
Propylene Polymerization and Deactivation Processes with Isoselective {Cp/Flu} Zirconocene Catalysts

 , and
, and
Abstract
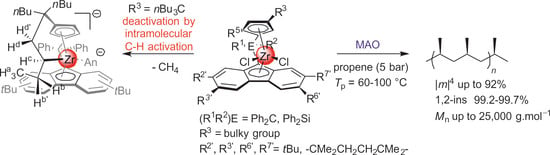
:
1. Introduction
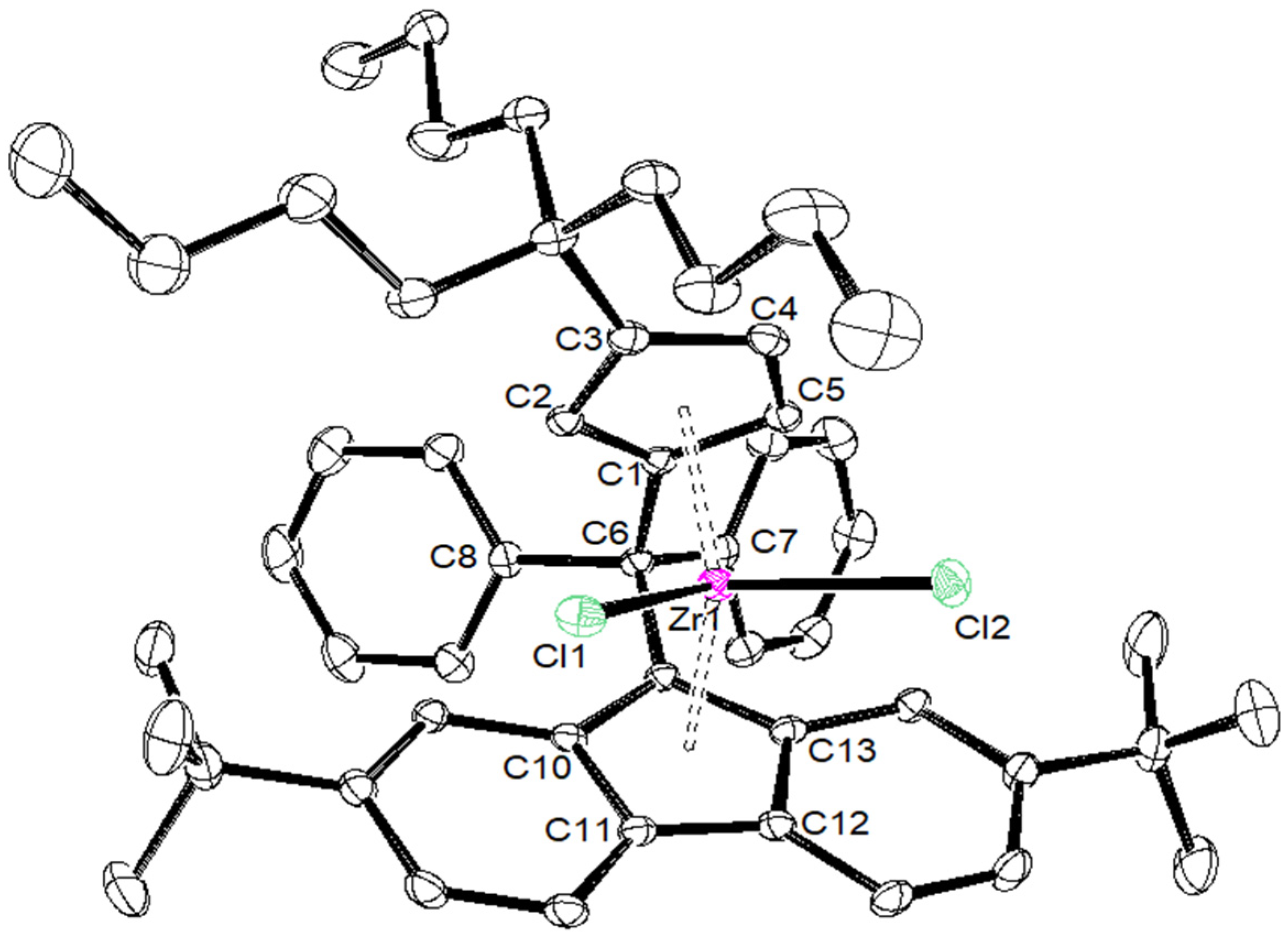
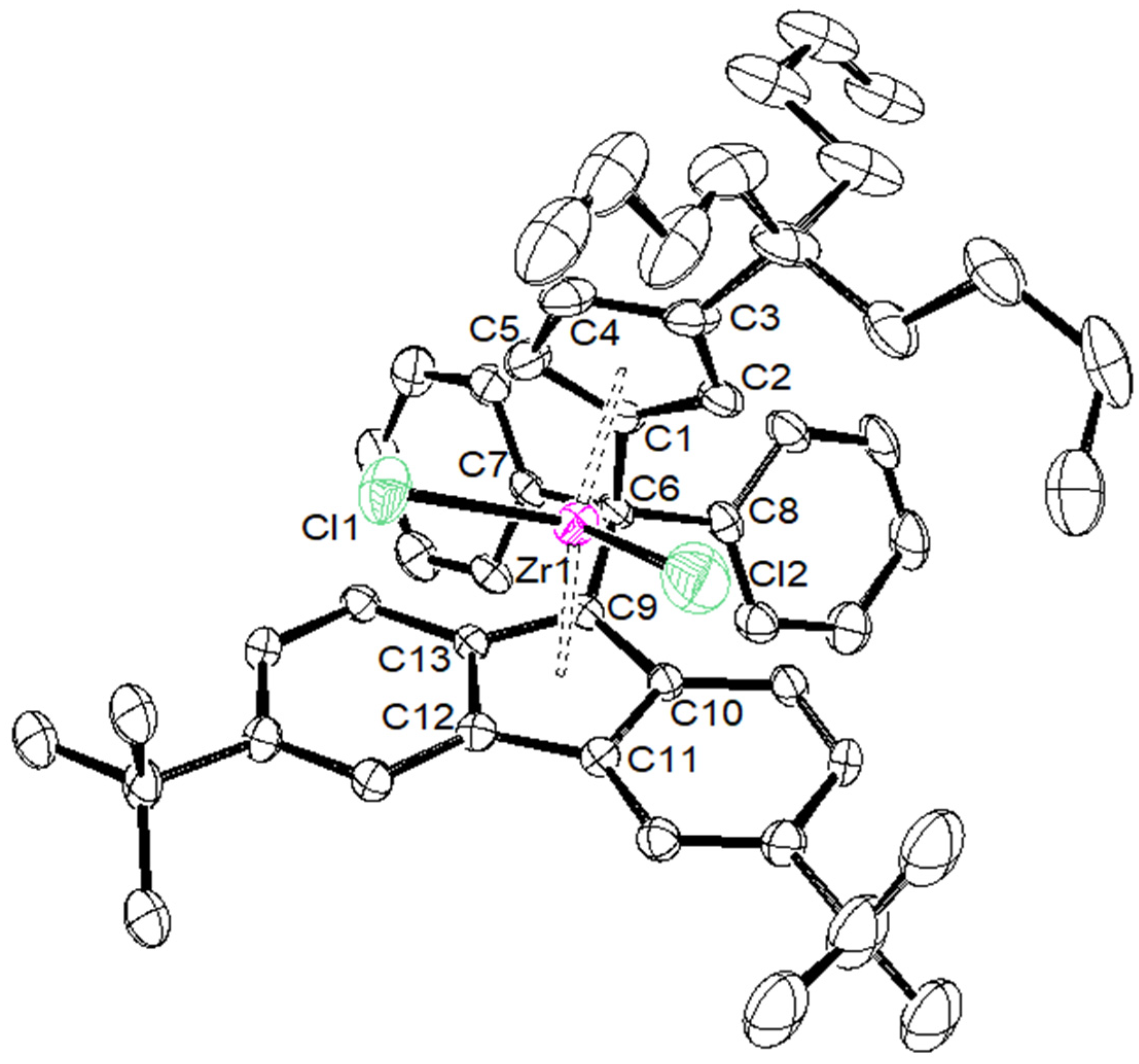
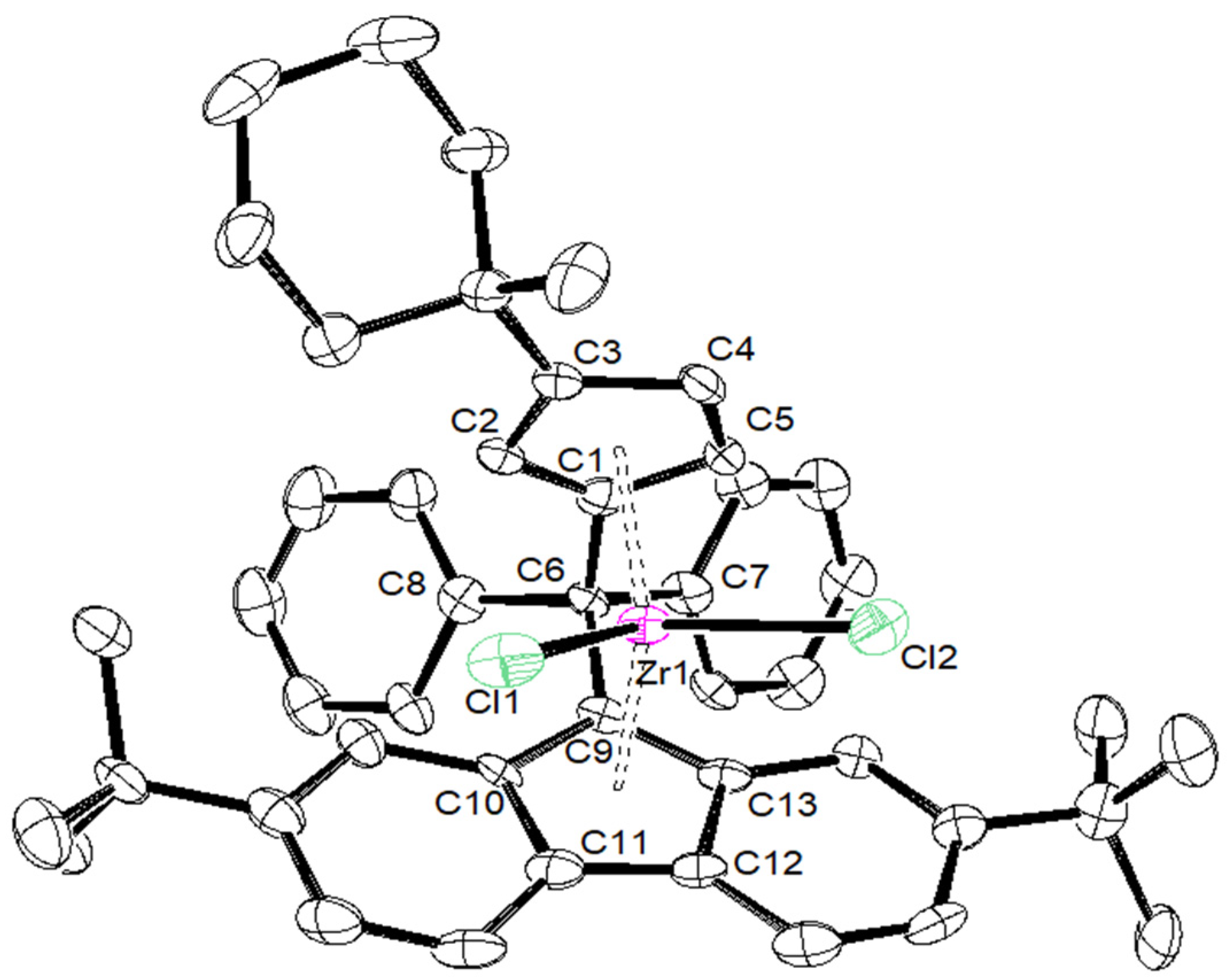
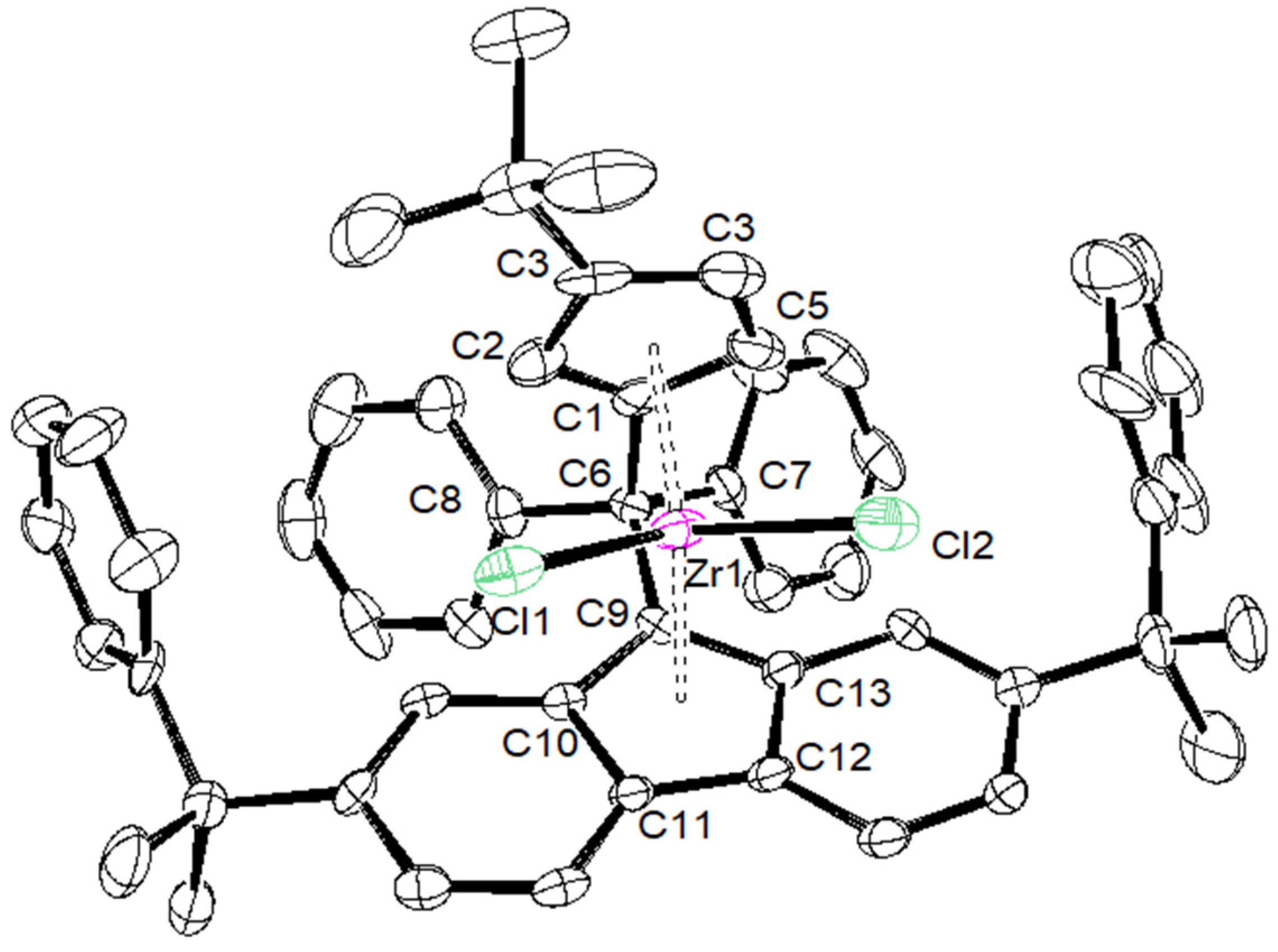
2. Results and Discussion
- -
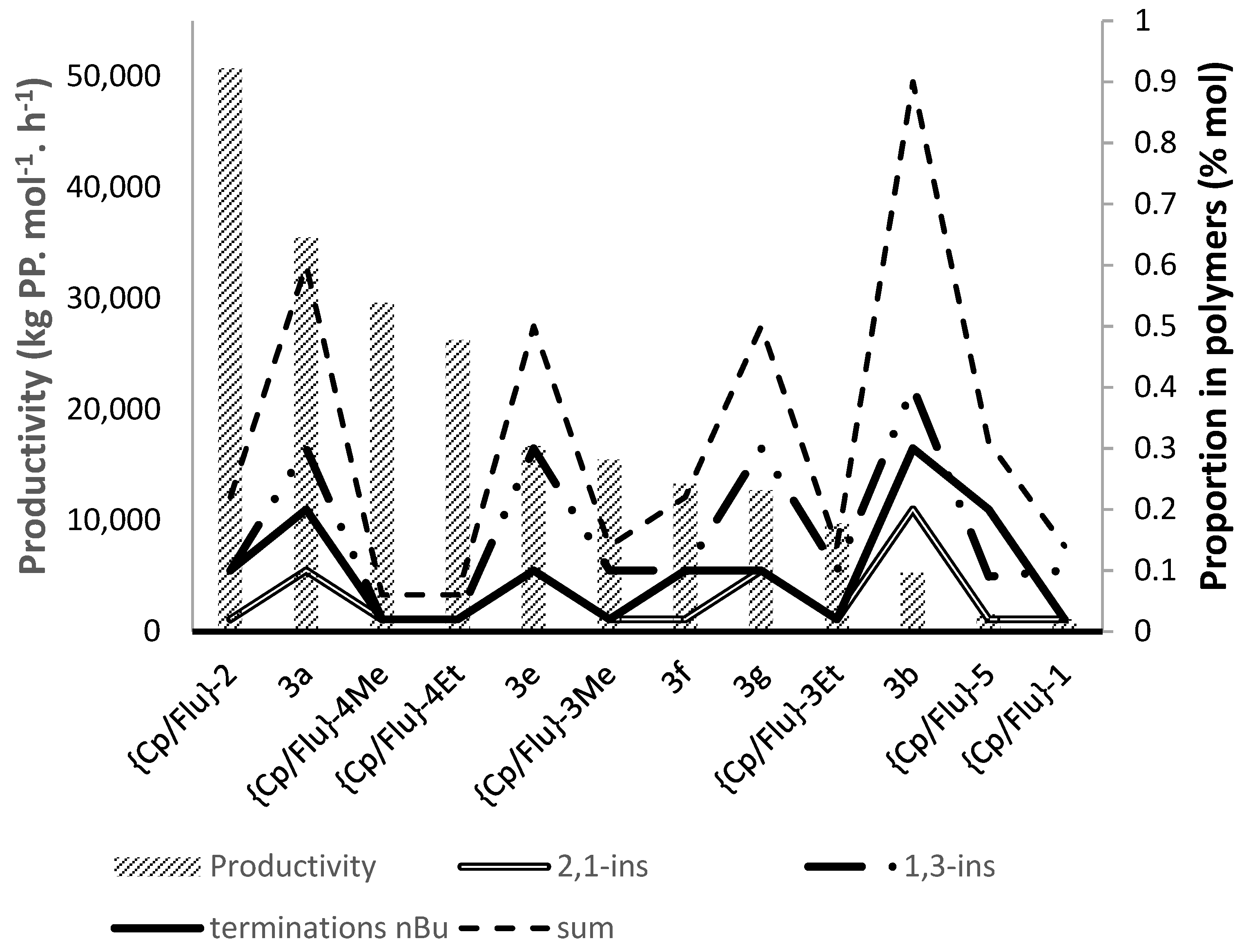
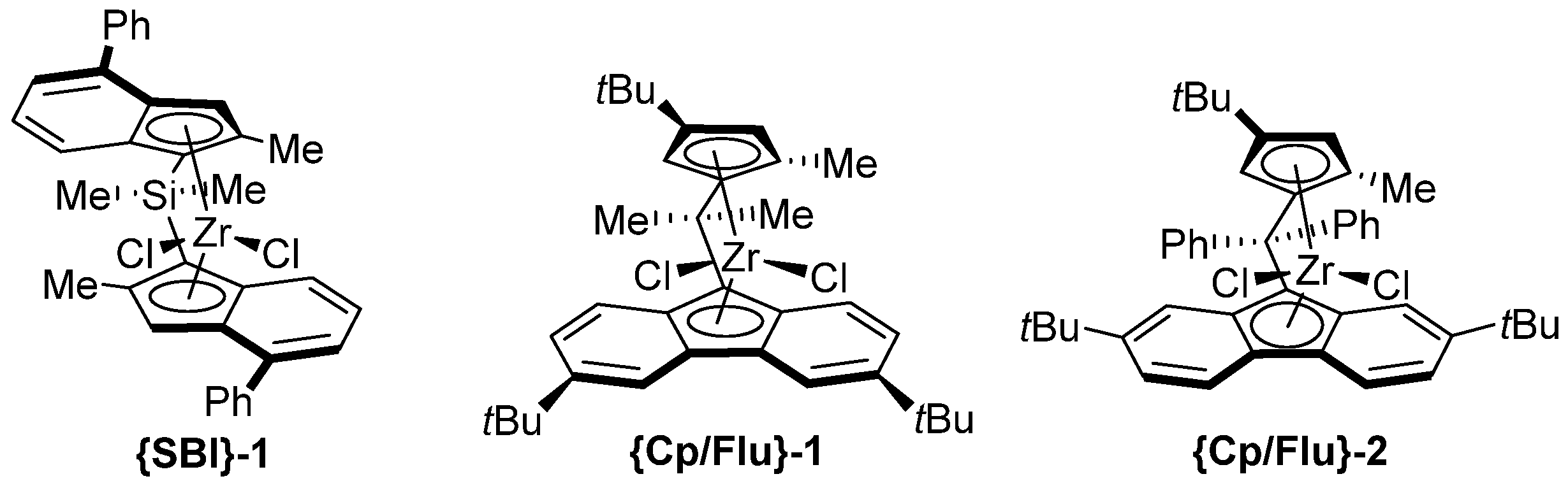
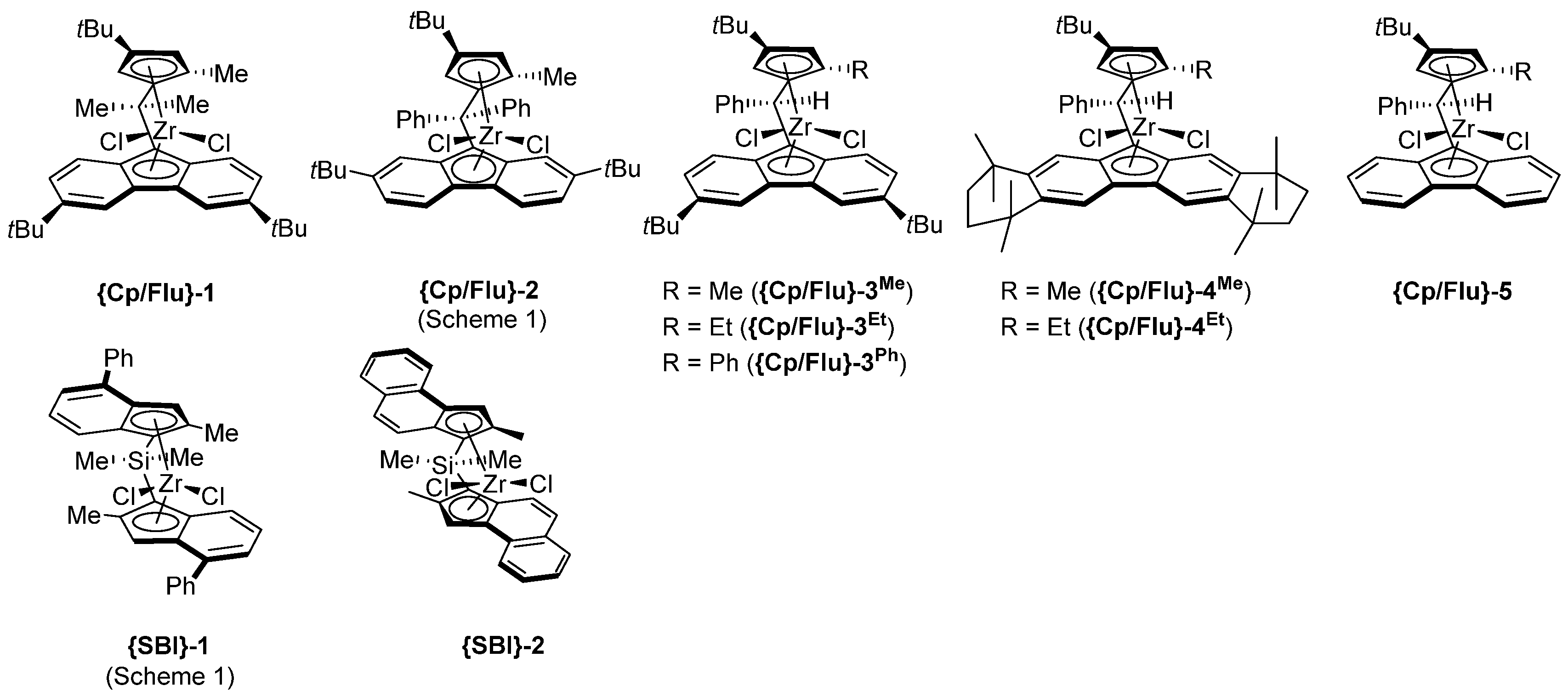
- The most active precatalyst within the given series of propylene polymerization precatalysts appeared to be the reference metallocene {Cp/Flu}-2. For instance, in a typical experiment performed at 60 °C (entry 1), the productivity of the latter catalyst was found to be 1.5–10 times higher (50,700 kg PP·mol−1·h−1) than those observed for all other precursors (up to 35,500 kg PP·mol−1·h−1).
- -
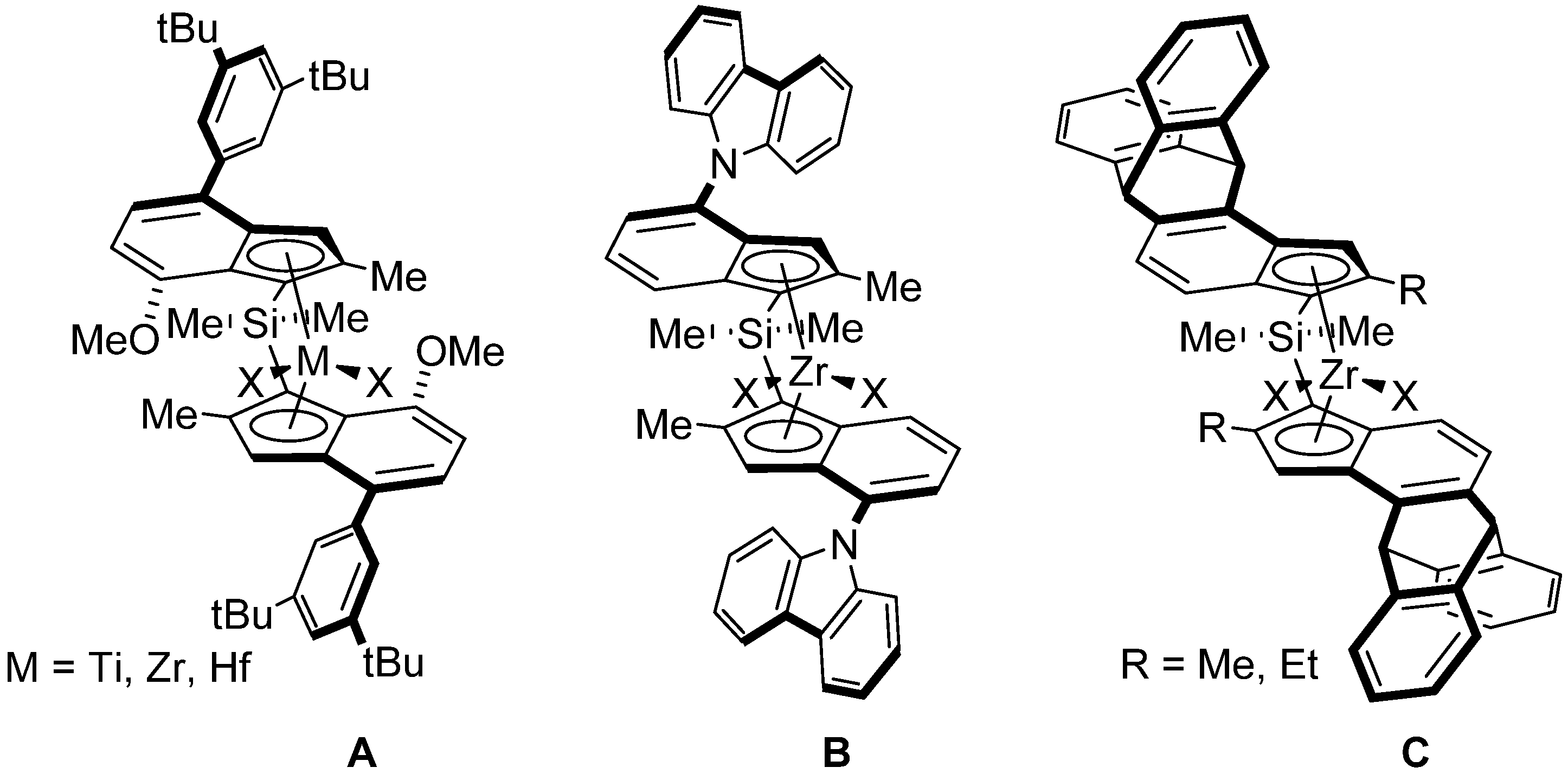
- The second by productivity (35,500 kg PP·mol−1·h−1) appeared to be the less sterically constrained system incorporating 3a, which is analogous to {Cp/Flu}-2 but has no Me substituent at the position 5 of the Cp ligand.
- -
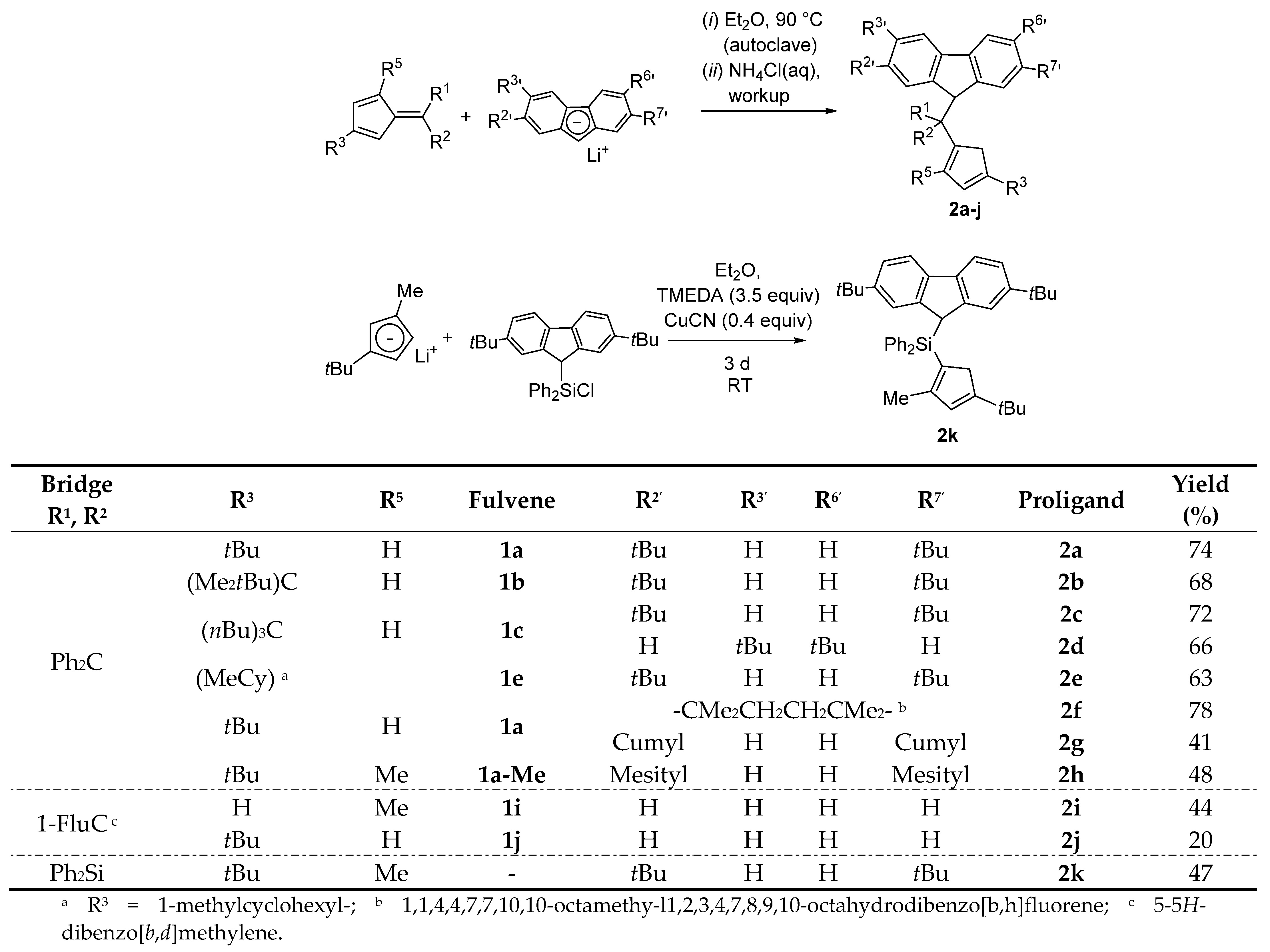
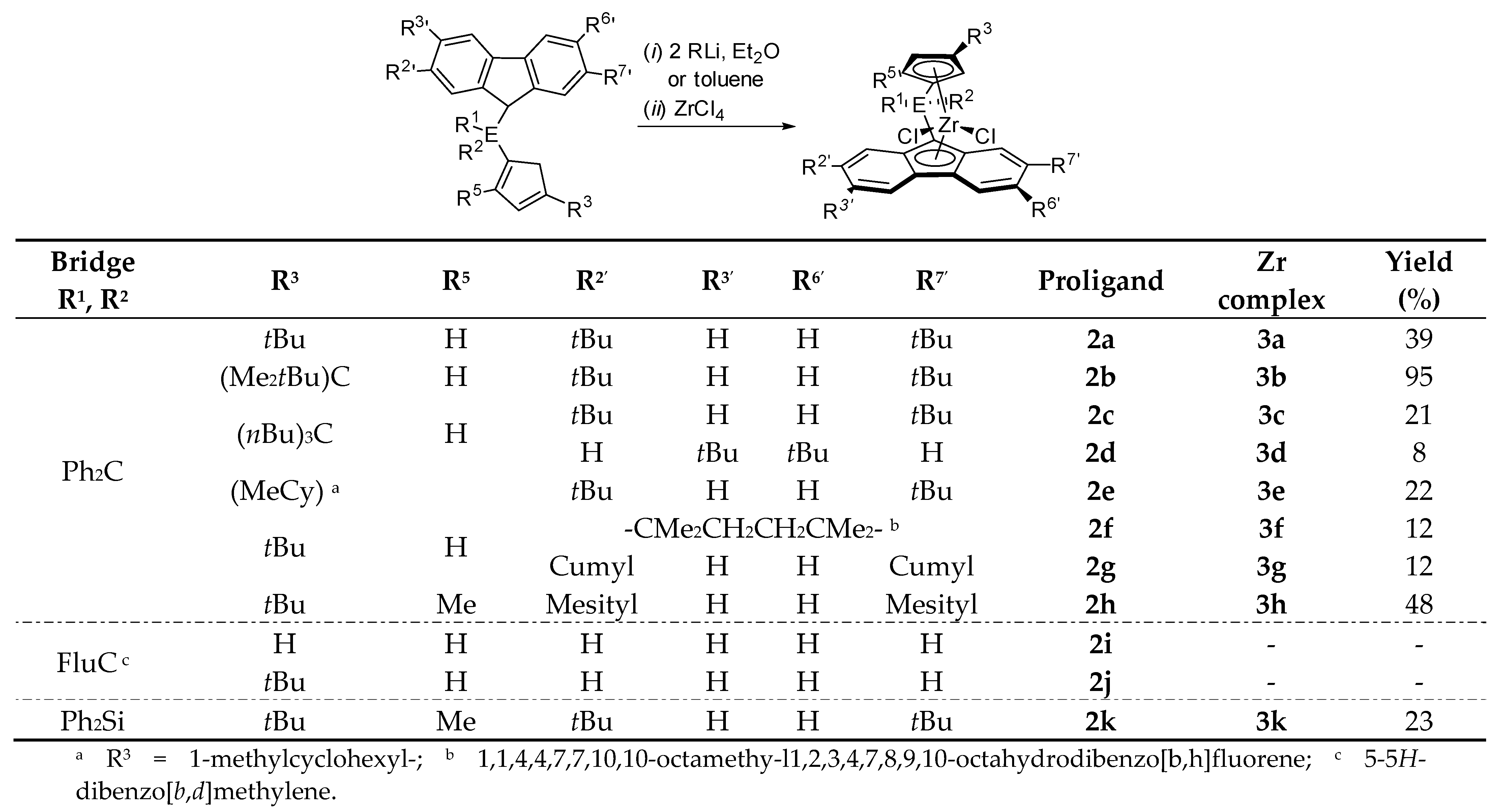
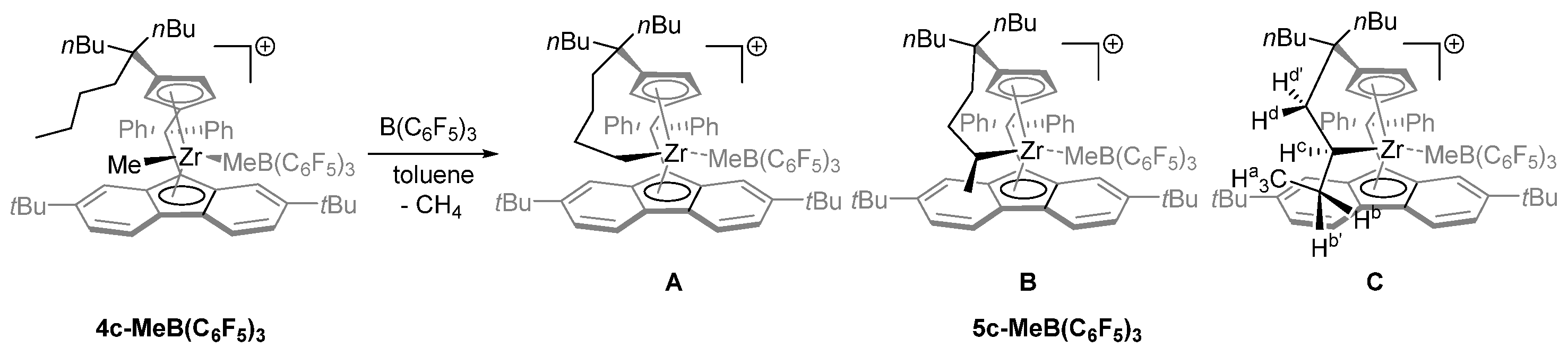
- Increasing the bulkiness of the 3-R substituent at the Cp ligand (R = tBuMe2C in 3b and R = 1-methylcyclohexyl in 3e) resulted in a significant drop in productivity (5300 kg PP·mol−1·h−1 and 16,700 kg PP·mol−1·h−1; entries 3 and 7, respectively). At the same time, metallocene systems 3c and 3d, both incorporating the bulky aliphatic 3-nBu3C substituent at the Cp ligand, were unexpectedly found to be very poorly or completely inactive (entries 4 and 5, respectively). The latter result can stem from deactivation involving a nBu group of the 3-nBu3C substituent (vide infra).
- -
- Keeping the 3-tBu substituent at the Cp ligand while altering the nature of the 2,7-/3,6- substituents also resulted in catalysts featuring inferior productivities. Thus, 3f–3h afforded comparable productivities in the range of 12,700–17,200 kg PP·mol−1·h−1 (entries 8–10).
- -
- Regarding the molecular weights of the PPs produced, all active systems yielded similar products, with Mn lying in the range of 11.4–24.9 kg·mol−1, showing no specific trend and being lower than that of the PP obtained with the reference metallocene catalyst {Cp/Flu}-2 (50.7 kg·mol−1). The polydispersity values (PDI = 2.1–2.5) were all consistent with the single-site behavior of these catalyst systems.
- -
- The stereoregularities of the PPs obtained from {Cp/Flu}-based catalysts can vary over a broad range, depending on the substituents both on the Cp and Flu moieties [10,11,18,19,20]. The new metallocenes with bulky 3-R substituents on the Cp ligand (3a,b,e) afforded polypropylenes featuring pentad [m]4 values (86.2–88.6%) higher than those observed with {Cp/Flu}-2 (82.1%). These stereoregularity levels are also reflected in the corresponding Tm values determined for these polymers (139.8–141.1 vs. 131.1, respectively). The highest stereoselectivity ([m]4 = 91.2%) within the series was achieved with metallocene catalyst 3f with the 2,3,6,7-tetra-substituted version of fluorenyl ligand (Oct), which afforded PP with Tm of 148.4.
- -
- Being the 2,7-Mes2-Flu-substituted analogue of {Cp/Flu}-2, system 3h quite unexpectedly afforded PP with a low isotacticity index ([m]4 = 54.0%; see the Supporting Information Table S4). This result cannot be explained by the higher bulkiness of the mesityl substituents.
- -
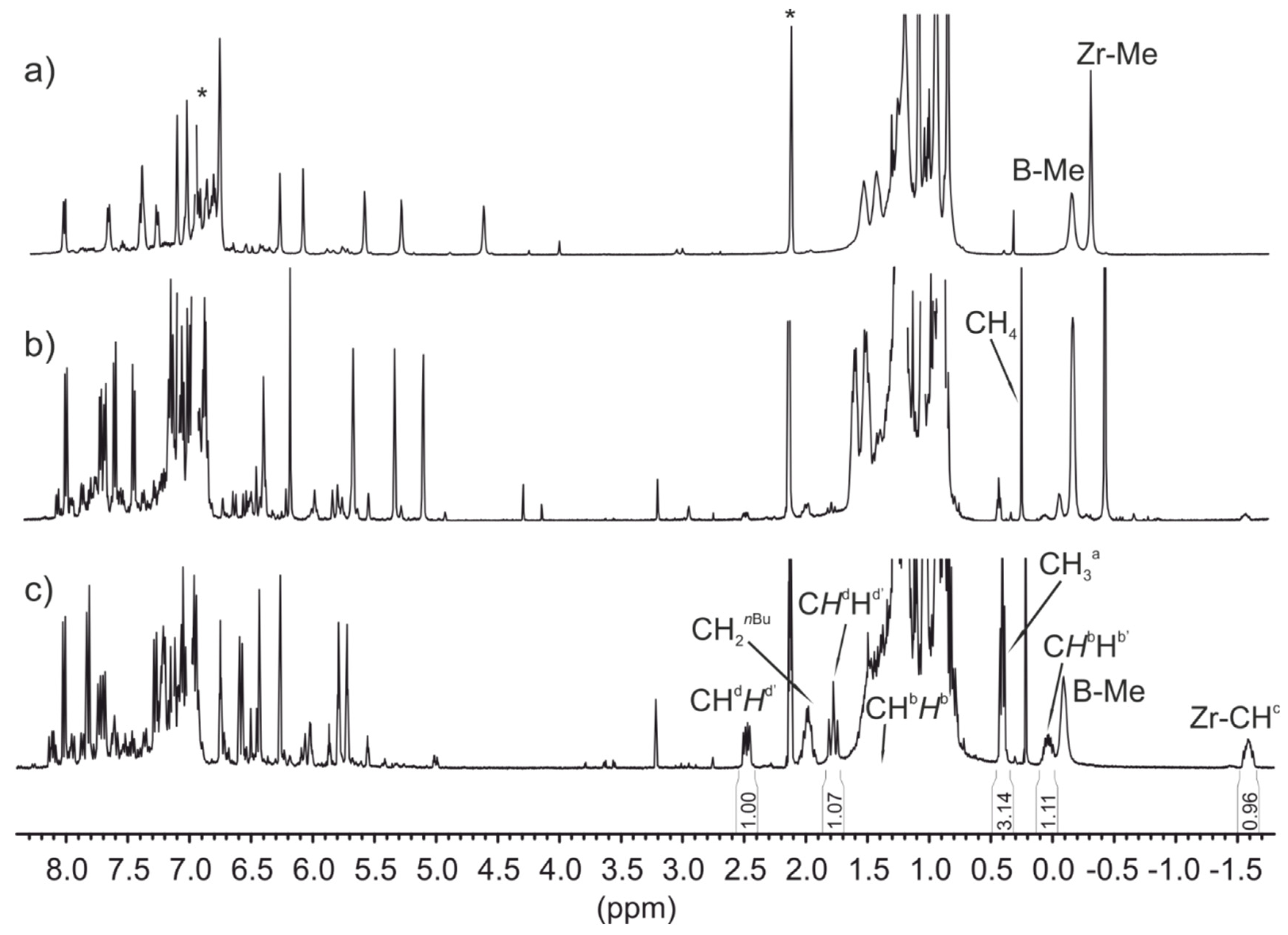
- The incorporation of a Si-bridge (entry 11) appeared to confer a negative effect on all parameters (productivity, molecular weight, stereoregularity (Table S4), and Tm), as evidenced upon comparing the results obtained with 3k and its Ph2C-bridged analogue {Cp/Flu}-2. In order to improve the productivity, a polymerization experiment was conducted at 100 °C (entry 12), but it resulted in an almost total loss of activity; this possibly reflects significant deactivation or even degradation of the catalyst at this temperature. The viscous material recovered from these attempts was found to be a mixture of oligomers exhibiting no melting transition. In a previous contribution, Chen and Rausch et al. [30] explained that the loss of syndioselectivity and poorer productivity of {Me2Si(Flu)(Cp)}ZrCl2 with respect to the Me2C-bridged congener were due to steric hindrance of metal center. Thus, the larger Cpcent-Si-Flucent bite angle, which is ca. 10° greater than that in the carbon-bridged counterpart, makes the coordination sphere both less accessible and less selective towards the coordination of monomer.
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Resconi, L.; Fritze, C. Polypropylene Handbook; Pasquini, N., Ed.; Hanser Publishers: Munich, Germany, 2005; pp. 107–147. [Google Scholar]
- Fink, G.; Brintzinger, H.H. Polymerization Reactions. In Metal-Catalysis in Industrial Organic Processes; Chiusoli, G.P., Maitlis, P.M., Eds.; Royal Society of Chemistry: Colchester, UK, 2006; pp. 218–254. [Google Scholar]
- Ewen, J.A.; Elder, M.J. Process and Catalyst for Producing Isotactic Polyolefins. Eurpean Patent EP0537130A1, 14 April 1993. [Google Scholar]
- Razavi, A.; Atwood, J.L. Preparation and crystal structures of the complexes (η5-C5H3Me-CMe2-η5-C13H8)MCl2 (M = Zr or Hf): Mechanistic aspects of the catalytic formation of a syndiotactic-isotactic stereoblock-type polypropylene. J. Organomet. Chem. 1995, 497, 105–111. [Google Scholar] [CrossRef]
- Resconi, L.; Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar] [CrossRef]
- Alt, H.G.; Köppl, A. Effect of the nature of metallocene complexes of group IV metals on their performance in catalytic ethylene and propylene polymerization. Chem. Rev. 2000, 100, 1205–1222. [Google Scholar] [CrossRef]
- Miller, S.A.; Bercaw, J.E. Isotactic−Hemiisotactic Polypropylene from C1-Symmetric ansa-Metallocene Catalysts: A New Strategy for the Synthesis of Elastomeric Polypropylene. Organometallics 2002, 21, 934–945. [Google Scholar] [CrossRef]
- Miller, S.A.; Bercaw, J.E. Highly Stereoregular Syndiotactic Polypropylene Formation with Metallocene Catalysts via Influence of Distal Ligand Substituents. Organometallics 2004, 23, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.A.; Bercaw, J.E. Mechanism of Isotactic Polypropylene Formation with C1-Symmetric Metallocene Catalysts. Organometallics 2006, 25, 3576–3592. [Google Scholar] [CrossRef]
- Castro, L.; Kirillov, E.; Miserque, O.; Welle, A.; Haspeslagh, L.; Carpentier, J.-F.; Maron, L. Are Solvent and Dispersion Effects Crucial in Olefin Polymerization DFT Calculations? Some Insights from Propylene Coordination and Insertion Reactions with Group 3 and 4 Metallocenes. ACS Catal. 2015, 5, 416–425. [Google Scholar] [CrossRef]
- Castro, L.; Theurkauff, G.; Vantomme, A.; Welle, A.; Haspeslagh, L.; Brusson, J.-M.; Maron, L.; Carpentier, J.-F.; Kirillov, E. A theoretical outlook on the stereoselectivity origins of isoselective zirconocene propylene polymerization catalysts. Chem. Eur. J. 2018, 24, 10784–10792. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, E.; Carpentier, J.-F. {Cyclopentadienyl/Fluorenyl}-Group 4 ansa-Metallocene Catalysts for Production of Tailor-Made Polyolefins. Chem. Rec. 2021, 21, 357–375. [Google Scholar] [CrossRef]
- Schoebel, A.; Herdtweck, E.; Parkinson, M.; Rieger, B. Ultra-Rigid Metallocenes for Highly Iso- and Regiospecific Polymerization of Propene: The Search for the Perfect Polypropylene Helix. Chem. Eur. J. 2012, 18, 4174–4178. [Google Scholar] [CrossRef]
- Tranchida, D.; Mileva, D.; Resconi, L.; Rieger, B.; Schoebel, A. Molecular and Thermal Characterization of a Nearly Perfect Isotactic Poly(propylene). Macromol. Chem. Phys. 2015, 216, 2171–2178. [Google Scholar] [CrossRef]
- Machat, M.R.; Lanzinger, D.; Poethig, A.; Rieger, B. Ultrarigid Indenyl-based Hafnocene Complexes for the Highly Isoselective Polymerization of Propene: Tunable Polymerization Performance Adopting Various Sterically Demanding 4-Aryl Substituents. Organometallics 2017, 36, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Machat, M.R.; Jandle, C.; Rieger, B. Titanocenes in Olefin Polymerization: Sustainable Catalyst System or an Extinct Species? Organometallics 2017, 36, 1408–1418. [Google Scholar] [CrossRef]
- Izmer, V.V.; Lebedev, A.Y.; Kononovich, D.S.; Borisov, I.S.; Kulyabin, P.S.; Goryunov, G.P.; Uborsky, D.V.; Canich, J.A.M.; Voskoboynikov, A.Z. Ansa-Metallocenes Bearing 4-(N-Azolyl)-2- methylindenyl and Related Ligands: Development of Highly Isoselective Catalysts for Propene Polymerization at Higher Temperatures. Organometallics 2019, 38, 4645–4657. [Google Scholar] [CrossRef]
- Kulyabin, P.S.; Goryunov, G.P.; Sharikov, M.I.; Izmer, V.V.; Vittoria, A.; Budzelaar, P.H.M.; Busico, V.; Voskoboynikov, A.Z.; Ehm, C.; Cipullo, R.; et al. ansa-Zirconocene Catalysts for Isotactic-Selective Propene Polymerization at High Temperature: A Long Story Finds a Happy Ending. J. Am. Chem. Soc. 2021, 143, 7641–7647. [Google Scholar] [CrossRef] [PubMed]
- Desert, X.; Proutiere, F.; Welle, A.; Den Dauw, K.; Vantomme, A.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Kirillov, E. Zirconocene-catalyzed polymerization of α-olefins: When intrinsic higher activity is flawed by rapid deactivation. Organometallics 2019, 38, 2664–2673. [Google Scholar] [CrossRef]
- Spaleck, W.; Kuber, F.; Winter, A.; Rohrmann, J.; Bachmann, B.; Antberg, M.; Dolle, V.; Paulus, E.F. The influence of aromatic substituents on the polymerization behavior of bridged zirconocene catalysts. Organometallics 1994, 13, 954–963. [Google Scholar] [CrossRef]
- Razavi, A.; Bellia, V.; Brauwer, Y.D.; Hortmann, K.; Peters, L.; Sirole, S.; Belle, S.V.; Thewalt, U. Syndiotactic-and Isotactic Specific Bridged Cyclopentadienyl-Fluorenyl Based Metallocenes; Structural Features, Catalytic Behavior. Macromol. Chem. Phys. 2004, 205, 347–356. [Google Scholar] [CrossRef]
- Razavi, A.; Bellia, V.; Baekelmans, D.; Slawinsky, M.; Sirol, S.; Peters, L.; Thewalt, U. Chain “stationary” insertion mechanism and production of isotactic polypropylene with C1-symmetric catalyst systems. Kinet. Catal. 2006, 47, 257–267. [Google Scholar] [CrossRef]
- Tomasi, S.; Razavi, A.; Ziegler, T. Stereoregularity, Regioselectivity, and Dormancy in Polymerizations Catalyzed by C1-Symmetric Fluorenyl-Based Metallocenes. A Theoretical Study Based on Density Functional Theory. Organometallics 2009, 28, 2609–2618. [Google Scholar] [CrossRef]
- Wondimagegn, T.; Wang, D.; Razavi, A.; Ziegler, T. In Silico Design of C1- and Cs-Symmetric Fluorenyl-Based Metallocene Catalysts for the Synthesis of High-Molecular-Weight Polymers from Ethylene/Propylene Copolymerization. Organometallics 2009, 28, 1383–1390. [Google Scholar] [CrossRef]
- Kirillov, E.; Marquet, N.; Razavi, A.; Belia, V.; Hampel, F.; Roisnel, T.; Gladysz, J.A.; Carpentier, J.-F. New C1-Symmetric Ph2C-Bridged Multisubstituted ansa-Zirconocenes for Highly Isospecific Propylene Polymerization: Synthetic Approach via Activated Fulvenes. Organometallics 2010, 29, 5073–5082. [Google Scholar] [CrossRef]
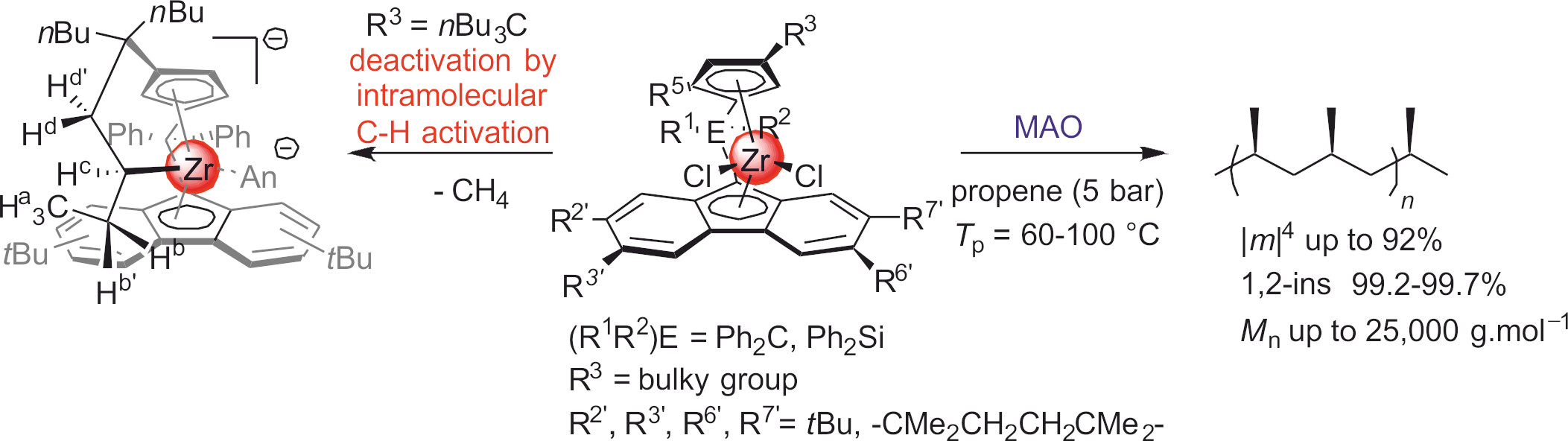
- Kirillov, E.; Marquet, N.; Bader, M.; Razavi, A.; Belia, V.; Hampel, F.; Roisnel, T.; Gladysz, J.A.; Carpentier, J.-F. Chiral-at-ansa-Bridged Group 4 Metallocene Complexes {(R1R2C)-(3,6-tBu2Flu)(3-R3-5-Me-C5H2)}MCl2: Synthesis, Structure, Stereochemistry, and Use in Highly Isoselective Propylene Polymerization. Organometallics 2011, 30, 263–272. [Google Scholar] [CrossRef]
- Bader, M.; Marquet, N.; Kirillov, E.; Roisnel, T.; Razavi, A.; Lhost, O.; Carpentier, J.-F. Old and New C1-Symmetric Group 4 Metallocenes {(R1R2C)-(R2′R3′R6′R7′-Flu)(3-R3-5-R4-C5H2)}ZrCl2: From Highly Isotactic Polypropylenes to Vinyl End-Capped Isotactic-Enriched Oligomers. Organometallics 2012, 31, 8375–8387. [Google Scholar] [CrossRef]
- Theurkauff, G.; Bader, M.; Marquet, N.; Bondon, A.; Roisnel, T.; Guegan, J.-P.; Amar, A.; Boucekkine, A.; Carpentier, J.-F.; Kirillov, E. Discrete ionic complexes of highly isoselective zirconocenes. solution dynamics, trimethylaluminum adducts, and implications in propylene polymerization. Organometallics 2016, 35, 258–276. [Google Scholar] [CrossRef]
- Theurkauff, G.; Bondon, A.; Dorcet, V.; Carpentier, J.-F.; Kirillov, E. Heterobi- and -trimetallic Ion Pairs of Zirconocene-Based Isoselective Olefin Polymerization Catalysts with AlMe3. Angew. Chem. Int. Ed. 2015, 127, 6343–6346. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Rausch, M.D.; Chien, J.C.W. Silylene-bridged fluorenyl-containing ligands and zirconium complexes with C1 and Cs symmetry: General synthesis and olefin polymerization catalysis. J. Organomet. Chem. 1995, 497, 1–9. [Google Scholar] [CrossRef]
- Patsidis, K.; Alt, H.G.; Milius, W.; Palackal, S.J. The synthesis, characterization and polymerization behavior of ansa cyclopentadienyl fluorenyl complexes; the X-ray structures of the complexes [(C13H8)SiR2(C5H4)]ZrCl2 (R = Me or Ph). J. Organomet. Chem. 1996, 509, 63–71. [Google Scholar] [CrossRef]
- Inazawa, S.; Okumura, Y.; Ono, M.; Sakuragi, T. Catalyst for Polyolefin Production and Process for Producing Polyolefin. International Patent WO1997040075A1, 30 October 1997. [Google Scholar]
- Izmer, V.V.; Agarkov, A.Y.; Nosova, V.M.; Kuz’mina, L.G.; Howard, J.A.K.; Beletskaya, I.P.; Voskoboynikov, A.Z. ansa-Metallocenes with a Ph2Si bridge: Molecular structures of HfCl2[Ph2Si(η5-C13H8)(η5-C5H4)] and HfCl2[Ph2Si(C13H9)(η5-C5H4)]2. J. Chem. Soc. Dalton Trans. 2001, 1131–1136. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Bagrov, V.V.; Okumura, Y.; Elder, M.; Churakov, A.V. Asymmetric ansa-Zirconocenes Containing a 2-Methyl-4-aryltetrahydroindacene Fragment: Synthesis, Structure, and Catalytic Activity in Propylene Polymerization and Copolymerization. Organometallics 2011, 30, 5744–5752. [Google Scholar] [CrossRef]
- Ivchenko, N.B.; Ivchenko, P.V.; Nifant’ev, I.E.; Bagrov, V.V.; Kuz’mina, L.G. Synthesis of bis-cyclopentadienyl compounds with a 9,9-fluorenylidene bridge. Crystal andmolecular structure of [μ-9,9-Flu(η5-Cp)2]ZrCI2. Russ. Chem. Bull. 2000, 49, 1287–1291. [Google Scholar] [CrossRef]
- Irwin, L.J.; Reibenspies, J.H.; Miller, S.A. Synthesis and characterization of sterically expanded ansa-η1-fluorenyl-amido complexes. Polyhedron 2005, 24, 1314–1324. [Google Scholar] [CrossRef]
- Drago, D.; Pregosin, P.S.; Razavi, A. 13C NMR Studies on Zr(ansa-Fluorenyl) Complexes. Molecules with Flexible Bonding Modes. Organometallics 2000, 19, 1802–1805. [Google Scholar] [CrossRef]
- Razavi, A.; Thewalt, U. Preparation and crystal structures of the complexes (η5-C5H3TMS–CMe2–η5-C13H8)MCl2 and [3,6-ditButC13H6–SiMe2–NtBu]MCl2 (M=Hf, Zr or Ti): Mechanistic aspects of the catalytic formation of a isotactic–syndiotactic stereoblock-type polypropylene. J. Organomet. Chem. 2001, 621, 267–276. [Google Scholar] [CrossRef]
- Razavi, A.; Thewalt, U. Site selective ligand modification and tactic variation in polypropylene chains produced with metallocene catalysts. Coord. Chem. Rev. 2006, 250, 155–169. [Google Scholar] [CrossRef]
- Won, Y.C.; Kwon, H.Y.; Lee, B.Y.; Park, Y.-W. Fulvene having substituents only on 1-, 4-, and 6-positions: A key intermediate for novel ansa-metallocene complexes. J. Organomet. Chem. 2003, 677, 133–139. [Google Scholar] [CrossRef]
- Yu, Y.; Busico, V.; Budzelaar, P.H.M.; Vittoria, A.; Cipullo, R. Of poisons and antidotes in polypropylene catalysis. Angew. Chem. Int. Ed. 2016, 55, 8590–8594. [Google Scholar] [CrossRef] [PubMed]
- Prosenc, M.-H.; Britzinger, H.-H. Zirconium−Alkyl Isomerizations in Zirconocene-Catalyzed Olefin Polymerization: A Density Functional Study. Organometallics 1997, 16, 3889–3894. [Google Scholar] [CrossRef]
- Rieger, B.; Mu, X.; Mallin, D.T.; Rausch, M.D.; Chien, J.C.W. Degree of Stereochemical Control of rac-Et[Ind]2ZrCl2/MAO Catalyst and Properties of Anisotactic Polypropylenes. Macromolecules 1990, 23, 3559–3568. [Google Scholar] [CrossRef]
- Carvill, A.; Zetta, L.; Zannoni, G.; Sacchi, M.C. ansa-Zirconocene-Catalyzed Solution Polymerization of Propene: Influence of Polymerization Conditions on the Unsaturated Chain-End Groups. Macromolecules 1998, 31, 3783–3789. [Google Scholar] [CrossRef]
- Shiono, T.; Azad, S.M.; Ikeda, T. Copolymerization of atactic polypropene macromonomer with propene by an isospecific metallocene catalyst. Macromolecules 1999, 32, 5723–5727. [Google Scholar] [CrossRef]
- Kawahara, N.; Kojoh, S.; Matsuo, S.; Kaneko, H.; Matsugi, T.; Toda, Y.; Mizuno, A.; Kashiwa, N. Study on chain end structures of polypropylenes prepared with different symmetrical metallocene catalysts. Polymer 2004, 45, 2883–2888. [Google Scholar]
- Quevedo-Sanchez, B.; Nimmons, J.F.; Coughlin, E.B.; Henson, M.A. Kinetic modeling of the effect of MAO/Zr ratio and chain transfer to aluminum in zirconocene catalyzed propylene polymerization. Macromolecules 2006, 39, 4306–4316. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Fraldi, N.; Budzelaar, P.H.M.; Busico, V.; Macchioni, A. Activation of a bis (phenoxy-amine) precatalyst for olefin polymerisation: First evidence for an outer sphere ion pair with the methylborate counterion. Dalton Trans. 2009, 8824–8827. [Google Scholar] [CrossRef]
- Bazinet, P.; Don Tilley, T. Octa- and Nonamethylfluorenyl Complexes of Zirconium(IV): Reactive Hydride Derivatives and Reversible Hydrogen Migration between the Metal and the Fluorenyl Ligand. Organometallics 2009, 28, 2285–2293. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
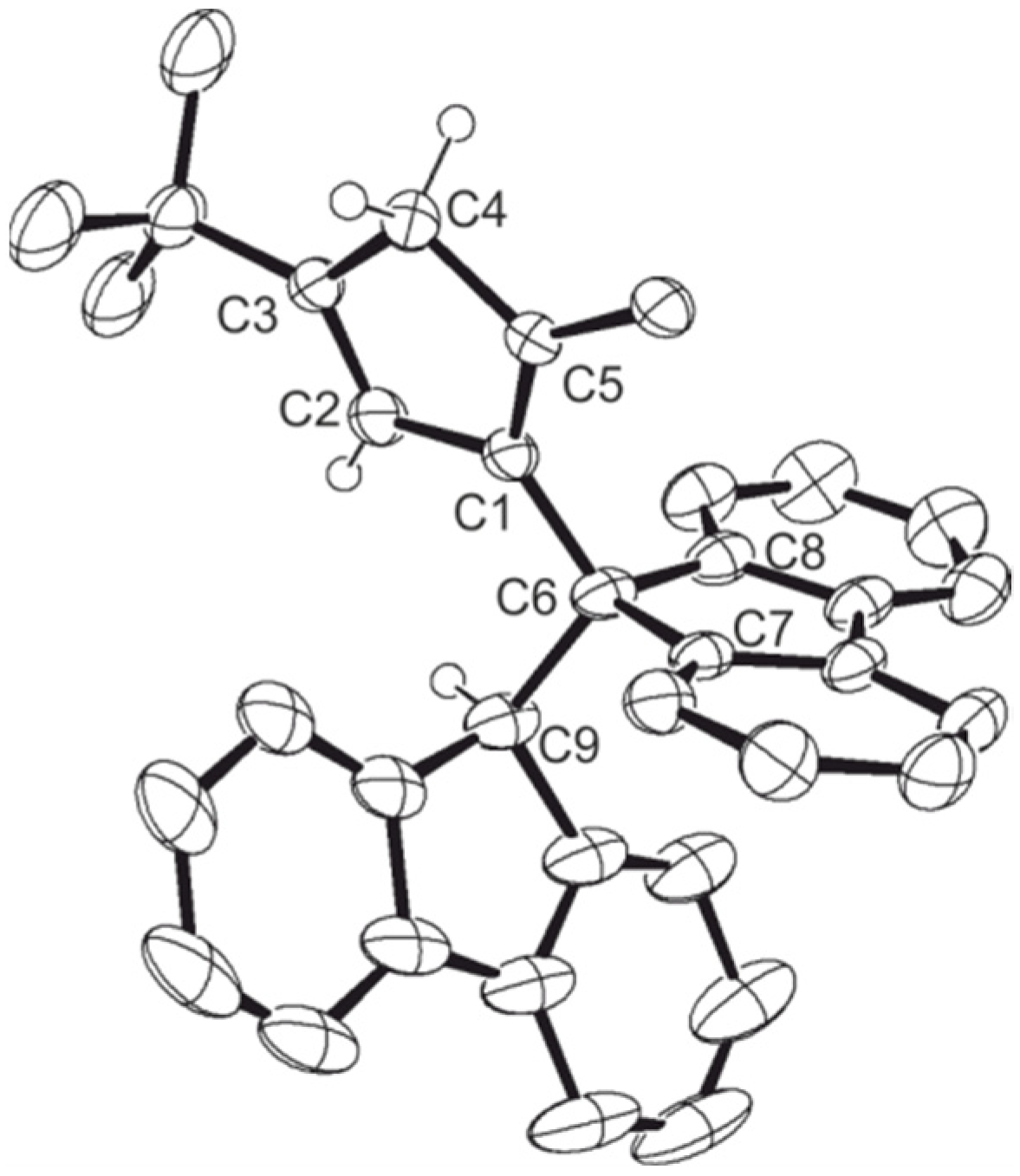
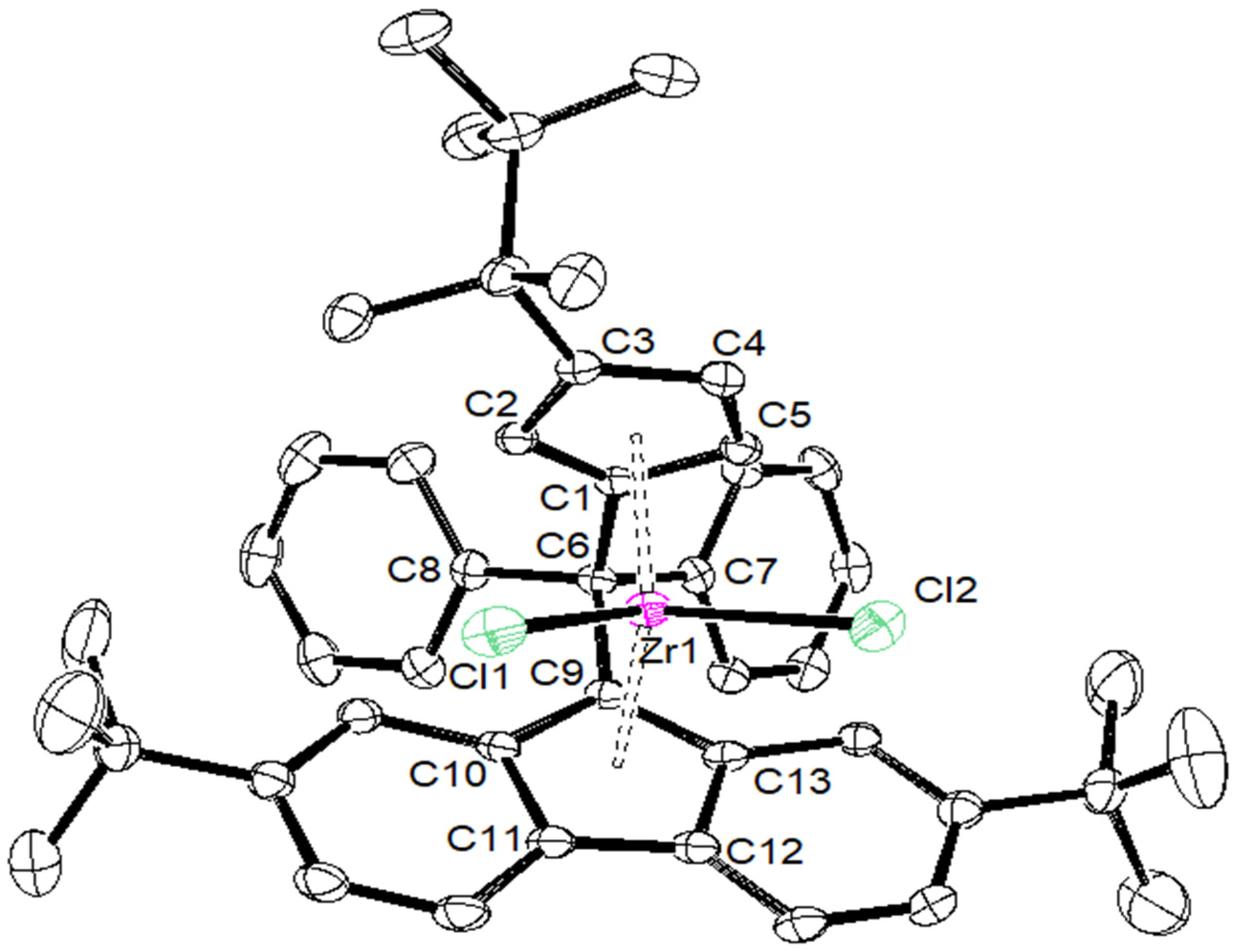
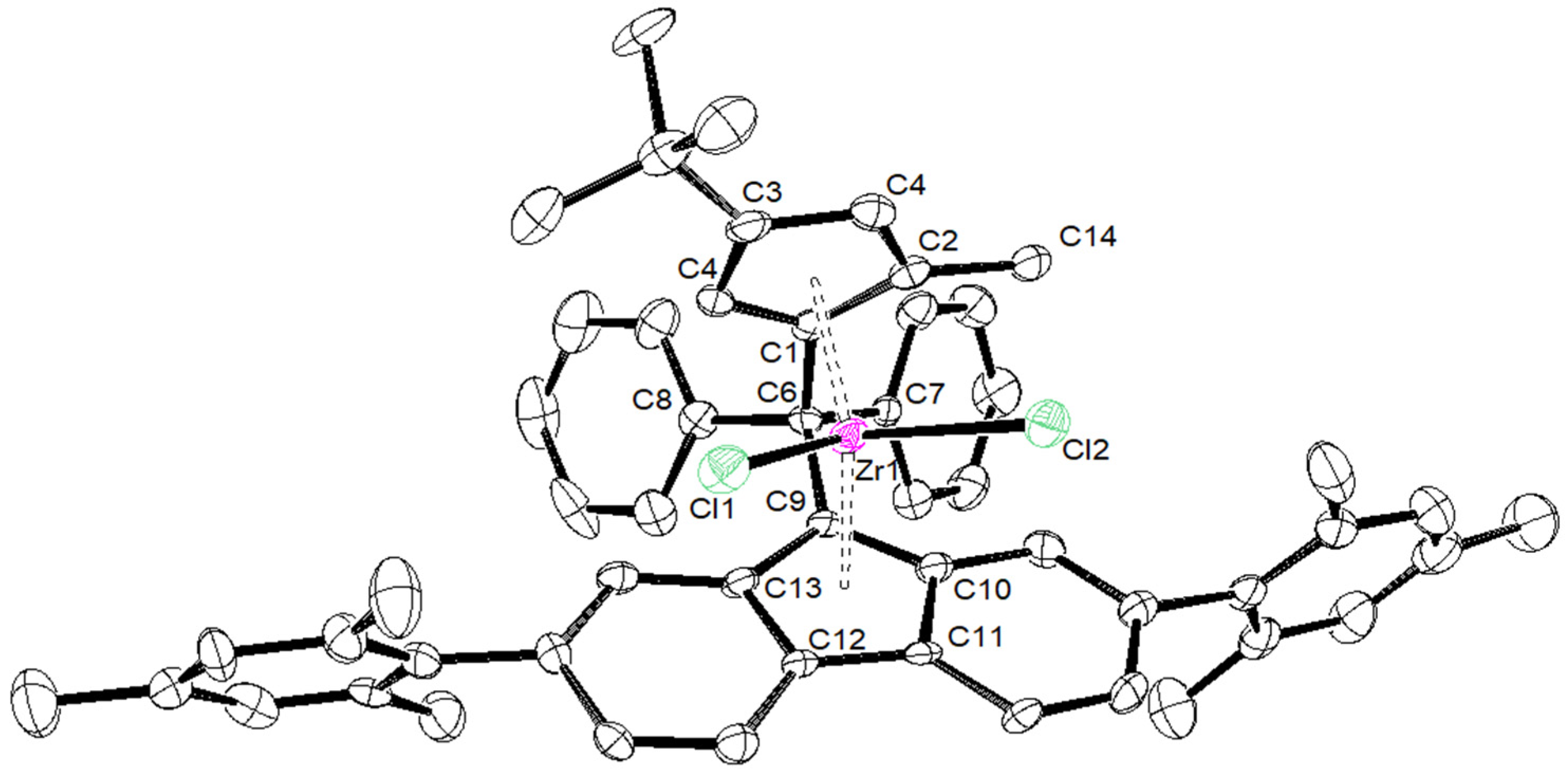
| {Cp/Flu}5H | 3b | 3c | 3d | 3e | 3g | 3h | |
|---|---|---|---|---|---|---|---|
| Zr–C(1) | 2.442 (4) | 2.4450 (17) | 2.4391 (14) | 2.451 (4) | 2.443 (8) | 2.440 (5) | 2.442 (6) |
| Zr–C(2) | 2.492 (3) | 2.4745 (18) | 2.4720 (15) | 2.484 (4) | 2.489 (8) | 2.459 (5) | 2.468 (6) |
| Zr–C(3) | 2.630 (4) | 2.6079 (18) | 2.6431 (15) | 2.645 (4) | 2.594 (8) | 2.582 (5) | 2.587 (6) |
| Zr–C(4) | 2.547 (4) | 2.5284 (18) | 2.5706 (15) | 2.558 (4) | 2.514 (8) | 2.554 (6) | 2.525 (6) |
| Zr–C(5) | 2.457 (4) | 2.4384 (18) | 2.4408 (15) | 2.433 (4) | 2.429 (8) | 2.463 (5) | 2.459 (6) |
| Zr–Cpcent | 2.204 | 2.242 | 2.204 | 2.209 | 2.180 | 2.190 | 2.181 |
| Zr–C(9) | 2.426 (3) | 2.4108 (18) | 2.4229 (15) | 2.445 (4) | 2.403 (8) | 2.407 (5) | 2.420 (6) |
| Zr–C(10) | 2.497 (4) | 2.5128 (18) | 2.5020 (15) | 2.510 (4) | 2.500 (8) | 2.505 (5) | 2.488 (6) |
| Zr–C(11) | 2.666 (4) | 2.6740 (18) | 2.6621 (15) | 2.683 (4) | 2.686 (8) | 2.681 (5) | 2.695 (6) |
| Zr–C(12) | 2.712 (4) | 2.6633 (18) | 2.6732 (15) | 2.707 (4) | 2.670 (8) | 2.688 (5) | 2.660 (6) |
| Zr–C(13) | 2.588 (4) | 2.5083 (18) | 2.5263 (15) | 2.552 (4) | 2.514 (8) | 2.524 (4) | 2.563 (6) |
| Zr–Flucent | 2.269 | 2.188 | 2.240 | 2.272 | 2.245 | 2.250 | 2.256 |
| Cpcent–Zr–Flucent | 118.03 | 118.15 | 117.80 | 116.85 | 117.93 | 118.45 | 118.61 |
| C(1)–C(6)–C(9) | 98.60 (3) | 99.13 (14) | 98.73 (11) | 99.20 (3) | 99.10 (6) | 99.20 (6) | 100.10 (5) |
| C(7)–C(6)–C(8) | 104.00 (4) | 104.33 (14) | 104.82 (12) | 105.60 (3) | 104.30 (7) | 104.50 (4) | 101.10 (5) |
| Entry | Precat. | Tpolymb | Prod (kgPP·mol·h−1) | Tm (°C) c | Tcrystc (°C) | Mnd (×103) | Mw/Mnd | [m]4 e (%) | 1,2-ins e (%) | 2,1-ins e (%) | 1,3-ins e (%) | nBu termini e (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | {Cp/Flu}-2 | 60 (68) | 50,700 f | 131.1 | 93.6 | 50.8 | 2.5 | 82.1 | 99.8 | <0.02 | 0.1 | 0.1 |
| 2 | 3a | 60 (66) | 35,500 | 135.8 | 102.6 | 11.4 | 2.1 | 86.5 | 99.3 | <0.1 | 0.3 | 0.2 |
| 3 | 3b | 60 (65) | 5300 | 141.1 | 106.2 | 15.4 | 2.1 | 88.6 | 99.2 | 0.2 | 0.4 | 0.3 |
| 4 | 3c | 60 (65) | 241 | 137.1 | 106.2 | - | - | - | - | - | - | - |
| 5 | 3d | 60 (64) | 0 | - | - | - | - | - | - | - | - | - |
| 6 | 3e | 60 (76) | 33,600 | 132.9 | 100.3 | 13.7 | 2.1 | 81.6 | 99.3 | <0.02 | 0.3 | 0.04 |
| 7 | 60 (67) | 16,700 g | 139.8 | 104.5 | 21.7 | 2.1 | 86.2 | 99.5 | <0.1 | 0.3 | ≈0.1 | |
| 8 | 3f | 60 (66) | 13,300 | 148.4 | 112.6 | 17.6 | 2.3 | 91.7 | 99.6 | <0.02 | 0.1 | < 0.1 |
| 9 | 3g | 60 (66) | 12 700 | 136.1 | 102.3 | 21.0 | 2.3 | 82.9 | 99.4 | 0.1 | 0.3 | 0.1 |
| 10 | 3h | 60 (68) | 17,200 | n.o. | n.o. | 24.9 | 2.1 | 54.0 | 99.7 | <0.02 | 0.1 | <0.02 |
| 11 | 3k | 60 (64) | 1870 | 131.7 | 119.8 | 13.4 | 2.2 | 31.9 | 99.6 | <0.02 | <0.01 | <0.02 |
| 12 | 100 (102) | 640 | n.o. | n.o. | 3.4 | 1.4 | - | - | - | - | - |
| Entry | Precat. | [Zr]0 (µmol.L−1) | [Al/[Zr] | Prod (kgPP·mol·h−1) | [m]4 b (%) | 1,2-ins b (%) | 2,1-ins b (%) | 1,3-ins b (%) | nBu Termini b (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | {Cp/Flu}-1 | 10.0 | 5000 | 1100 | 90.5 | 99.8 | <0.02 | 0.1 | <0.02 |
| 2 | {Cp/Flu}-3Me | 10.0 | 5000 | 15,470 | 88.1 | 99.8 | <0.02 | <0.1 | <0.02 |
| 3 | {Cp/Flu}-3Et | 10.0 | 5000 | 9730 | 89.5 | 99.8 | <0.02 | <0.1 | <0.02 |
| 4 | {Cp/Flu}-3Ph | 20.0 | 5000 | 2000 | 53.8 | 99.5 | <0.02 | <0.01 | <0.02 |
| 5 | {Cp/Flu}-4Me | 10.0 | 5000 | 29,600 | 90.7 | 99.8 | <0.02 | 0.02 | <0.02 |
| 6 | {Cp/Flu}-4Et | 10.0 | 5000 | 26,260 | 90.8 | 99.7 | <0.02 | 0.02 | <0.02 |
| 7 | {Cp/Flu}-5 | 10.0 | 5000 | 1550 | 80.9 | 99.6 | <0.02 | 0.09 | 0.2 |
| 8 | {SBI}-1 | 2.0 | 25,000 | 62,700 | 93.6 | 98.6 | 0.26 | 0.5 | 0.05 |
| 9 | {SBI}-2 | 2.0 | 25,000 | 27,900 | 83.4 | 99.2 | 0.45 | 0.05 | <0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desert, X.; Roisnel, T.; Dorcet, V.; Den Dauw, K.; Vantomme, A.; Welle, A.; Carpentier, J.-F.; Kirillov, E. Propylene Polymerization and Deactivation Processes with Isoselective {Cp/Flu} Zirconocene Catalysts. Catalysts 2021, 11, 959. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080959
Desert X, Roisnel T, Dorcet V, Den Dauw K, Vantomme A, Welle A, Carpentier J-F, Kirillov E. Propylene Polymerization and Deactivation Processes with Isoselective {Cp/Flu} Zirconocene Catalysts. Catalysts. 2021; 11(8):959. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080959
Chicago/Turabian StyleDesert, Xavier, Thierry Roisnel, Vincent Dorcet, Katty Den Dauw, Aurélien Vantomme, Alexandre Welle, Jean-François Carpentier, and Evgueni Kirillov. 2021. "Propylene Polymerization and Deactivation Processes with Isoselective {Cp/Flu} Zirconocene Catalysts" Catalysts 11, no. 8: 959. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11080959