
Magnesium as a Methanation Suppressor for Iron- and Cobalt-Based Oxide Catalysts during the Preferential Oxidation of Carbon Monoxide


Abstract
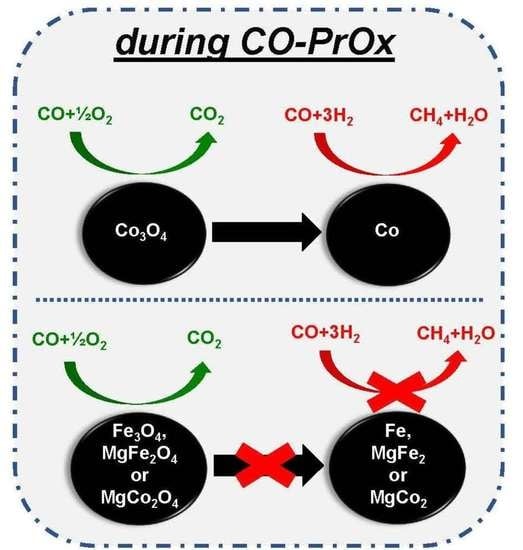
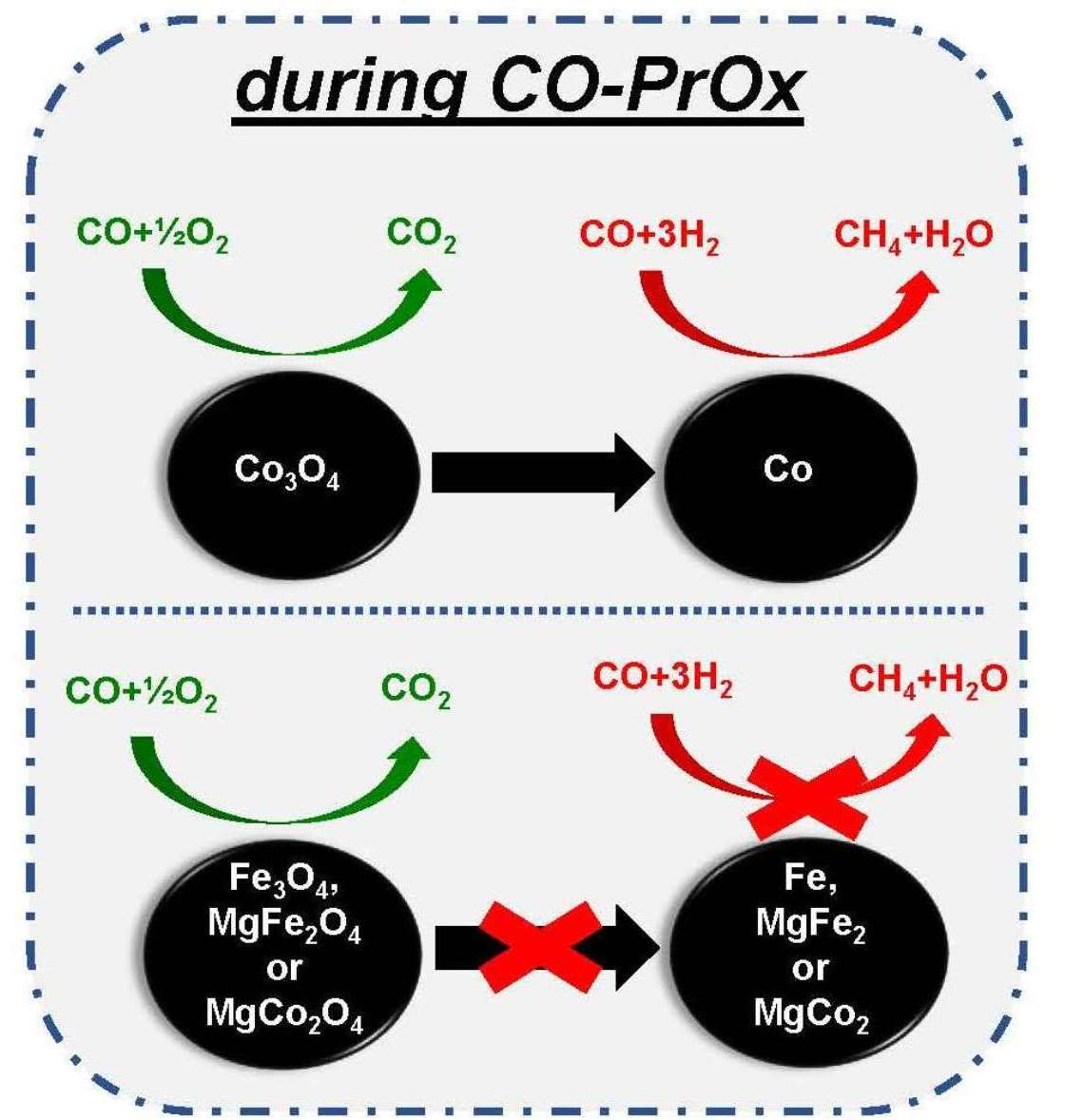
:
1. Introduction
2. Results and Discussion
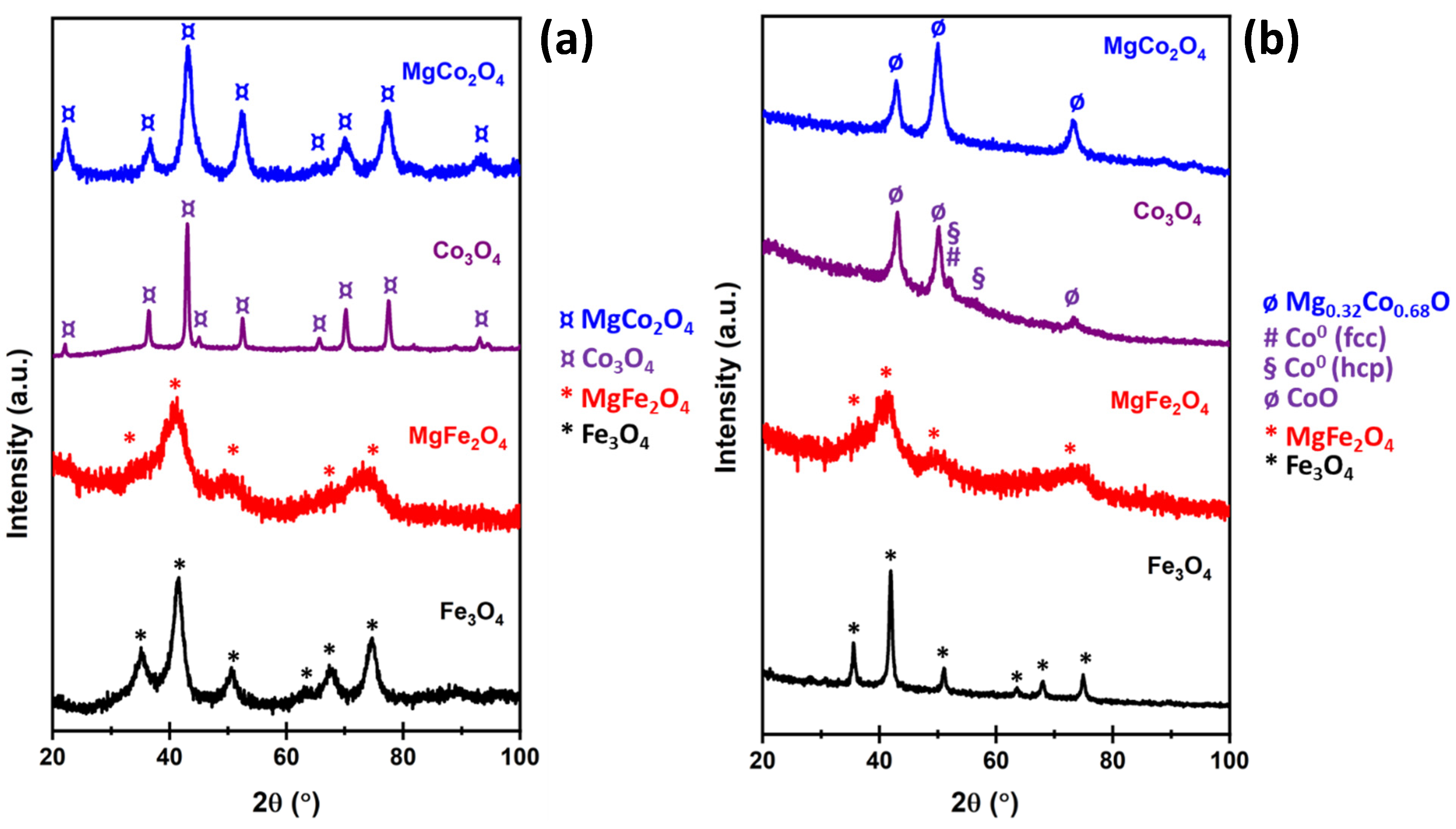
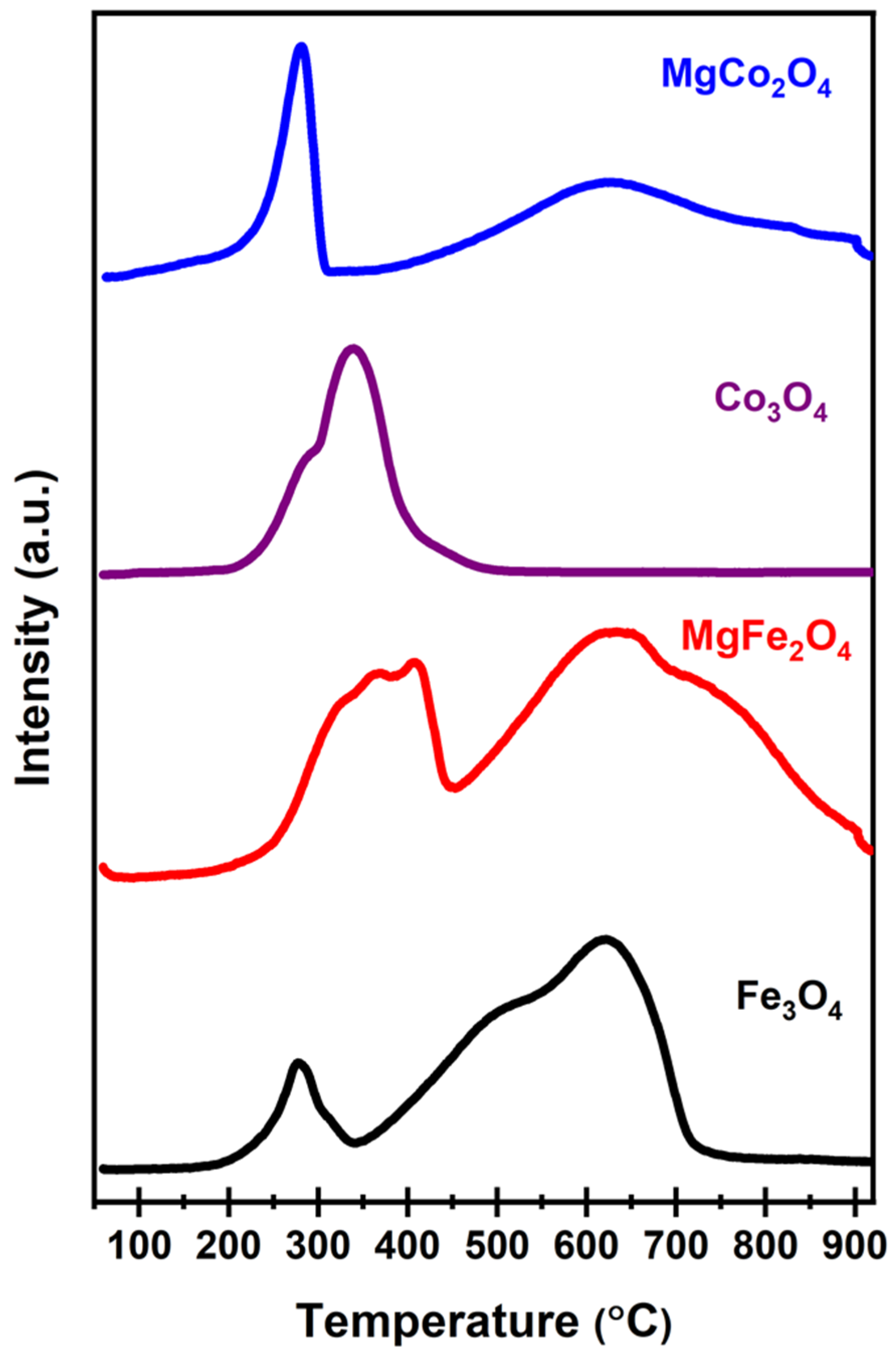
2.1. Fresh Catalyst Characterisation
2.2. Spent Catalyst Characterisation
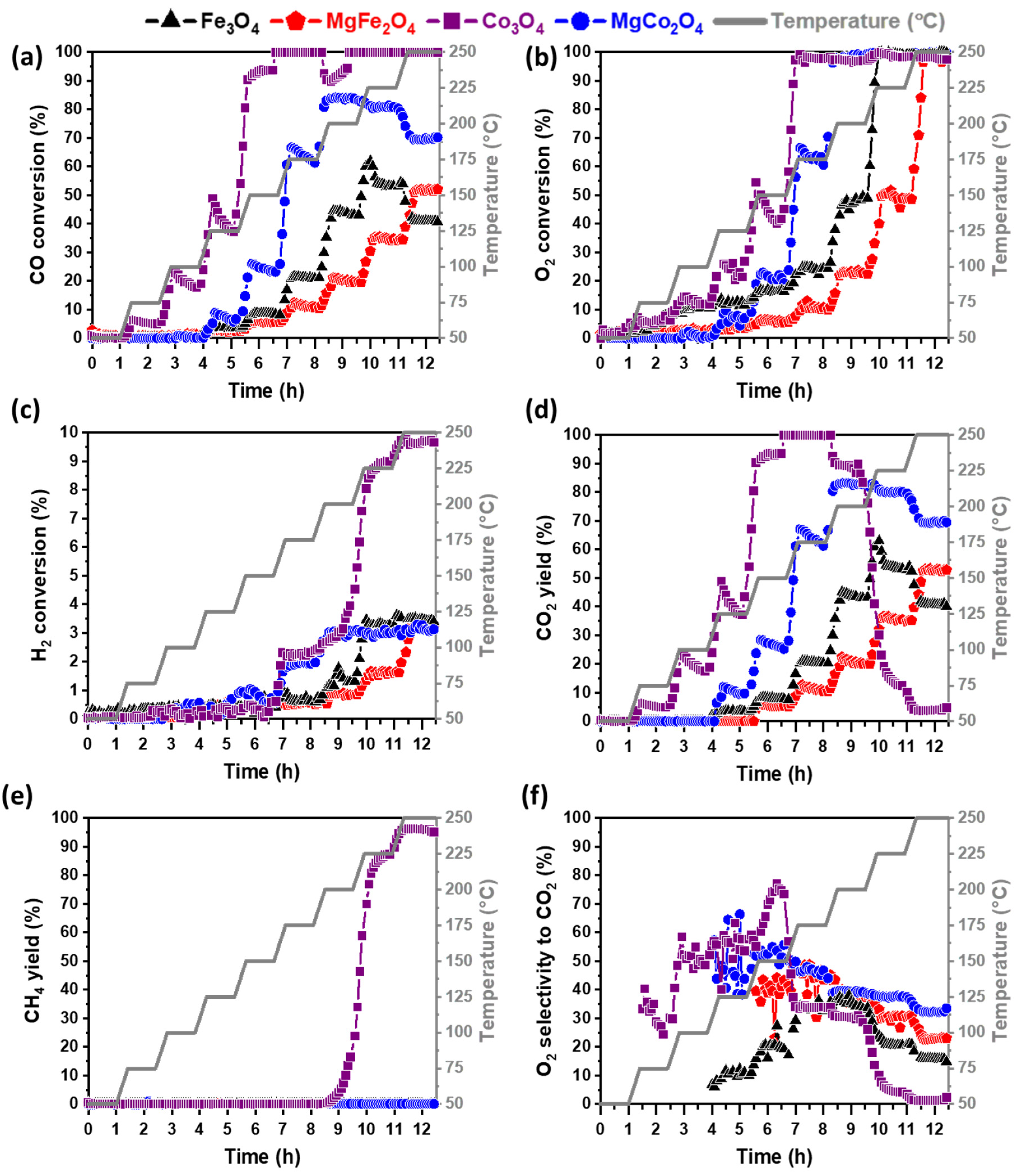
2.3. CO-PrOx Performance Evaluation
3. Experimental Section
3.1. Catalyst Synthesis
3.2. Catalyst Characterisation
3.3. CO-PrOx Catalyst Evaluation
4. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations. Department of Economic and Social Affairs Sustainable Development, Goal 7. Available online: https://sdgs.un.org/goals/goal7 (accessed on 13 December 2021).
- Santos, P.; Costa, A.; Kiminami, R.; Andrade, H.M.C.; Lira, H.L.; Gama, L. Synthesis of a NiFe2O4 catalyst for the preferential oxidation of carbon monoxide (PROX). J. Alloys Compd. 2009, 483, 399–401. [Google Scholar] [CrossRef]
- Jing, P.; Gong, X.; Liu, B.; Zhang, J. Recent advances in synergistic effect promoted catalysts for preferential oxidation of carbon monoxide. Catal. Sci. Technol. 2020, 10, 919–934. [Google Scholar] [CrossRef]
- Zhou, G.; Jiang, Y.; Xie, H.; Qiu, F. Non-noble metal catalyst for carbon monoxide selective oxidation in excess hydrogen. Chem. Eng. J. 2005, 109, 141–145. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Goodman, D.W. CO-free fuel processing for fuel cell applications. Catal. Today 2002, 77, 65–78. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Teng, Y.; Sakurai, H.; Ueda, A.; Kobayashi, T. Oxidative removal of CO contained in hydrogen by using metal oxide catalysts. Int. J. Hydrogen Energy 1999, 24, 355–358. [Google Scholar] [CrossRef]
- Qwabe, L.Q.; Friedrich, H.B.; Singh, S. Preferential oxidation of CO in a hydrogen rich feed stream using Co–Fe mixed metal oxide catalysts prepared from hydrotalcite precursors. J. Mol. Catal. A Chem. 2015, 404–405, 167–177. [Google Scholar] [CrossRef]
- Malwadkar, S.; Bera, P.; Hegde, M.S.; Satyanarayana, C.V.V. Preferential oxidation of CO on Ni/CeO2 catalysts in the presence of excess H2 and CO2. React. Kinet. Mech. Catal. 2012, 107, 405–419. [Google Scholar] [CrossRef]
- Khasu, M.; Nyathi, T.; Morgan, D.J.; Hutchings, G.J.; Claeys, M.; Fischer, N. Co3O4 morphology in the preferential oxidation of CO. Catal. Sci. Technol. 2017, 7, 4806–4817. [Google Scholar] [CrossRef]
- Nyathi, T.M.; Fischer, N.; York, A.P.E.; Morgan, D.J.; Hutchings, G.J.; Gibson, E.K.; Wells, P.P.; Catlow, C.R.A.; Claeys, M. Impact of nanoparticle–Support interactions in Co3O4/Al2O3 catalysts for the preferential Oxidation of carbon monoxide. ACS Catal. 2019, 9, 7166–7178. [Google Scholar] [CrossRef] [Green Version]
- Lukashuk, L.; Föttinger, K.; Kolar, E.; Rameshan, C.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Yigit, N.; Li, H.; McDermott, E. Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts. J. Catal. 2016, 344, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Barreau, M.; Chen, D.; Caps, V.; Haevecker, M.; Teschner, D.; Simonne, D.H.; Borfecchia, E.; Baaziz, W.; Šmíd, B.; et al. Effect of manganese promotion on the activity and selectivity of cobalt catalysts for CO preferential oxidation. Appl. Catal. B 2021, 297, 120397. [Google Scholar] [CrossRef]
- Park, J.-W.; Jeong, J.-H.; Yoon, W.-L.; Jung, H.; Lee, H.-T.; Lee, D.-K.; Park, Y.-K.; Rhee, Y.-W. Activity and characterization of the Co-promoted CuO–CeO2/γ-Al2O3 catalyst for the selective oxidation of CO in excess hydrogen. Appl. Catal. A 2004, 274, 25–32. [Google Scholar] [CrossRef]
- Ratnasamy, P.; Srinivas, D.; Satyanarayana, C.V.V.; Manikandan, P.; Kumaran, R.S.S.; Sachin, M.; Shetti, V.N. Influence of the support on the preferential oxidation of CO in hydrogen-rich steam reformates over the CuO–CeO2–ZrO2 system. J. Catal. 2004, 221, 455–465. [Google Scholar] [CrossRef]
- Nyathi, T.M.; Fadlalla, M.I.; Fischer, N.; York, A.P.E.; Olivier, E.J.; Gibson, E.K.; Wells, P.P.; Claeys, M. Support and gas environment effects on the preferential oxidation of carbon monoxide over Co3O4 catalysts studied in situ. Appl. Catal. B 2021, 297, 120450. [Google Scholar] [CrossRef]
- Nyathi, T.M.; Fischer, N.; York, A.P.E.; Claeys, M. Environment-dependent catalytic performance and phase stability of Co3O4 in the preferential oxidation of carbon monoxide studied in situ. ACS Catal. 2020, 10, 11892–11911. [Google Scholar] [CrossRef]
- Nyathi, T.M.; Fischer, N.; York, A.P.E.; Claeys, M. Effect of crystallite size on the performance and phase transformation of Co3O4/Al2O3 catalysts during CO-PrOx—an in situ study. Faraday Discuss. 2017, 197, 269–285. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, D.; Hammache, S.; Goodwin, J.G. Effect of water vapor on the reduction of Ru-promoted Co/Al2O3. J. Catal. 1999, 188, 281–290. [Google Scholar] [CrossRef]
- Fischer, N.; Clapham, B.; Feltes, T.; van Steen, E.; Claeys, M. Size-Dependent phase transformation of catalytically active nanoparticles captured in situ. Angew. Chem. Int. Ed. 2014, 53, 1342–1345. [Google Scholar] [CrossRef]
- Wolf, M.; Gibson, E.K.; Olivier, E.J.; Neethling, J.H.; Catlow, C.R.A.; Fischer, N.; Claeys, M. Water-induced formation of cobalt-support compounds under simulated high conversion Fischer–Tropsch environment. ACS Catal. 2019, 9, 4902–4918. [Google Scholar] [CrossRef]
- González-Carballo, J.M.; Sadasivan, S.; Landon, P.; Tooze, R.P. Synthesis of cobalt nanodumbbells and their thermal stability under H2, H2/CO and O2 atmospheres. Mater. Charact. 2016, 118, 519–526. [Google Scholar] [CrossRef]
- Lu, J.; Wang, J.; Zou, Q.; He, D.; Zhang, L.; Xu, Z.; He, S.; Luo, Y. Unravelling the nature of the active species as well as the doping effect over Cu/Ce-based catalyst for carbon monoxide preferential oxidation. ACS Catal. 2019, 9, 2177–2195. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Bharuth-Ram, K.; Hanzel, D.; Likozar, B. Heterogeneous Cu–Fe oxide catalysts for preferential CO oxidation (PROX) in H2-rich process streams. RSC Adv. 2020, 10, 35792–35802. [Google Scholar] [CrossRef]
- Li, S.; Zhu, H.; Qin, Z.; Zhang, Y.; Wang, G.; Wu, Z.; Fan, W.; Wang, J. Catalytic performance of gold supported on Mn, Fe and Ni doped ceria in the preferential oxidation of CO in H2-rich stream. Catalysts 2018, 8, 469. [Google Scholar] [CrossRef] [Green Version]
- Qwabe, L.Q.; Friedrich, H.B.; Singh, S. Remediation of CO by oxidation over Au nanoparticles supported on mixed metal oxides. J. Environ. Chem. Eng. 2019, 7, 102827. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent advances in preferential oxidation of CO reaction over platinum group metal catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Dongil, A.B.; Bachiller-Baeza, B.; Castillejos, E.; Escalona, N.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Promoter effect of alkalis on CuO/CeO2/carbon nanotubes systems for the PROx reaction. Catal. Today 2018, 301, 141–146. [Google Scholar] [CrossRef]
- Dongil, A.B.; Bachiller-Baeza, B.; Castillejos, E.; Escalona, N.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. The promoter effect of potassium in CuO/CeO2 systems supported on carbon nanotubes and graphene for the CO-PROX reaction. Catal. Sci. Technol. 2016, 6, 6118–6127. [Google Scholar] [CrossRef]
- Kuriyama, M.; Tanaka, H.; Ito, S.-i.; Kubota, T.; Miyao, T.; Naito, S.; Tomishige, K.; Kunimori, K. Promoting mechanism of potassium in preferential CO oxidation on Pt/Al2O3. J. Catal. 2007, 252, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-H.; Park, J.-S.; Choi, S.-H.; Kim, S.-H. Effect of magnesium on preferential oxidation of carbon monoxide on platinum catalyst in hydrogen-rich stream. J. Power Sources 2006, 156, 260–266. [Google Scholar] [CrossRef]
- Kharisov, B.I.; Dias, H.V.R.; Kharissova, O.V. Mini-review: Ferrite nanoparticles in the catalysis. Arab. J. Chem. 2019, 12, 1234–1246. [Google Scholar] [CrossRef] [Green Version]
- Fadlalla, M.I.; Babu, S.G.; Nyathi, T.M.; Weststrate, C.J.K.-J.; Fischer, N.; Niemantsverdriet, J.W.H.; Claeys, M. Enhanced oxygenates formation in the Fischer–Tropsch synthesis over Co- and/or Ni-containing Fe alloys: Characterization and 2D gas chromatographic product analysis. ACS Catal. 2020, 10, 14661–14677. [Google Scholar] [CrossRef]
- Chonco, Z.H.; Lodya, L.; Claeys, M.; van Steen, E. Copper ferrites: A model for investigating the role of copper in the dynamic iron-based Fischer–Tropsch catalyst. J. Catal. 2013, 308, 363–373. [Google Scholar] [CrossRef]
- Rethwisch, D.G.; Phillips, J.; Chen, Y.; Hayden, T.F.; Dumesic, J.A. Water-gas shift over magnetite particles supported on graphite: Effects of treatments in CO/CO2 and H2/H2O gas mixtures. J. Catal. 1985, 91, 167–180. [Google Scholar] [CrossRef]
- Coelho, A. Indexing of powder diffraction patterns by iterative use of singular value decomposition. J. Appl. Crystallogr. 2003, 36, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sun, Y.; Tang, Y.; Liu, Y.; Wang, H.; Tian, L.; Wang, H.; Zhang, Z.; Xiang, H.; Li, Y. Effect of magnesium promoter on iron-based catalyst for Fischer–Tropsch synthesis. J. Mol. Catal. A Chem. 2006, 245, 26–36. [Google Scholar] [CrossRef]
- Newbury, D.E.; Ritchie, N.W.M. Is scanning electron microscopy/energy dispersive X-ray spectrometry (SEM/EDS) quantitative? Scanning 2013, 35, 141–168. [Google Scholar] [CrossRef] [PubMed]
- Farahani, M.D.; Dasireddy, V.D.; Friedrich, H.B. Oxidative dehydrogenation of n-Octane over Niobium-Doped NiAl2O4: An example of beneficial coking in catalysis over spinel. ChemCatChem 2018, 10, 2059–2069. [Google Scholar] [CrossRef]
- Ghenciu, A.F. Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr. Opin. Solid State Mater. Sci. 2002, 6, 389–399. [Google Scholar] [CrossRef]
- Mishra, A.; Prasad, R. A review on preferential oxidation of carbon monoxide in hydrogen rich gases. Bull. Chem. React. Eng. Catal. 2011, 6, 1–14. [Google Scholar] [CrossRef]
- Jiang, J.; Wen, C.; Tian, Z.; Wang, Y.; Zhai, Y.; Chen, L.; Li, Y.; Liu, Q.; Wang, C.; Ma, L. Manganese-promoted Fe3O4 microsphere for efficient conversion of CO2 to light olefins. Ind. Eng. Chem. Res. 2020, 59, 2155–2162. [Google Scholar] [CrossRef]
- Duvenhage, D.J.; Coville, N.J. Deactivation of a precipitated iron Fischer–Tropsch catalyst—A pilot plant study. Appl. Catal. A 2006, 298, 211–216. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, A.; Li, L.; Wang, X.; Zhang, T. Promoting role of Fe in the preferential oxidation of CO over Ir/Al2O3. Catal. Lett. 2008, 121, 319–323. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Perti, D.; Kabel, R.; McCarthy, G. Kinetics of CO oxidation over Co3O4/γ-Al2O3. Part III: Mechanism. AlChE J. 1985, 31, 1435–1440. [Google Scholar] [CrossRef]
- Iablokov, V.; Barbosa, R.; Pollefeyt, G.; van Driessche, I.; Chenakin, S.; Kruse, N. Catalytic CO oxidation over well-defined cobalt oxide nanoparticles: Size-reactivity correlation. ACS Catal. 2015, 5, 5714–5718. [Google Scholar] [CrossRef]
- Lukashuk, L.; Yigit, N.; Rameshan, R.; Kolar, E.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Föttinger, K.; Rupprechter, G.N. Operando insights into CO oxidation on cobalt oxide catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 2018, 8, 8630–8641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yung, M.M.; Ozkan, U.S. Effect of support on the preferential oxidation of CO over cobalt catalysts. Catal. Commun. 2008, 9, 1465–1471. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | dcrys, PXRD, Fresh (nm) | Relative Phase Abundance, Fresh (wt.%) | BET Surface Area, Fresh (m2/g) | dcrys, PXRD, Spent (nm) | Relative Phase Abundance, Spent (wt.%) | Ratio of Fe or Co:Mg, Fresh * |
|---|---|---|---|---|---|---|
| Fe3O4 | 6.9 ± 0.1 | 100 | 159 | 19.3 ± 0.4 | 100 | - |
| MgFe2O4 | 2.1 ± 0.0 | 100 | 189 | 2.2 ± 0.0 | 100 | 2.2 |
| Co3O4 | 28.2 ± 0.3 | 100 | 32 | 6.6 ± 0.2 (CoO), 25.7 ± 7.1 (fcc Co0), 3.0 ± 0.2 (hcp Co0) | 72.3 ± 2.5 (CoO), 2.5 ± 0.6 (fcc Co0), 25.2 ± 2.6 (hpc Co0) | - |
| MgCo2O4 | 7.2 ± 0.1 | 100 | 77 | 7.7 ± 0.1 (CoO) | 100 | 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadlalla, M.I.; Nyathi, T.M.; Claeys, M. Magnesium as a Methanation Suppressor for Iron- and Cobalt-Based Oxide Catalysts during the Preferential Oxidation of Carbon Monoxide. Catalysts 2022, 12, 118. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020118
Fadlalla MI, Nyathi TM, Claeys M. Magnesium as a Methanation Suppressor for Iron- and Cobalt-Based Oxide Catalysts during the Preferential Oxidation of Carbon Monoxide. Catalysts. 2022; 12(2):118. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020118
Chicago/Turabian StyleFadlalla, Mohamed I., Thulani M. Nyathi, and Michael Claeys. 2022. "Magnesium as a Methanation Suppressor for Iron- and Cobalt-Based Oxide Catalysts during the Preferential Oxidation of Carbon Monoxide" Catalysts 12, no. 2: 118. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020118