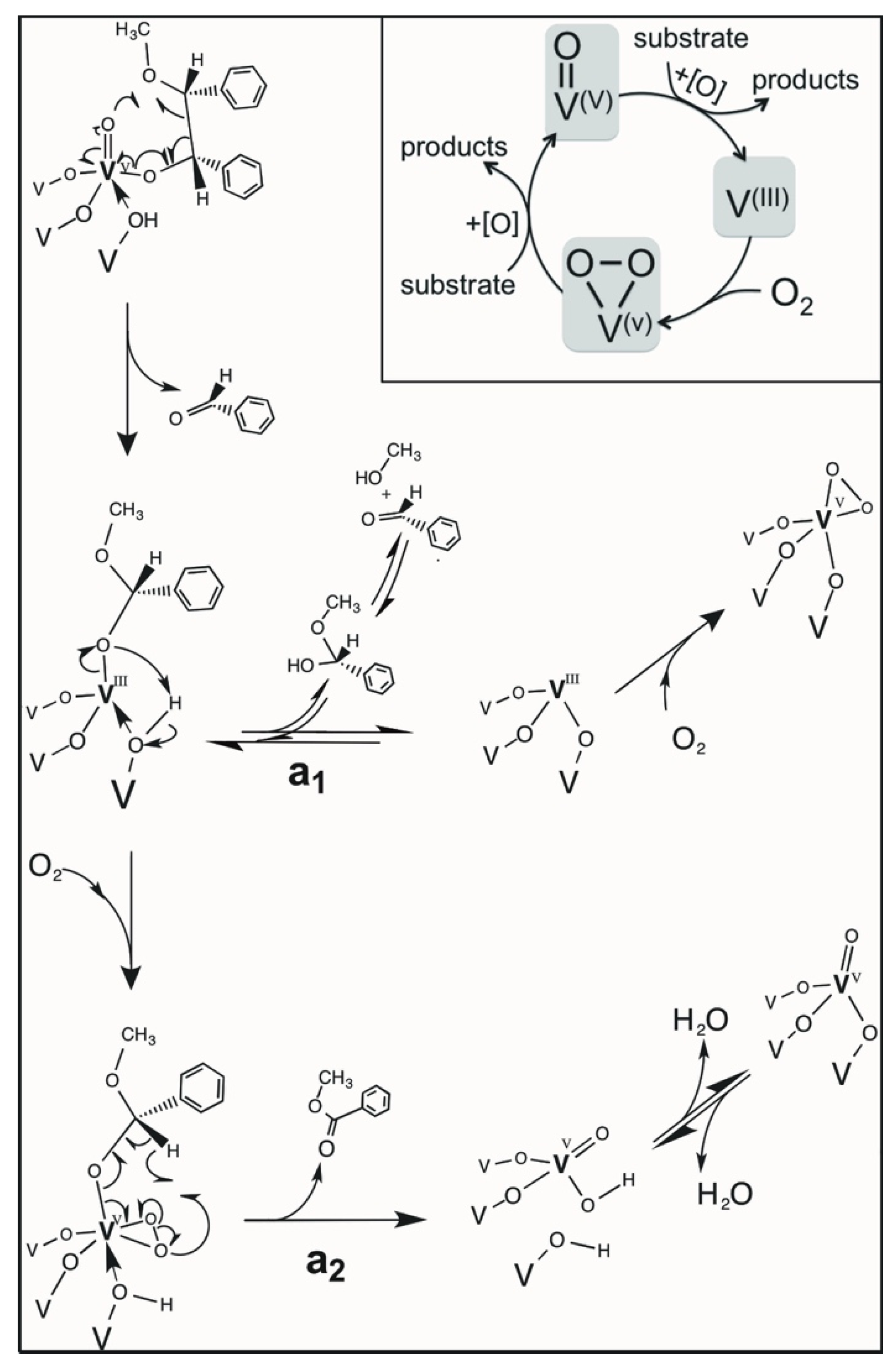
2.12. Mechanistic Considerations
2.13. Catalytic Cycle
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Synthesis
4.2.1. Preparation of Mesoporous Silica MS (MS = Microwave Synthetic Mesoporous Silica)
4.2.2. Preparation of 1V-MS-L
4.2.3. Preparation of VTi-MS-T
- Solution A: 25.52 g of tetramethylammonium hydroxyde (TMAOH, 25 wt%) was dissolved in 90 g of deionized water, then 41.66 g of tetraethoxysilane (TEOS) was added dropwise under stirring. The mixture was stirred at room temperature overnight until it was clear.
- Solution B: 0.68 g of tetrabutyl orthotitanate (TBOT) was dissolved in 25 mL isopropanol.
- Solution C: solution B was added dropwise to solution A with stirring at 0 °C. The mixture was stirred at 0 °C until clear, then solution C was separated into 4 aliquots, each containing about 44.5 g.
- Solution D: four surfactant solutions were prepared in parallel, i.e., D1, D2, D3, and D4. Each one contained 2.05 g CTATos dissolved in 44.8 g deionized water and stirred at 60 °C for 1 h. Solution D1 was a clear surfactant solution without additional vanadyl sulfate. Vanadyl sulfate (0.106 g, 0.266 g, and 0.531 g) was added in solutions D2, D3, and D4 separately, and stirred for another 1 h at 60 °C.
4.2.4. Preparation of VAl-MS-L
4.2.5. Preparation of VZr-MS-L and VZr-MS-L
4.2.6. Preparation of VCe-MS-T
4.2.7. Synthesis of Lignin Model 1,2-Diphenyl-2-methoxyethanol (β-Methoxy-α-phenylphenethyl Alcohol)
4.3. Catalytic Tests Using High-Throughput Screening (HTS)
4.4. Leaching Tests of Vanadium-Containing MCM-41 Catalysts
4.5. Recycling Test of the Catalyst
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [Green Version]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Sudasanam, P.; Peeters, E.; Makshina, E.V.; Parvulescu, V.I.; Sels, B.F. Advances in porous and nanoscale catalysts for viable biomass conversion. Chem. Soc. Rev. 2019, 48, 2366–2421. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bouxin, F.P.; Fan, J.; Budarin, V.L.; Hu, C.; Clark, J.H. Recent advances in the catalytic depolymerization of lignin towards phenolic chemicals: A review. ChemSusChem 2020, 13, 4296–4317. [Google Scholar] [CrossRef] [PubMed]
- Brosse, N.; Ibrahim, M.N.M.; Rahim, A.A. Biomass to bioethanol: Initiatives of the future for lignin. ISRN Mater. Sci. 2011, 2011, 461482. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Dong, L.; Guo, Y.; Liu, X.; Wang, Y. Chemicals from Lignin: A review of catalytic conversion involving hydrogen. ChemSusChem 2020, 13, 4181–4198. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Yan, L.; Yu, H.-Z.; Zhang, Q.; Fu, Y. Mechanism of vanadium-catalyzed selective C–O and C–C cleavage of lignin model compound. ACS Catal. 2016, 6, 4399–4410. [Google Scholar] [CrossRef]
- Hanson, S.K.; Wu, R.; Silks, L.A. C–C or C–O Bond Cleavage in a Phenolic Lignin Model Compound: Selectivity Depends on Vanadium Catalyst. Angew. Chem. Int. Ed. 2012, 124, 3466–3469. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Thorn, D.L. Aerobic oxidation of lignin models using a base metal vanadium catalyst. Inorg. Chem. 2010, 49, 5611–5618. [Google Scholar] [CrossRef]
- Sedai, B.; Díaz-Urrutia, C.; Baker, R.T.; Wu, R.; Silks, L.A.; Hanson, S.K. Aerobic oxidation of β-1 lignin model compounds with copper and oxovanadium catalysts. ACS Catal. 2013, 3, 3111–3122. [Google Scholar] [CrossRef]
- Díaz-Urrutia, C.; Hurisso, B.B.; Gauthier, P.M.; Sedai, B.; Singer, R.D.; Baker, R.T. Catalytic aerobic oxidation of lignin-derived bio-oils using oxovanadium and copper complex catalysts and ionic liquids. J. Mol. Catal. A Chem. 2016, 423, 414–422. [Google Scholar] [CrossRef]
- Díaz-Urrutia, C.; Chen, W.-C.; Crites, C.-O.; Daccache, J.; Korobkov, I.; Baker, R.T. Towards lignin valorisation: Comparing homogeneous catalysts for the aerobic oxidation and depolymerisation of organosolv lignin. RSC Adv. 2015, 5, 70502–70511. [Google Scholar] [CrossRef]
- Sedai, B.; Baker, R.T. Copper catalysts for selective C-C bond cleavage of β-O-4 lignin model compounds. Adv. Synth. Catal. 2014, 356, 3563–3574. [Google Scholar] [CrossRef]
- Mottweiler, J.; Puche, M.; Räuber, C.; Schmidt, T.; Concepción, P.; Corma, A.; Bolm, C. Copper-and vanadium-catalyzed oxidative cleavage of lignin using dioxygen. ChemSusChem 2015, 8, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, I.; Van Der Voort, P. Supported vanadium oxide in heterogeneous catalysis: Elucidating the structure-activity relationship with spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 2826–2832. [Google Scholar] [CrossRef]
- Gao, X.T.; Bare, S.R.; Fierro, J.L.G.; Wachs, I.E. Structural characteristics and reactivity/reducibility properties of dispersed and bilayered V2O5/TiO2/SiO2 catalysts. J. Phys. Chem. B 1999, 103, 618–629. [Google Scholar] [CrossRef]
- Fang, L. Surface Engineering of Mesoporous Silica for Ti-Based Epoxidation Catalysts. Ph.D. Thesis, Ecole Normale Supérieure de Lyon—ENS Lyon, Lyon, France, 2012. [Google Scholar]
- Zheng, Y. Synthesis and Characterization of Vanadium-Containing Mesoporous Silica and Its Application in the Catalysis of Oxidation Reaction. Ph.D. Thesis, Ecole Normale Supérieure de Lyon—ENS Lyon, Lyon, France, 2014. [Google Scholar]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical properties and electronic structure of amorphous germanium. Phys. Status Solidi 1966, 15, 627–637. [Google Scholar] [CrossRef]
- Swanepoel, R. Determination of the thickness and optical constants of amorphous silicon. J. Phys. E Sci. Inst. 1983, 16, 1214. [Google Scholar] [CrossRef]
- Weber, R.S. Effect of local-structure on the UV-visible absorption edges of molybdenum oxide clusters and supported molybdenum oxides. J. Catal. 1995, 151, 470–474. [Google Scholar] [CrossRef]
- Wu, C.-G.; Bein, T. Microwave synthesis of molecular sieve MCM-41. Chem. Commun. 1996, 925–926. [Google Scholar] [CrossRef]
- Tompsett, G.A.; Conner, W.C.; Yngvesson, K.S. Microwave synthesis of nanoporous materials. ChemPhysChem 2006, 7, 296–319. [Google Scholar] [CrossRef] [PubMed]
- Chaignon, J.; Bouizi, Y.; Davin, L.; Calin, N.; Albela, B.; Bonneviot, L. Minute-made and low carbon fingerprint microwave synthesis of high quality templated mesoporous silica. Green Chem. 2015, 17, 3130–3140. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular-sieves prepared with liquid-crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Kresge, C.T.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E. The discovery of ExxonMobil’s M41S family of mesoporous molecular sieves. In Studies in Surface Science and Catalysis; Osamu, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 148, pp. 53–72. [Google Scholar]
- Busca, G.; Giamello, E. Electron spin resonance of V (4+) centers in V-Ti complex oxide powders. Mater. Chem. Phys. 1990, 25, 475–485. [Google Scholar] [CrossRef]
- Busca, G.; Centi, G.; Marchetti, L.; Trifiro, F. Chemical and spectroscopic study of the nature of a vanadium oxide monolayer supported on a high-surface-area TiO2 anatase. Langmuir 1986, 2, 568–577. [Google Scholar] [CrossRef]
- Dutoit, D.; Schneider, M.; Fabrizioli, P.; Baiker, A. Vanadia—Silica low-temperature aerogels: Influence of aging and vanadia loading on structural and chemical properties. Chem. Mater. 1996, 8, 734–743. [Google Scholar] [CrossRef]
- Chao, K.; Wu, C.; Chang, H.; Lee, L.; Hu, S.-F. Incorporation of vanadium in mesoporous MCM-41 and microporous AFI zeolites. J. Phys. Chem. B 1997, 101, 6341–6349. [Google Scholar] [CrossRef]
- Gao, X.T.; Wachs, I.E. Investigation of surface structures of supported vanadium oxide catalysts by UV-vis-NIR diffuse reflectance spectroscopy. J. Phys. Chem. B 2000, 104, 1261–1268. [Google Scholar] [CrossRef]
- Lee, E.L.; Wachs, I.E. Molecular design and in situ spectroscopic investigation of multilayered supported M1Ox/M2Ox/SiO2 catalysts. J. Phys. Chem. C 2008, 112, 20418–20428. [Google Scholar] [CrossRef]
- Lee, E.L.; Wachs, I.E. Surface chemistry and reactivity of well-defined multilayered supported M1Ox/M2Ox/SiO2 catalysts. J. Catal. 2008, 258, 103–110. [Google Scholar] [CrossRef]
- Moussa, N.; Ghorbel, A. UV–vis–DR study of VOx/SiO2 catalysts prepared by sol–gel method. Appl. Surf. Sci. 2008, 255, 2270–2275. [Google Scholar] [CrossRef]
- Gao, X.; Bare, S.R.; Weckhuysen, B.M.; Wachs, I.E. In situ spectroscopic investigation of molecular structures of highly dispersed vanadium oxide on silica under various conditions. J. Phys. Chem. B 1998, 102, 10842–10852. [Google Scholar] [CrossRef] [Green Version]
- Bulanek, R.; Capek, L.; Setnicka, M.; Cicmanec, P. DR UV-vis Study of the Supported Vanadium Oxide Catalysts. J. Phys. Chem. C 2011, 115, 12430–12438. [Google Scholar] [CrossRef]
- Bulanek, R.; Cicmanec, P.; Sheng-Yang, H.; Knotek, P.; Capek, L.; Setnicka, M. Effect of preparation method on nature and distribution of vanadium species in vanadium-based hexagonal mesoporous silica catalysts: Impact on catalytic behavior in propane ODH. Appl. Catal. A-General 2012, 415, 29–39. [Google Scholar] [CrossRef]
- He, J.; Yang, X.; Men, B.; Wang, D. Interfacial Mechanisms of Heterogeneous Fenton Reactions Catalyzed by Iron-Based Materials: A Review. J. Environ. Sci. 2016, 39, 97–109. [Google Scholar] [CrossRef]
- Wigington, B.N.; Drummond, M.L.; Cundari, T.R.; Thorn, D.L.; Hanson, S.K.; Scott, S.L. A Biomimetic Pathway for Vanadium-Catalyzed Aerobic Oxidation of Alcohols: Evidence for a Base-Assisted Dehydrogenation Mechanism. Chem. Eur. J. 2012, 18, 14981–14988. [Google Scholar] [CrossRef]
- Khenkin, A.M.; Neumann, R. Oxidative C−C bond cleavage of primary alcohols and vicinal diols catalyzed by H5PV2Mo10O40 by an electron transfer and oxygen transfer reaction mechanism. J. Am. Chem. Soc. 2008, 130, 14474–14476. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Sutton, A.D.; Thorn, D.L. Aerobic oxidation of pinacol by vanadium (V) dipicolinate complexes: Evidence for reduction to vanadium (III). J. Am. Chem. Soc. 2009, 131, 428–429. [Google Scholar] [CrossRef]
- Ohler, N.; Bell, A.T. Study of the elementary processes involved in the selective oxidation of methane over MoOx/SiO2. J. Phys. Chem. B 2006, 110, 2700–2709. [Google Scholar] [CrossRef]
- Chempath, S.; Bell, A.T. A DFT study of the mechanism and kinetics of methane oxidation to formaldehyde occurring on silica-supported molybdena. J. Catal. 2007, 247, 119–126. [Google Scholar] [CrossRef]
- Getsoian, A.B.; Zhai, Z.; Bell, A.T. Band-gap energy as a descriptor of catalytic activity for propene oxidation over mixed metal oxide catalysts. J. Am. Chem. Soc. 2014, 136, 13684–13697. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Innocenti, G.; Oulego, P.; Gitis, V.; Wu, H.-H.; Ensing, B.; Cavani, F.; Rothenberg, G.; Shiju, N.R. Highly selective oxidation of ethyl lactate to ethyl pyruvate catalysed by mesoporous vanadia–titania. ACS Catal. 2018, 8, 2365–2374. [Google Scholar] [CrossRef] [Green Version]
- Yun, D.; Wang, Y.; Herrera, J.E. Ethanol partial oxidation over VOx/TiO2 catalysts: The role of titania surface oxygen on vanadia reoxidation in the Mars–van Krevelen mechanism. ACS Catal. 2018, 8, 4681–4693. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Catalyst | V 1 mol% | M 1 mol% | V 2 % | M 2 % |
|---|---|---|---|---|---|
| 1 | (2.5–2.5)VTi-MS-T-cal | 0.69 | 2.8 | 83 | 100 |
| 2 | (2.5–2.5)VAl-MS-L-cal | 2.2 | 4.8 | 49 | 93 |
| 3 | (2.5–2.5)VZr-MS-L-cal | 2.2 | 4.1 | 50 | 100 |
| 4 | (2.5–2.5)VCe-MS-T-cal | 1.2 | 2.5 | 60 | 61 |
| 5 | (5–1)VTi-MS-T-cal | 2.21 | 1.12 | - | - |
| 6 | (5–1)VAl-MS-L-cal | 3.90 | 2.11 | - | - |
| 7 | (1–0)V-MS-L-cal | 0.7 | - | 27 | - |
| Catalysts | Conversion | - | Selectivity | (%) | - |
|---|---|---|---|---|---|
| - | (%) | Benzaldehyde | Methyl Benzoate | Methanol | Benzoin Methyl Ether |
| 1Al-MS-L-cal | 0 | - | - | - | - |
| (1–1)VAl-MS-L-cal | 14 | 100 | 0 | 0 | 0 |
| (2.5–1)VAl-MS-L-cal | 53 | 86 | 7 | 7 | 0 |
| (5–1)VAl-MS-L-cal | 72 | 89 | 5 | 6 | 0 |
| 2.5Al-MS-L-cal | 0 | - | - | - | - |
| (2.5–2.5)VAl-MS-L-cal | 49 | 84 | 10 | 6 | 0 |
| (5–2.5)VAl-MS-L-cal | 63 | 88 | 6 | 6 | 0 |
| 5Al-MS-L-cal | 0 | - | - | - | - |
| (1–5)VAl-MS-L-cal | 7 | 100 | 0 | 0 | 0 |
| (2.5–5)VAl-MS-L-cal | 35 | 93 | 0 | 0 | 7 |
| (5–5)VAl-MS-L-cal | 43 | 85 | 8 | 7 | 0 |
| 10Al-MS-L-cal | 0 | - | - | - | - |
| (1–10)VAl-MS-L-cal | 5 | 100 | 0 | 0 | 0 |
| (2.5–10)VAl-MS-L-cal | 33 | 83 | 10 | 0 | 7 |
| (5–10)VAl-MS-L-cal | 41 | 94 | 0 | 0 | 6 |
| Catalysts | Conversion (%) |
|---|---|
| (5–1)VTi-MS-T-cal | 69 |
| (5–1)VAl-MS-L-cal | 82 |
| (5–2.5)VZr-MS-L-cal | 74 |
| (5–2.5)VCe-MS-T-cal | 77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Clément, R.; Lu, Y.; Albela, B.; Baker, R.T.; Bonneviot, L. Selective C–C Bond Cleavage in Diols and Lignin Models: High-Throughput Screening of Metal Oxide-Anchored Vanadium in Mesoporous Silica. Catalysts 2021, 11, 901. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080901
Lu X, Clément R, Lu Y, Albela B, Baker RT, Bonneviot L. Selective C–C Bond Cleavage in Diols and Lignin Models: High-Throughput Screening of Metal Oxide-Anchored Vanadium in Mesoporous Silica. Catalysts. 2021; 11(8):901. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080901
Chicago/Turabian StyleLu, Xinnan, Roxanne Clément, Yong Lu, Belén Albela, R. Tom Baker, and Laurent Bonneviot. 2021. "Selective C–C Bond Cleavage in Diols and Lignin Models: High-Throughput Screening of Metal Oxide-Anchored Vanadium in Mesoporous Silica" Catalysts 11, no. 8: 901. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080901