
Post-Plasma Catalysis for Trichloroethylene Abatement with Ce-Doped Birnessite Downstream DC Corona Discharge Reactor

 ,
,  ,
,
Abstract
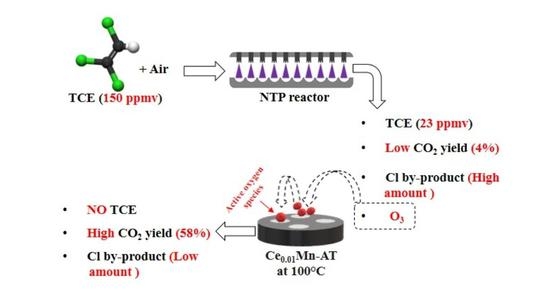
:
1. Introduction
2. Results and Discussion
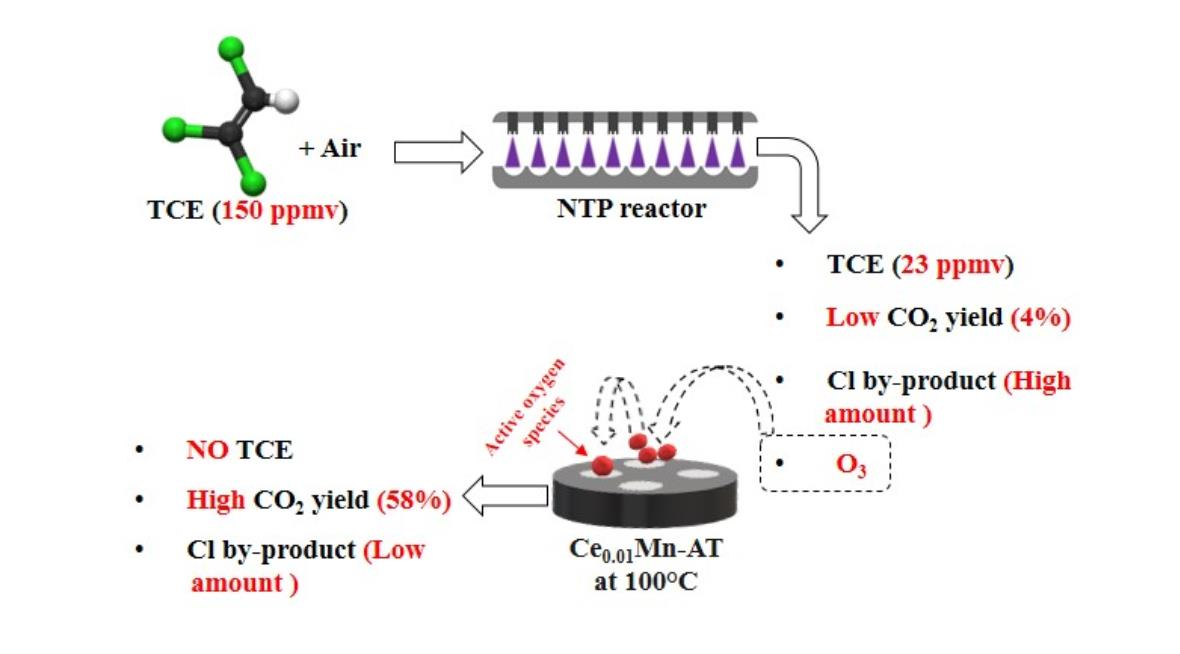
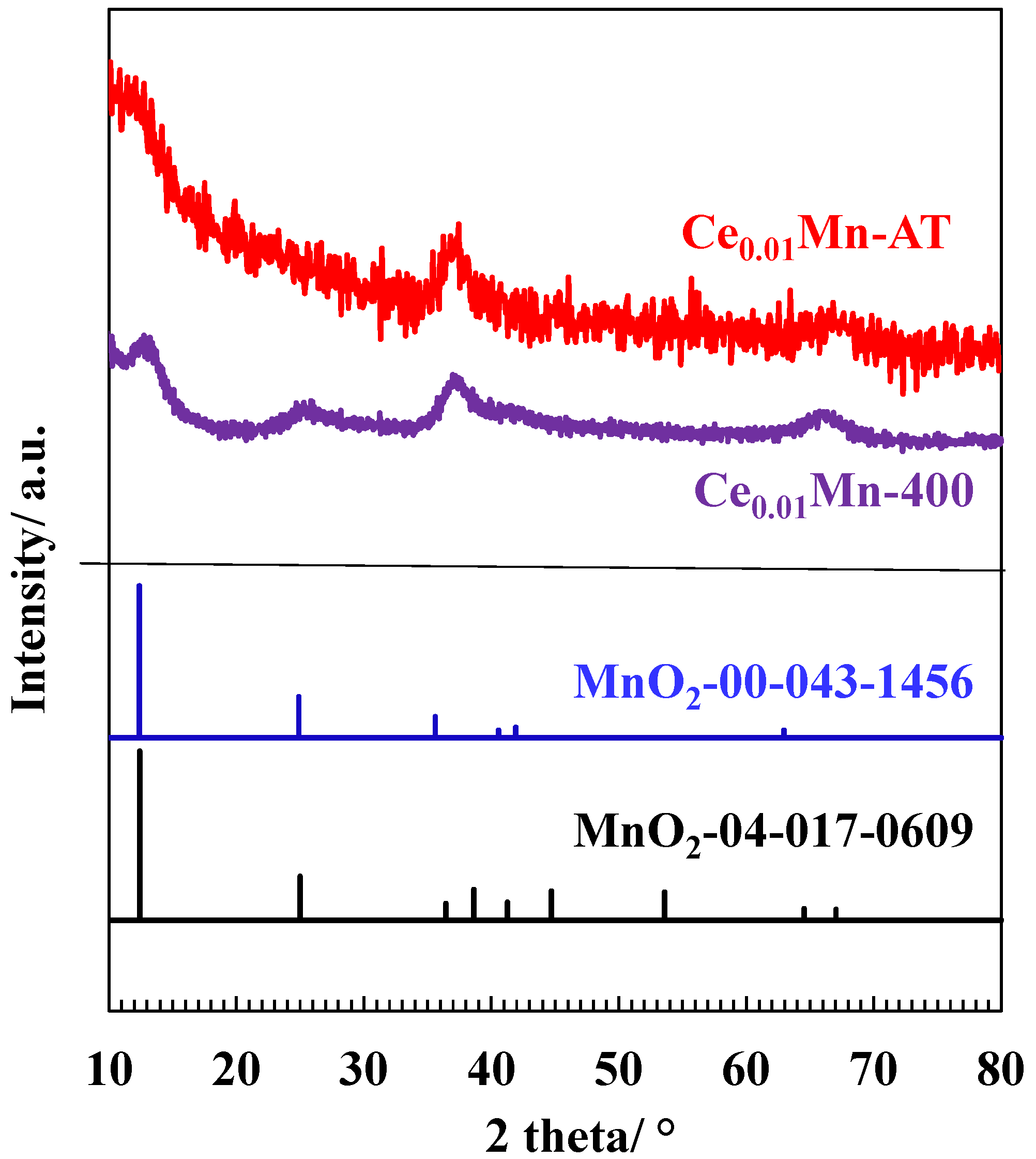
2.1. Main Physicochemical Characteristics of the Fresh Materials
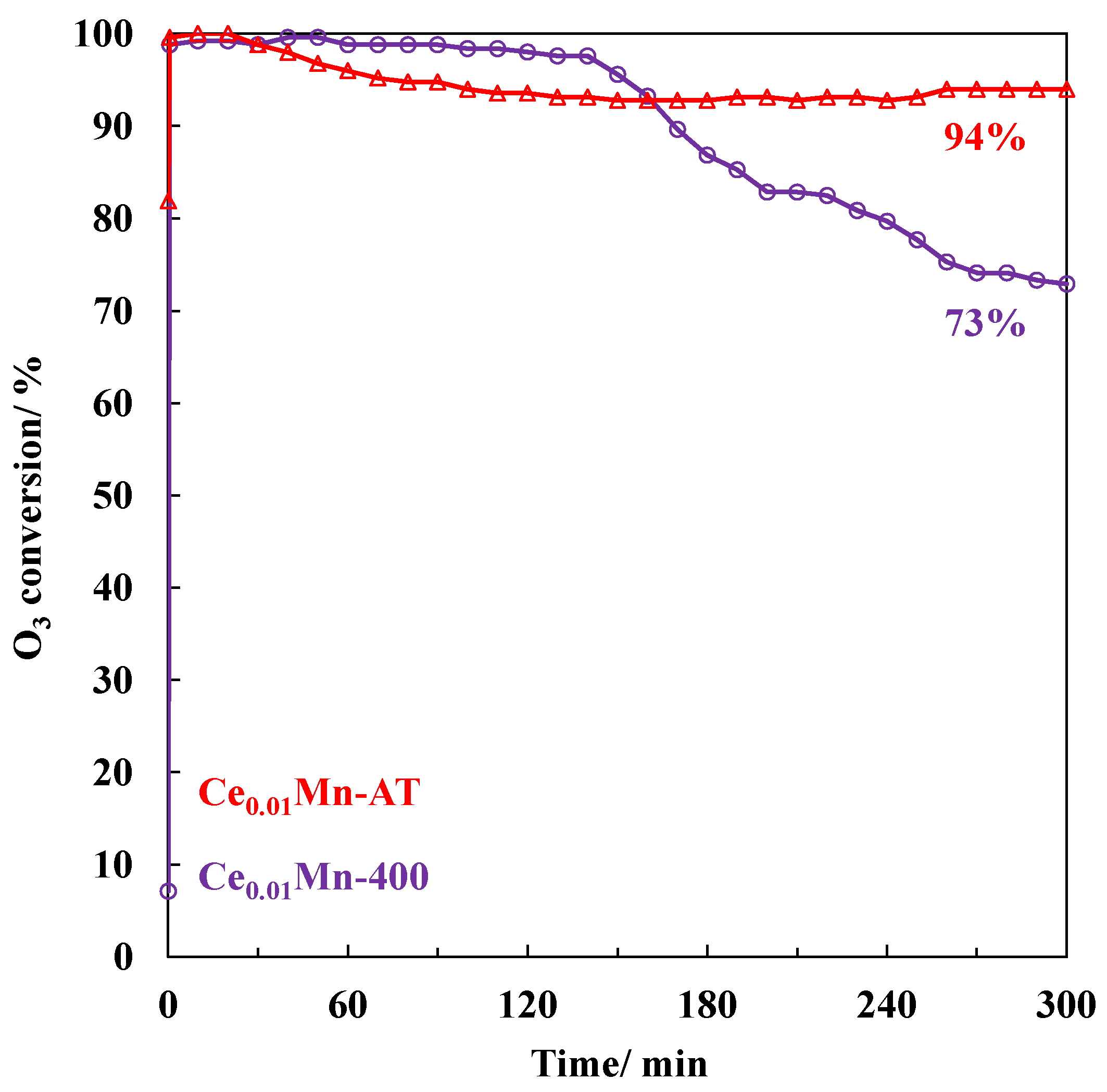
2.2. O3 Decomposition in Ce0.01Mn-400 and Ce0.01Mn-AT Samples Using Non-Thermal Plasma (NTP) as Ozoner
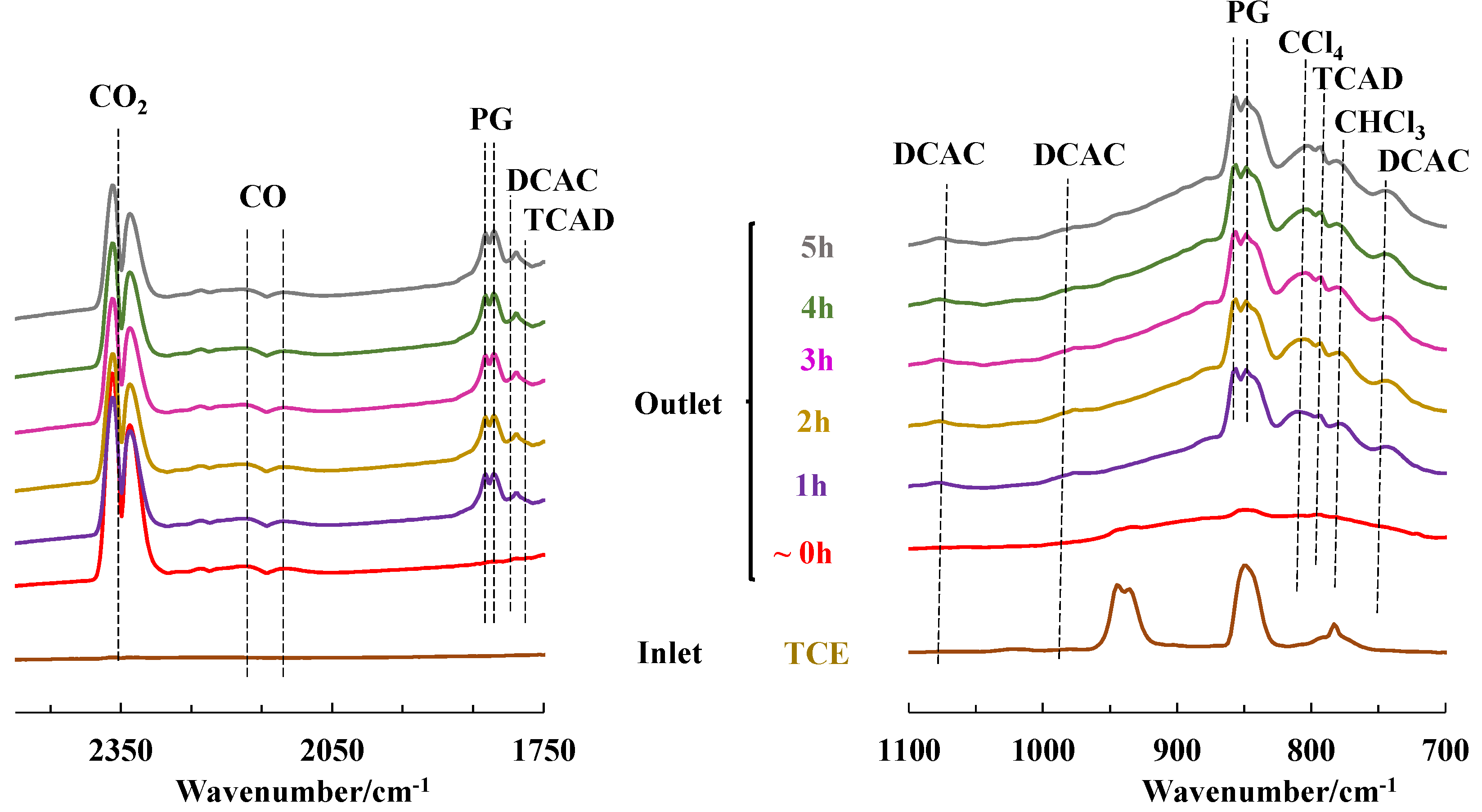
2.3. TCE Abatement by Non-Thermal Plasma (NTP)
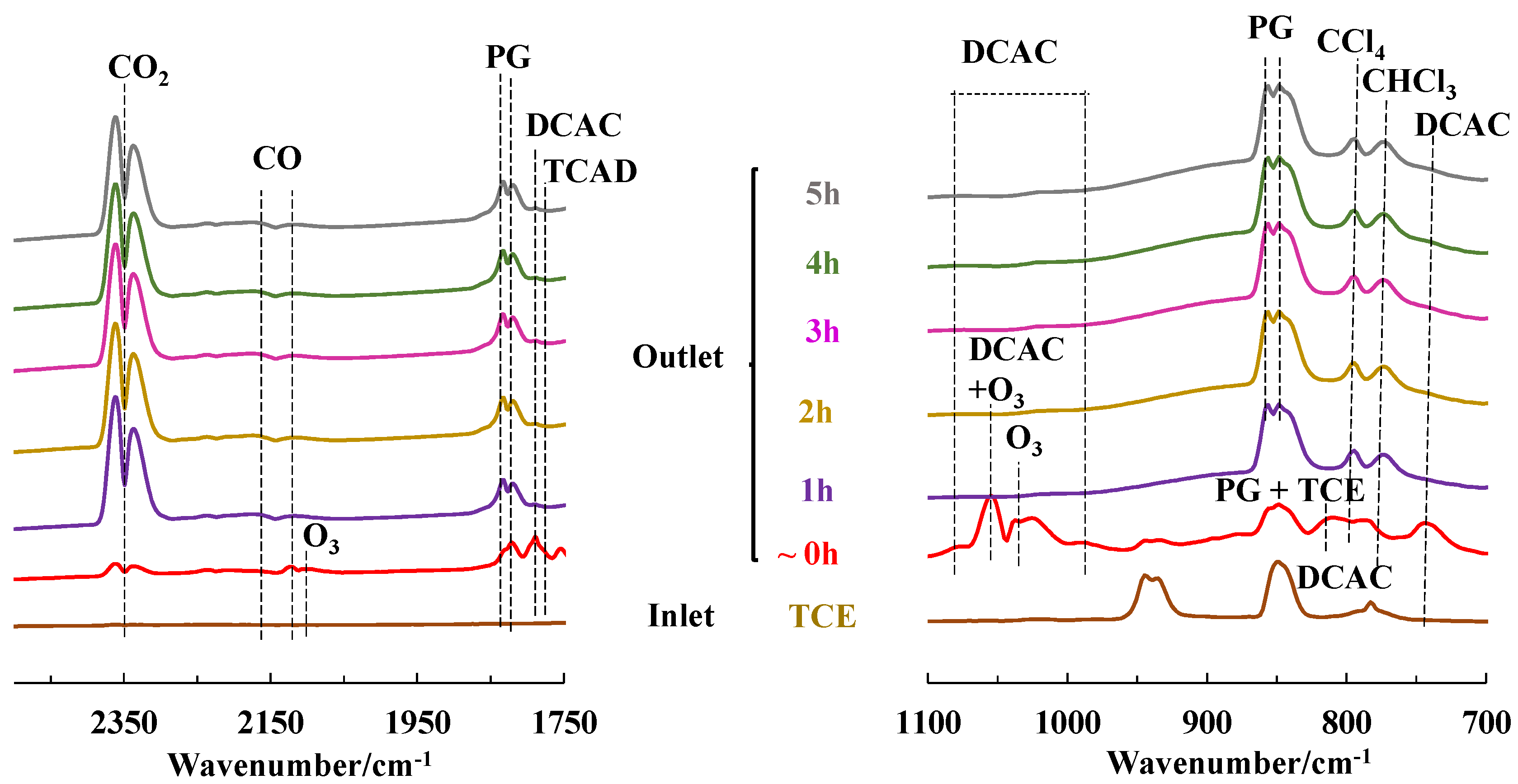
2.4. TCE Abatement by Non-Thermal Plasma (NTP)
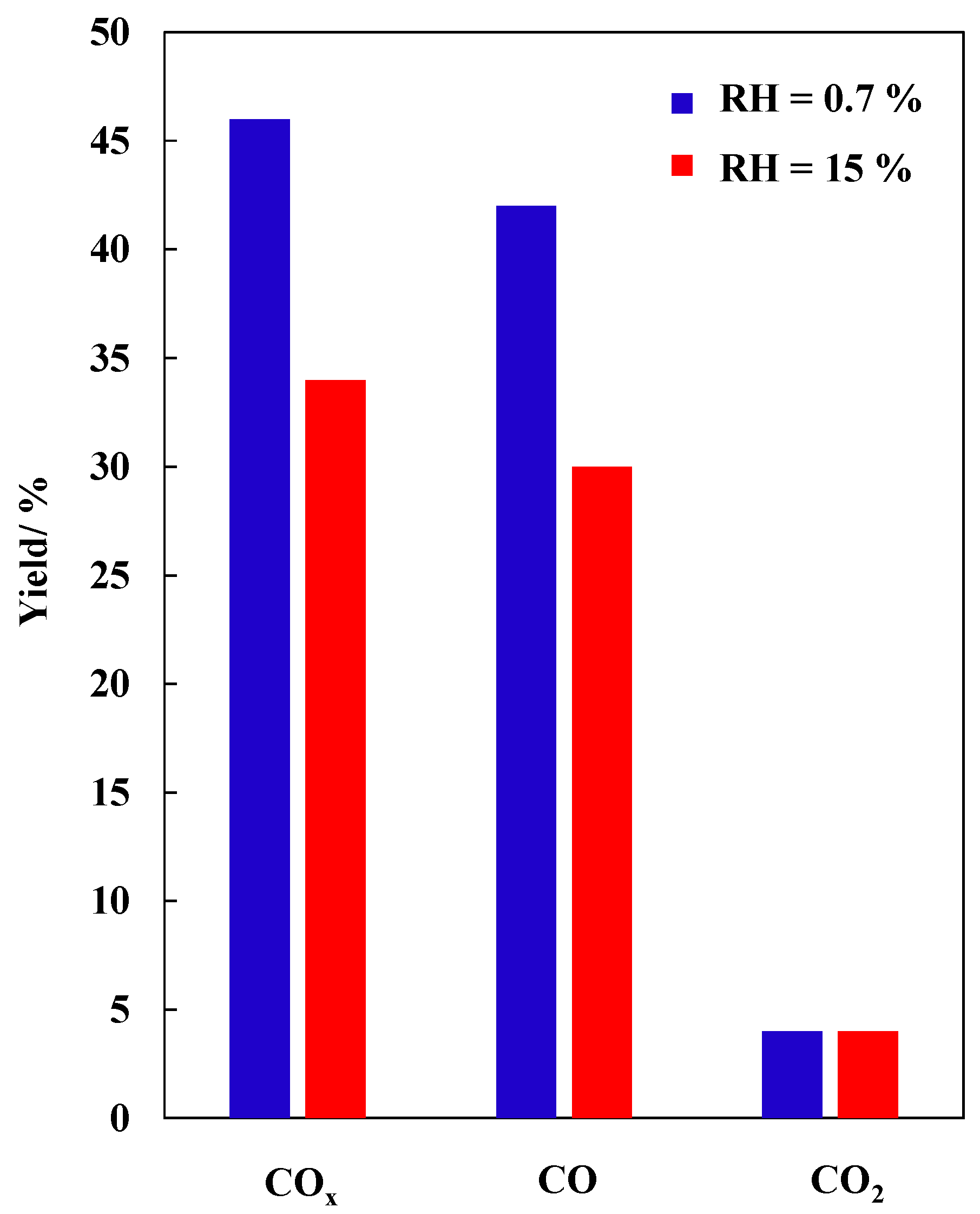
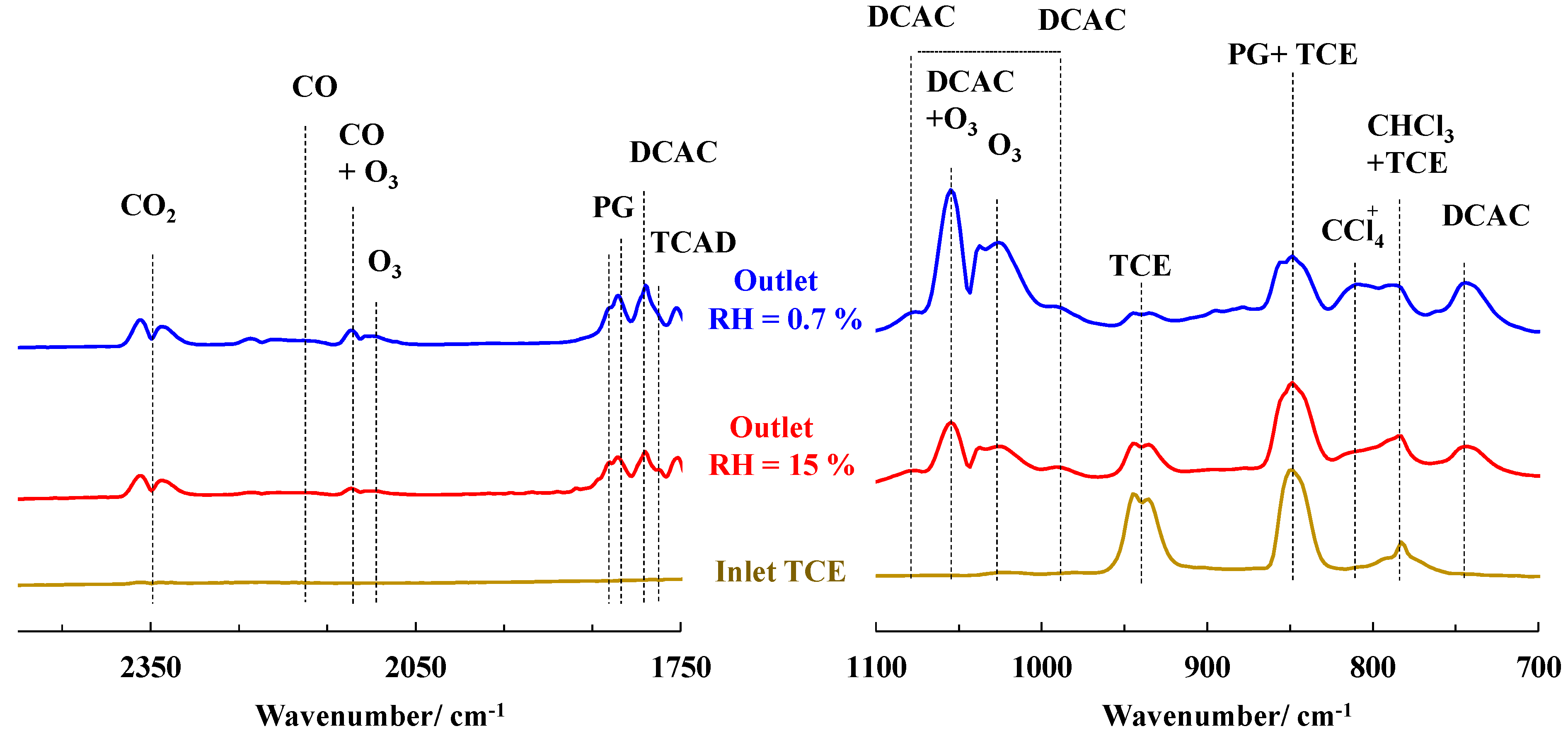
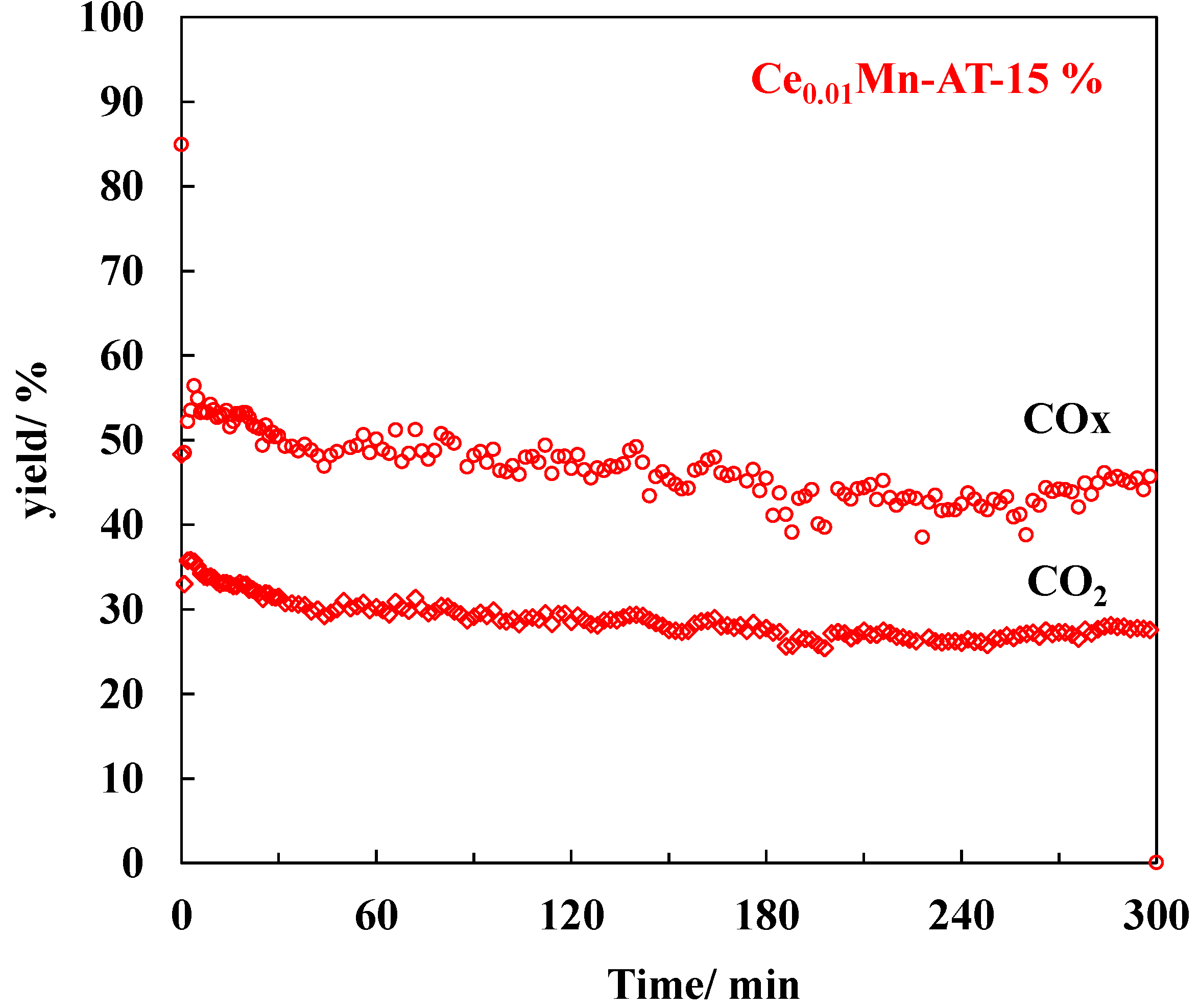
2.5. TCE Abatement in Post-Plasma Catalysis (PPC) Configuration—RH = 15%
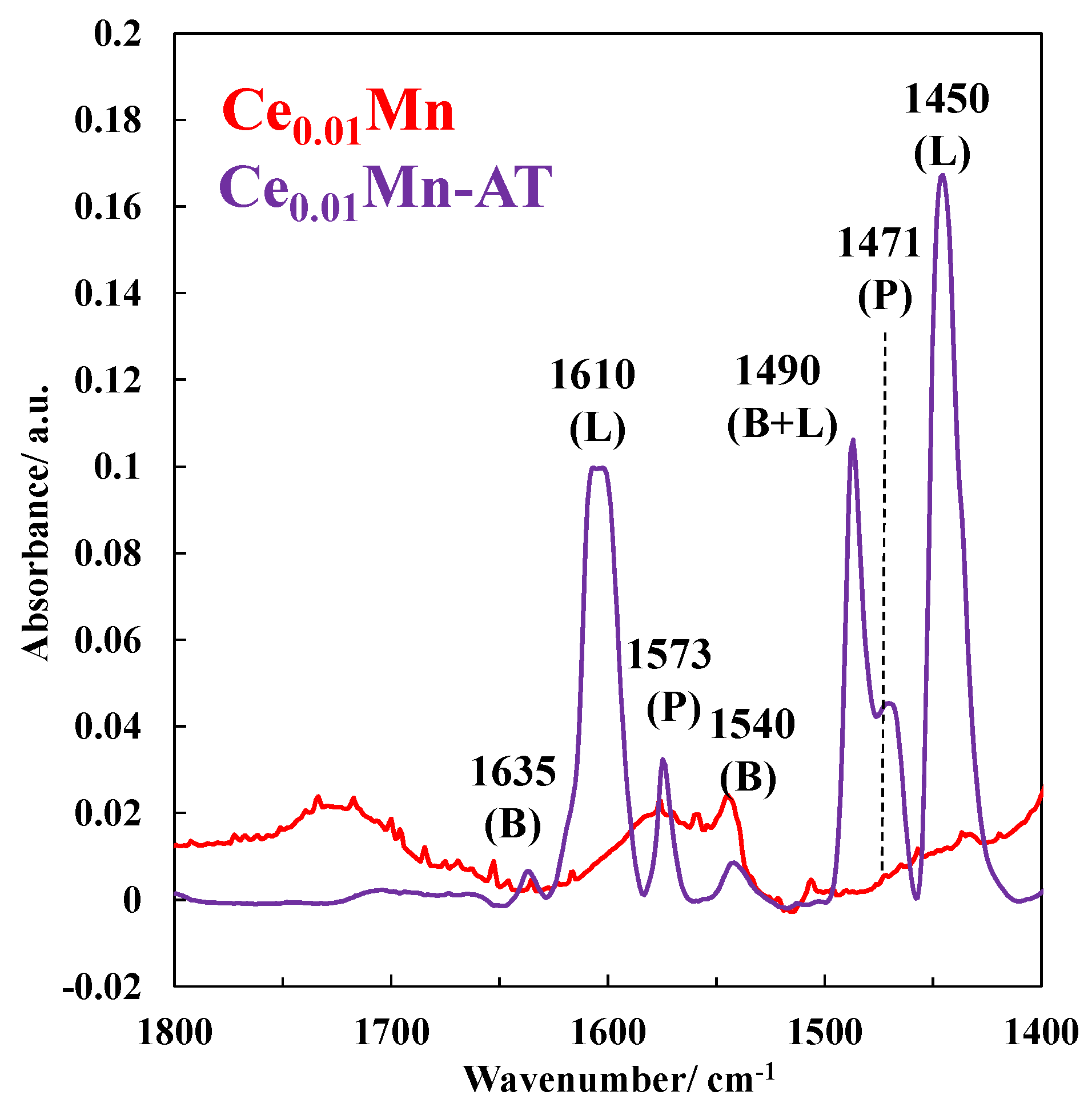
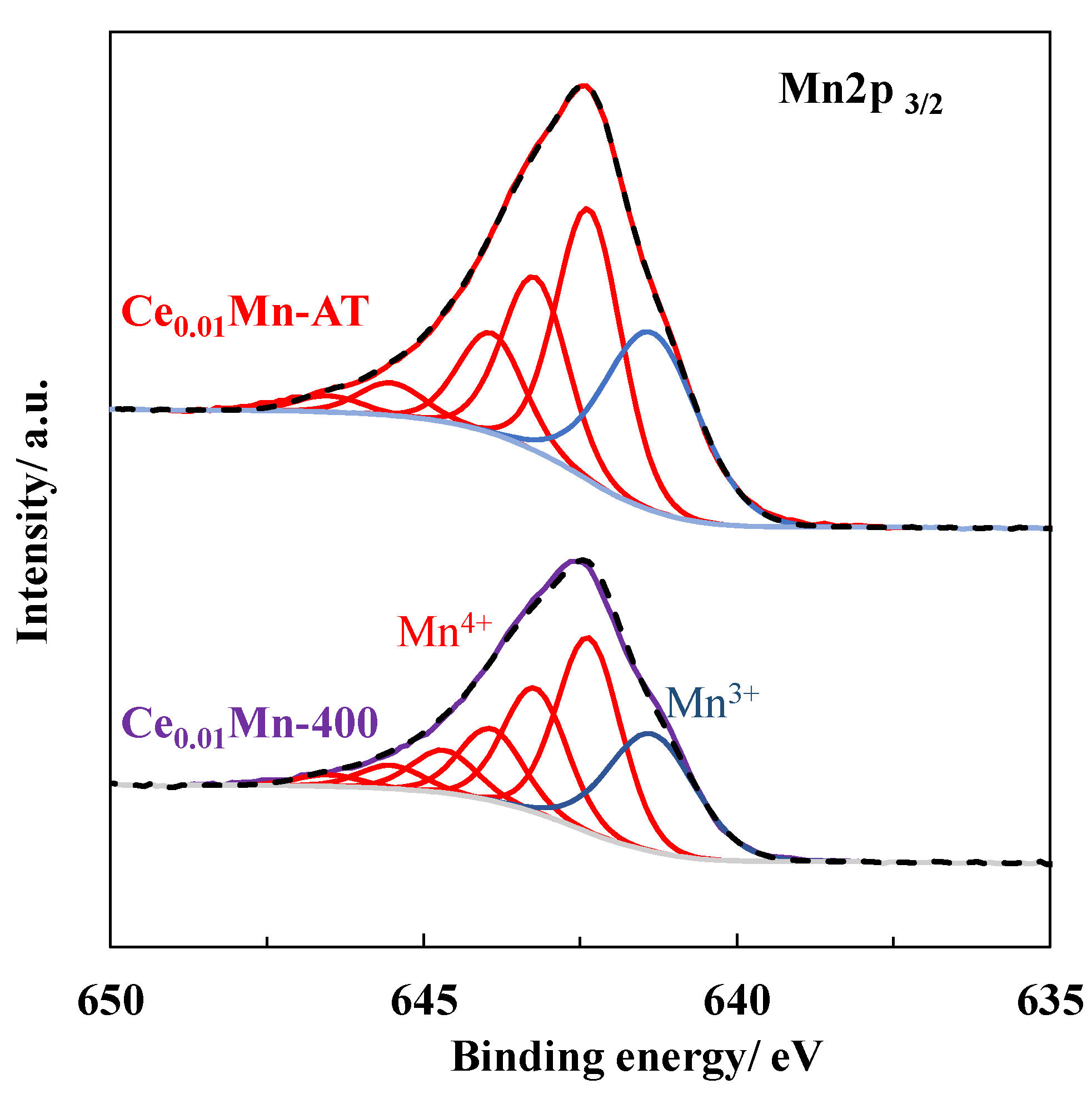
2.6. XPS Characterization of the Used Catalysts after the PPC Process
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Ozone Decomposition Using Non-Thermal Plasma (NTP)
3.4. Post-Plasma Catalysis for TCE Abatement
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated Volatile Organic Compounds (Cl-VOCs) in Environment-Sources, Potential Human Health Impacts, and Current Remediation Technologies. Environ. Inter. 2014, 71, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Schaum, J. Exposure Assessment of Trichloroethylene. Environ. Health Perspect. 2000, 108, 359–363. [Google Scholar] [CrossRef] [Green Version]
- Doherty, R.E. A History of the Production and Use of Carbon Tetrachloride, Tetrachloroethylene, Trichloroethylene and 1,1,1-Trichloroethane in the United States: Part 1--Historical Background; Carbon Tetrachloride and Tetrachloroethylene. Environ. Forensics 2000, 1, 69–81. [Google Scholar] [CrossRef]
- Rusyn, I.; Chiu, W.A.; Lash, L.H.; Kromhout, H.; Hansen, J.; Guyton, K.Z. Trichloroethylene: Mechanistic, Epidemiologic and Other Supporting Evidence of Carcinogenic Hazard. Pharmacol. Ther. 2014, 141, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, F.; Kopinke, F.D.; Roland, U. Influence of Ferroelectric Materials and Catalysts on the Performance of Non-Thermal Plasma (NTP) for the Removal of Air Pollutants. Plasma Chem. Plasma Process. 2005, 25, 595–611. [Google Scholar] [CrossRef]
- Schiavon, M.; Torretta, V.; Casazza, A.; Ragazzi, M. Non-Thermal Plasma as an Innovative Option for the Abatement of Volatile Organic Compounds: A Review. Water Air Soil Pollut. 2017, 228, 388. [Google Scholar] [CrossRef]
- Thevenet, F.; Sivachandiran, L.; Guaitella, O.; Barakat, C.; Rousseau, A. Plasma–Catalyst Coupling for Volatile Organic Compound Removal and Indoor Air Treatment: A Review. J. Phys. D Appl. Phys. 2014, 47, 224011. [Google Scholar] [CrossRef]
- Chen, H.L.; Lee, H.M.; Chen, S.H.; Chang, M.B.; Yu, S.J.; Li, S.N.I. Removal of Volatile Organic Compounds by Single-Stage and Two-Stage Plasma Catalysis Systems: A Review of the Performance Enhancement Mechanisms, Current Status, and Suitable Applications. Environ. Sci. Technol. 2009, 43, 2216–2227. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Renken, A.; Kiwi-Minsker, L. Catalytic Non-Thermal Plasma Reactor for Abatement of Toluene. Chem. Eng. J. 2010, 160, 677–682. [Google Scholar] [CrossRef]
- Wu, J.; Xia, Q.; Wang, H.; Li, Z. Catalytic Performance of Plasma Catalysis System with Nickel Oxide Catalysts on Different Supports for Toluene Removal: Effect of Water Vapor. App. Catal. B Environ. 2014, 156–157, 265–272. [Google Scholar] [CrossRef]
- Tu, X.; Gallon, H.J.; Whitehead, J.C. Electrical and Spectroscopic Diagnostics of a Single-Stage Plasma-Catalysis System: Effect of Packing with TiO2. J. Phys. D Appl. Phys. 2011, 44, 482003. [Google Scholar] [CrossRef]
- Sultana, S.; Ye, Z.; Veerapandian, S.K.P.; Löfberg, A.; De Geyter, N.; Morent, R.; Giraudon, J.M.; Lamonier, J.F. Synthesis and Catalytic Performances of K-OMS-2, Fe/K-OMS-2 and Fe-K-OMS-2 in Post Plasma-Catalysis for Dilute TCE Abatement. Catal. Today 2018, 307, 20–28. [Google Scholar] [CrossRef]
- Veerapandian, S.K.P.; Ye, Z.; Giraudon, J.-M.; De Geyter, N.; Morent, R.; Lamonier, J.-F. Plasma Assisted Cu-Mn Mixed Oxide Catalysts for Trichloroethylene Abatement in Moist Air. J. Hazar. Mater. 2019, 379, 120781. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P. Catalytic Decomposition of Gaseous Ozone over Todorokite-Type Manganese Dioxides at Room Temperature: Effects of Cerium Modification. Appl. Catal. A Gen. 2017, 530, 102–110. [Google Scholar] [CrossRef]
- Ma, J.; Wang, C.; He, H. Transition Metal Doped Cryptomelane-Type Manganese Oxide Catalysts for Ozone Decomposition. Appl. Catal. B Environ. 2017, 201, 503–510. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; Yang, L.; He, G.; Zhang, C.; Zhang, R.; He, H. Oxygen Vacancies Induced by Transition Metal Doping in γ-MnO2 for Highly Efficient Ozone Decomposition. Environ. Sci. Technol. 2018, 52, 12685–12696. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, X.; Zhang, C.; Ma, Q.; He, H. Novel CeMnaOx Catalyst for Highly Efficient Catalytic Decomposition of Ozone. Appl. Catal. B Environ. 2020, 264, 118498. [Google Scholar] [CrossRef]
- Tang, X.; Li, Y.; Huang, X.; Xu, Y.; Zhu, H.; Wang, J.; Shen, W. MnOx–CeO2 Mixed Oxide Catalysts for Complete Oxidation of Formaldehyde: Effect of Preparation Method and Calcination Temperature. Appl. Catal. B Environ. 2006, 62, 265–273. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Cao, R.; Sun, T.; Zong, W.; Zhan, J.; Liu, L. Different Behaviors of Birnessite-Type MnO2 Modified by Ce and Mo for Removing Carcinogenic Airborne Benzene. Mater. Chem. Phys. 2019, 221, 457–466. [Google Scholar] [CrossRef]
- Chen, J.; Chen, X.; Yan, D.; Jiang, M.; Xu, W.; Yu, H.; Jia, H. A Facile Strategy of Enhancing Interaction between Cerium and Manganese Oxides for Catalytic Removal of Gaseous Organic Contaminants. Appl. Catal. B Environ. 2019, 250, 396–407. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Rong, S.; Wang, H.; Zhang, P. Cerium Modified Birnessite-Type MnO2 for Gaseous Formaldehyde Oxidation at Low Temperature. Appl. Catal. B Environ. 2017, 211, 212–221. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, W.; Zhang, P.; Zhang, J. Nitric Acid-Treated Birnessite-Type MnO2: An Efficient and Hydrophobic Material for Humid Ozone Decomposition. Appl. Surf. Sci. 2018, 442, 640–649. [Google Scholar] [CrossRef]
- Gopi, T.; Swetha, G.; Chandra Shekar, S.; Ramakrishna, C.; Saini, B.; Krishna, R.; Rao, P.V.L. Catalytic Decomposition of Ozone on Nanostructured Potassium and Proton Containing α-MnO2 catalysts. Catal. Comm. 2017, 92, 51–55. [Google Scholar] [CrossRef]
- Lin, F.; Zhang, Z.; Li, N.; Yan, B.; He, C.; Hao, Z.; Chen, G. How to Achieve Complete Elimination of Cl-VOCs: A Critical Review on Byproducts Formation and Inhibition Strategies during Catalytic Oxidation. Chem. Eng. J. 2021, 404, 126534. [Google Scholar] [CrossRef]
- Abedi, K.; Ghorbani-Shahna, F.; Jaleh, B.; Bahrami, A.; Yarahmadi, R.; Haddadi, R.; Gandomi, M. Decomposition of Chlorinated Volatile Organic Compounds (CVOCs) Using NTP Coupled with TiO2/GAC, ZnO/GAC, and TiO2–ZnO/GAC in a Plasma-Assisted Catalysis System. J. Electrost. 2015, 73, 80–88. [Google Scholar] [CrossRef]
- Drits, V.A.; Silvester, E.; Gorshkov, A.I.; Manceau, A. Structure of Synthetic Monoclinic Na-Rich Birnessite and Hexagonal Birnessite; I, Results from X-Ray Diffraction and Selected-Area Electron Diffraction. Am. Mineral. 1997, 82, 946–961. [Google Scholar] [CrossRef]
- Gaillot, A.-C. Caractérisation Structurale de La Birnessite: Influence Du Protocole de Synthèse. Ph.D. Thesis, Université Joseph-Fourier-Grenoble I, Grenoble, France, 2002. [Google Scholar]
- Villalobos, M.; Lanson, B.; Manceau, A.; Toner, B.; Sposito, G. Structural Model for the Biogenic Mn Oxide Produced by Pseudomonas Putida. Am. Mineral. 2006, 91, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, P.; Cao, R. Porous Manganese Oxides Synthesized with Natural Products at Room Temperature: A Superior Humidity-Tolerant Catalyst for Ozone Decomposition. Catal. Sci. Technol. 2020, 10, 2254–2267. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, P.; Liu, Y.; Zheng, X. Ammonium-Treated Birnessite-Type MnO2 to Increase Oxygen Vacancies and Surface Acidity for Stably Decomposing Ozone in Humid Condition. Appl. Surf. Sci. 2019, 495, 143607. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Cao, R.; Liu, X.; Zhang, P.; Zhan, J.; Liu, L. Facile and Green Synthetic Strategy of Birnessite-Type MnO2 with High Efficiency for Airborne Benzene Removal at Low Temperatures. Appl. Catal. B Environ. 2019, 245, 569–582. [Google Scholar] [CrossRef]
- De Rivas, B.; López-Fonseca, R.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I. On the Mechanism of the Catalytic Destruction of 1,2-Dichloroethane over Ce/Zr Mixed Oxide Catalysts. J. Mol. Catal. A Chem. 2007, 278, 181–188. [Google Scholar] [CrossRef]
- Eren, E.; Gumus, H.; Eren, B.; Sarihan, A. Surface Acidity of H-Birnessite: Infrared Spectroscopic Study of Formic Acid Decomposition. Spectrosc. Lett. 2013, 46, 60–66. [Google Scholar] [CrossRef]
- Tang, X.; Li, J.; Sun, L.; Hao, J. Origination of N2O from NO Reduction by NH3 over β-MnO2 and α-Mn2O3. Appl. Catal. B Environ. 2010, 99, 156–162. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, B.; Liu, H.; He, B.; Ye, F.; Yu, L.; Sun, C.; Wen, H. The Effect of Acid/Alkali Treatment on the Catalytic Combustion Activity of Manganese Oxide Octahedral Molecular Sieves. RSC Adv. 2017, 7, 3958–3965. [Google Scholar] [CrossRef] [Green Version]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Galakhov, V.R.; Demeter, M.; Bartkowski, S.; Neumann, M.; Ovechkina, N.A.; Kurmaev, E.Z.; Lobachevskaya, N.I.; Mukovskii, Y.M.; Mitchell, J.; Ederer, D.L. Mn 3 s Exchange Splitting in Mixed-Valence Manganites. Phys. Rev. B 2002, 65, 113102. [Google Scholar] [CrossRef]
- Santos, V.P.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. Catalytic Oxidation of Ethyl Acetate over a Cesium Modified Cryptomelane Catalyst. Appl. Catal. B Environ. 2009, 88, 550–556. [Google Scholar] [CrossRef]
- Dhandapani, B.; Oyama, S.T. Gas Phase Ozone Decomposition Catalysts. Appl. Catal. B Environ. 1997, 11, 129–166. [Google Scholar] [CrossRef]
- Li, W.; Oyama, S.T. Mechanism of Ozone Decomposition on a Manganese Oxide Catalyst. 2. Steady-State and Transient Kinetic Studies. J. Am. Chem. Soc 1998, 120, 9047–9052. [Google Scholar] [CrossRef]
- Li, W.; Gibbs, G.V.; Oyama, S.T.; April, R.V. Mechanism of Ozone Decomposition on a Manganese Oxide Catalyst. 1. In Situ Raman Spectroscopy and Ab Initio Molecular Orbital Calculations. J. Am. Chem. Soc. 1998, 120, 9041–9046. [Google Scholar] [CrossRef]
- Zhu, G.; Zhu, J.; Jiang, W.; Zhang, Z.; Wang, J.; Zhu, Y.; Zhang, Q. Surface Oxygen Vacancy Induced A-MnO2 nanofiber for Highly Efficient Ozone Elimination. Appl. Catal. B Environ. 2017, 209, 729–737. [Google Scholar] [CrossRef]
- Gopi, T.; Swetha, G.; Chandra Shekar, S.; Ramakrishna, C.; Saini, B.; Krishna, R.; Rao, P.V.L. Catalytic Decomposition of Gaseous Ozone over Manganese Dioxides with Different Crystal Structures. Catal. Comm. 2016, 189, 210–218. [Google Scholar]
- Li, X.; Ma, J.; He, H. Recent Advances in Catalytic Decomposition of Ozone. J. Environ. Sci. 2020, 94, 14–31. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Dinh, M.T.N.; Giraudon, J.M.; Morent, R.; De Geyter, N.; Lamonier, J.F.; Leys, C. Qualitative By-Product Identification of Plasma-Assisted TCE Abatement by Mass Spectrometry and Fourier-Transform Infrared Spectroscopy. Plasma Chem. Plasma Process. 2011, 31, 707–718. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Decomposition of Trichloroethylene with Plasma-Catalysis: A Review. J. Adv. Oxid. Technol. 2011, 14, 165–175. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Aerts, R.; Van Gaens, W.; De Geyter, N.; Leys, C.; Morent, R.; Bogaerts, A. Modeling and Experimental Study of Trichloroethylene Abatement with a Negative Direct Current Corona Discharge. Plasma Chem. Plasma Process. 2015, 35, 217–230. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Nguyen Dinh, M.T.; Nuns, N.; Giraudon, J.-M.; De Geyter, N.; Leys, C.; Lamonier, J.-F.; Morent, R. Combination of Non-Thermal Plasma and Pd/LaMnO3 for Dilute Trichloroethylene Abatement. Chem. Eng. J. 2016, 283, 668–675. [Google Scholar] [CrossRef]
- Peyrous, R. The Effect of Relative Humidity on Ozone Production by Corona Discharge in Oxygen or Air-A Numerical Simulation-Part II: Air. Ozone Sci. Eng. 1990, 12, 41–64. [Google Scholar] [CrossRef]
- Chen, J.; Wang, P. Effect of Relative Humidity on Electron Distribution and Ozone Production by DC Coronas in Air. IEEE trans. Plasma Sci. 2005, 33, 808–812. [Google Scholar] [CrossRef]
- Dinh, M.T.N.; Giraudon, J.-M.; Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Lamonier, J.-F. Post Plasma-Catalysis for Total Oxidation of Trichloroethylene over Ce–Mn Based Oxides Synthesized by a Modified “Redox-Precipitation Route”. App. Catal. B Environ. 2015, 172, 65–72. [Google Scholar] [CrossRef]
- Dinh, M.T.N.; Giraudon, J.M.; Lamonier, J.F.; Vandenbroucke, A.; De Geyter, N.; Leys, C.; Morent, R. Plasma-Catalysis of Low TCE Concentration in Air Using LaMnO3+δ as Catalyst. Appl. Catal. B Environ. 2014, 147, 904–911. [Google Scholar] [CrossRef]
- Vandenbroucke, A. Abatement of Volatile Organic Compounds by Combined Use of Non-Thermal Plasma and Heterogeneous catalysis. Ph.D. Thesis, University of Ghent, Ghent, Belgium, 2015. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
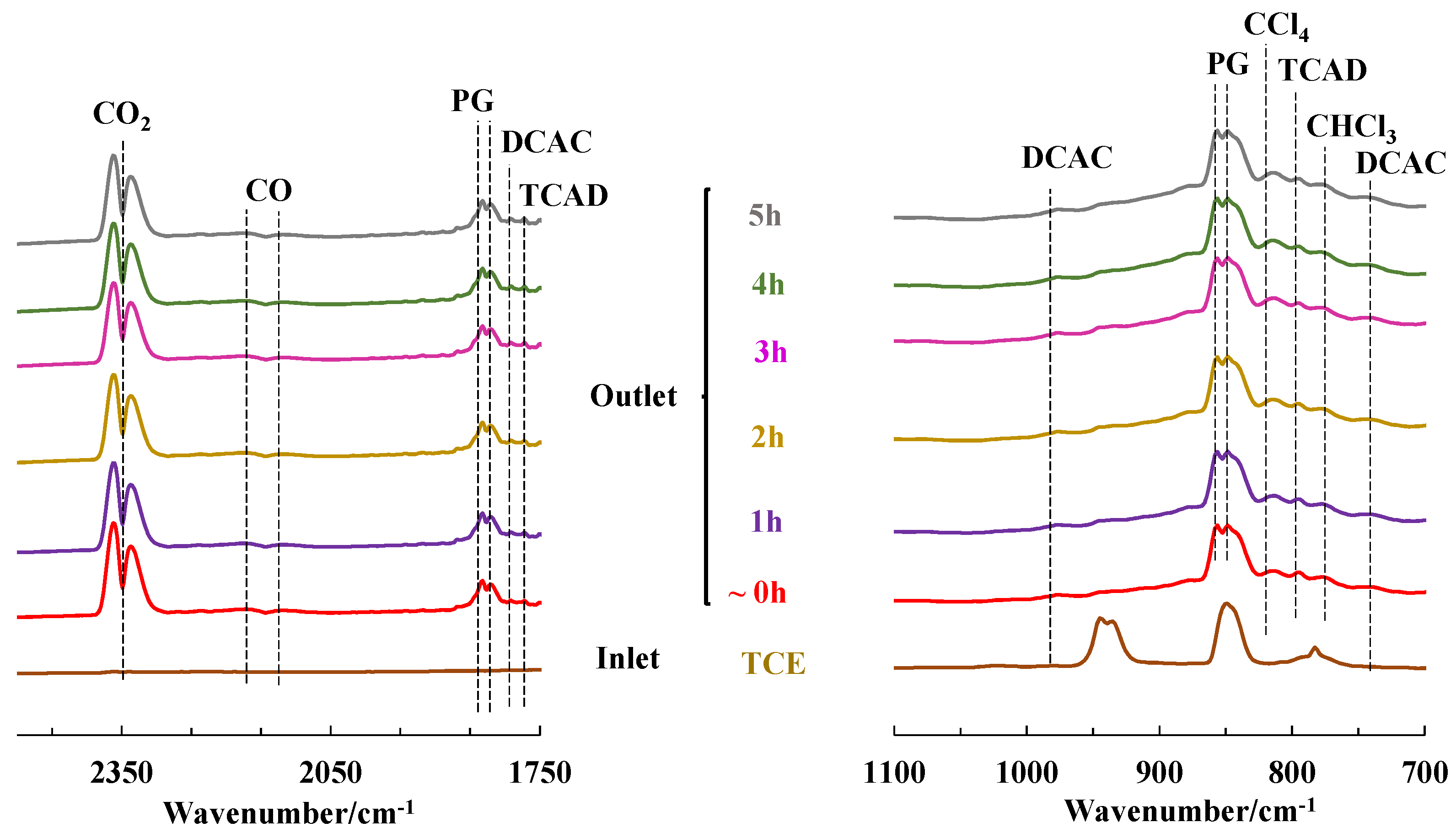{kind=link}
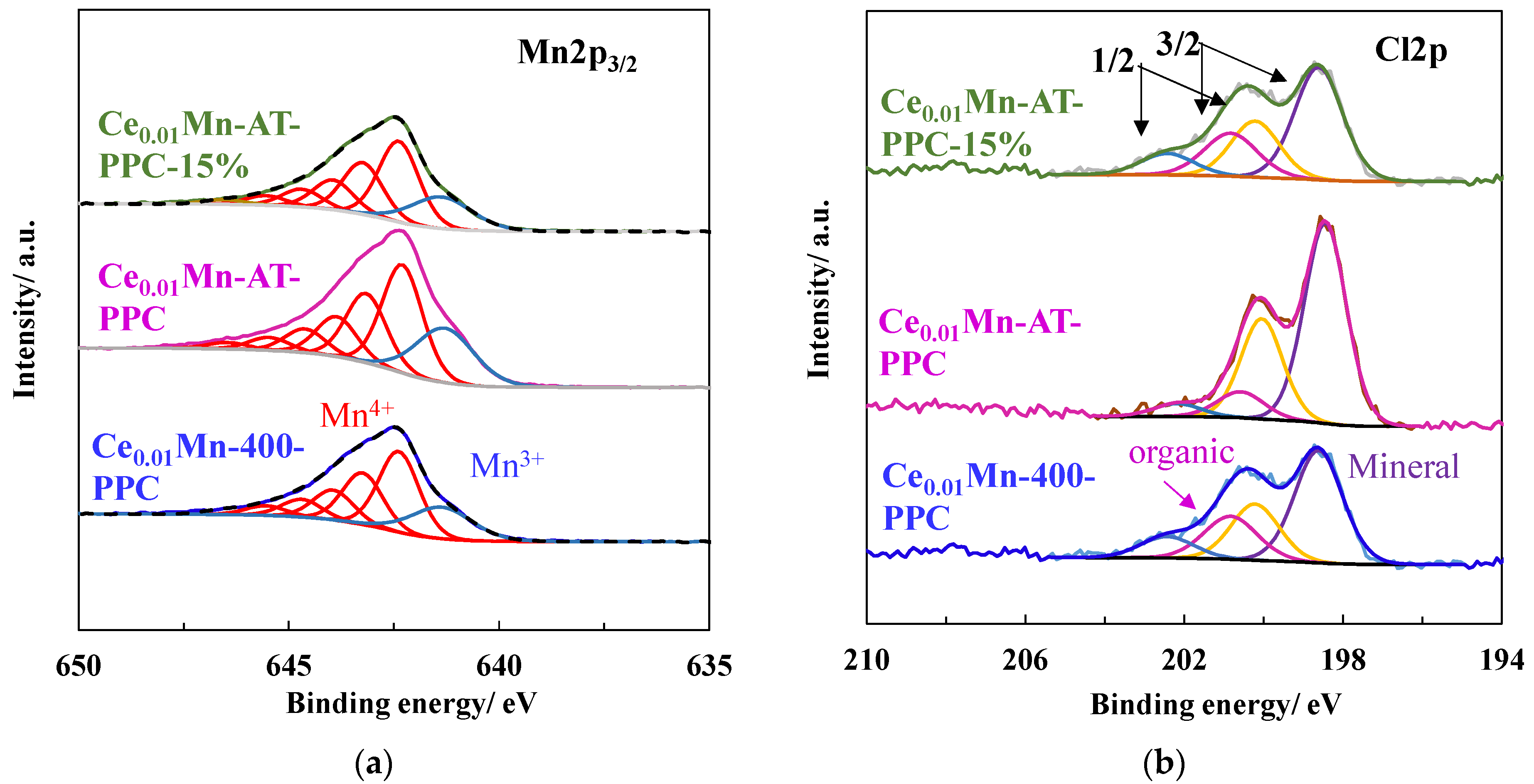{kind=link}
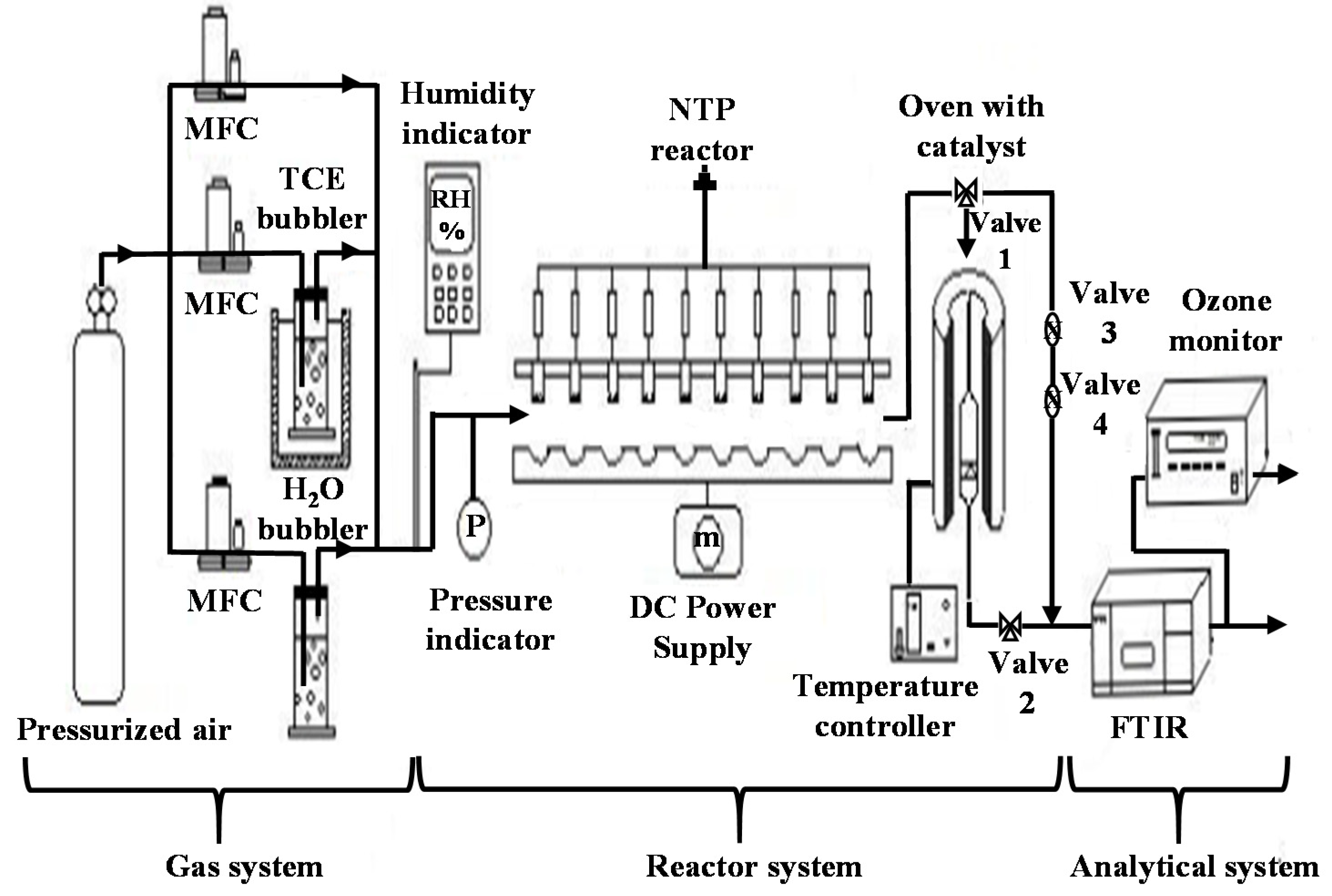{kind=link}
| Sample | Weight/% | Atomic Ratio | |||||
|---|---|---|---|---|---|---|---|
| K | Na | Ce | Mn | K/Mn | Na/Mn | Ce/Mn | |
| Ce0.01Mn-400 | 7.10 | 1.99 | 1.18 | 49.20 | 0.340 | 0.057 | 0.009 |
| Ce0.01Mn-AT | 1.30 | 0.01 | 1.06 | 52.01 | 0.059 | 0.004 | 0.008 |
| Sample | SBET/m2/g | Dp max/nm | Vp tot/cm3/g |
|---|---|---|---|
| Ce0.01Mn-400 | 132 | 4.0 | 0.22 |
| Ce0.01Mn-AT | 385 | 5.3 | 0.37 |
| Catalyst | O/Mn | K/Mn | Na/Mn | Ce/Mn | Cl/Mn | Mn3+/Mn4+ | Mn AOS from Mn 3s Splitting |
|---|---|---|---|---|---|---|---|
| Ce0.01Mn-400 | 2.07 | 0.18 | 0.05 | 0.015 | 0.34 | 3.44 | |
| Ce0.01Mn-400-PPC | 2.34 | 0.26 | 0.05 | 0.024 | 0.18 | 0.21 | 3.56 |
| Ce0.01Mn-AT | 1.97 | 0.06 | - | 0.018 | 0.38 | 3.32 | |
| Ce0.01Mn-AT-PPC | 1.91 | 0.04 | - | 0.028 | 0.16 | 0.29 | 3.42 |
| Ce0.01Mn-AT-PPC-15% | 1.81 | 0.04 | - | 0.025 | 0.22 | 0.28 | 3.41 |
| Air source | Air liquid, Alphagaz 1 |
| Flow rate | 1 L/min |
| Relative humidity (RH) | 0.7%/15% |
| Energy density | 150 J/L |
| Initial TCE concentration | 150 ppmv |
| Catalyst | Ce0.01Mn-400 and Ce0.01Mn-AT 0.1 g of catalyst mixed with 1 g of SiC |
| Activation conditions | 200 mL/min, 100 °C for 1 h |
| Catalyst temperature | 100 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdallah, G.; Giraudon, J.-M.; Bitar, R.; Geyter, N.D.; Morent, R.; Lamonier, J.-F. Post-Plasma Catalysis for Trichloroethylene Abatement with Ce-Doped Birnessite Downstream DC Corona Discharge Reactor. Catalysts 2021, 11, 946. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080946
Abdallah G, Giraudon J-M, Bitar R, Geyter ND, Morent R, Lamonier J-F. Post-Plasma Catalysis for Trichloroethylene Abatement with Ce-Doped Birnessite Downstream DC Corona Discharge Reactor. Catalysts. 2021; 11(8):946. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080946
Chicago/Turabian StyleAbdallah, Grêce, Jean-Marc Giraudon, Rim Bitar, Nathalie De Geyter, Rino Morent, and Jean-François Lamonier. 2021. "Post-Plasma Catalysis for Trichloroethylene Abatement with Ce-Doped Birnessite Downstream DC Corona Discharge Reactor" Catalysts 11, no. 8: 946. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080946