
Cr(III) Complexes Bearing a β-Ketoimine Ligand for Olefin Polymerization: Are There Differences between Coordinative and Covalent Bonding?

 , ,
, ,  , , , , and
, , , , and
Abstract
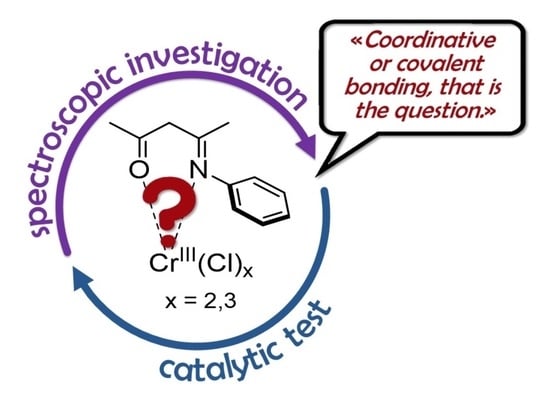
:
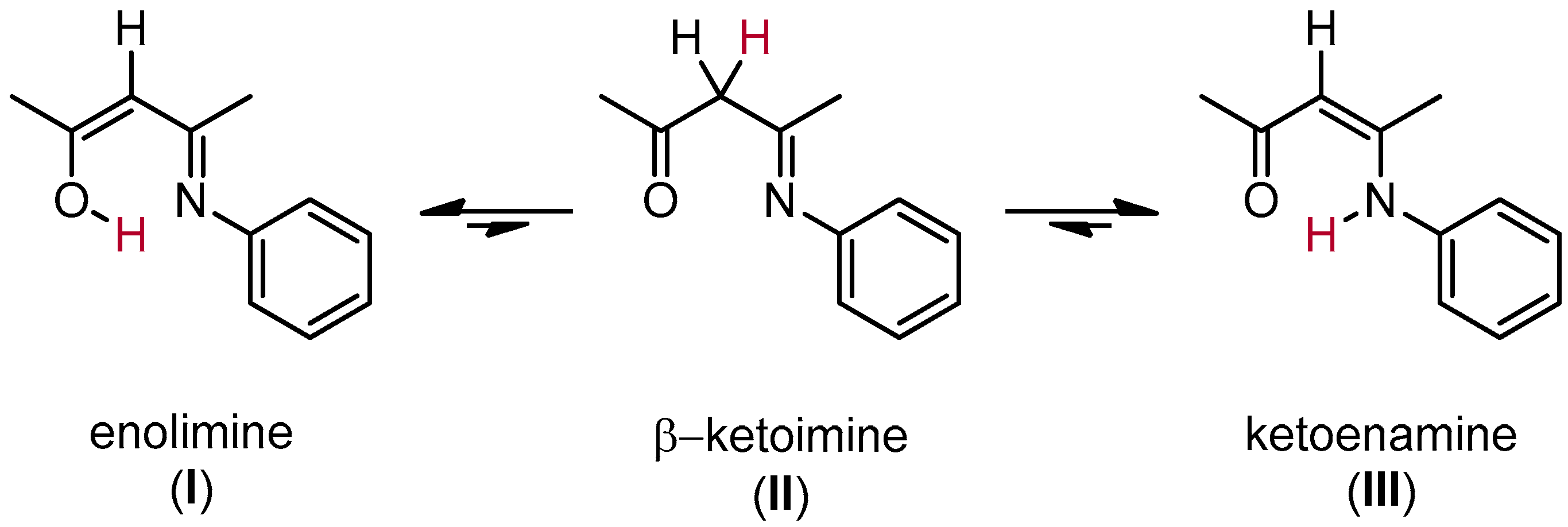
1. Introduction
2. Results and Discussion
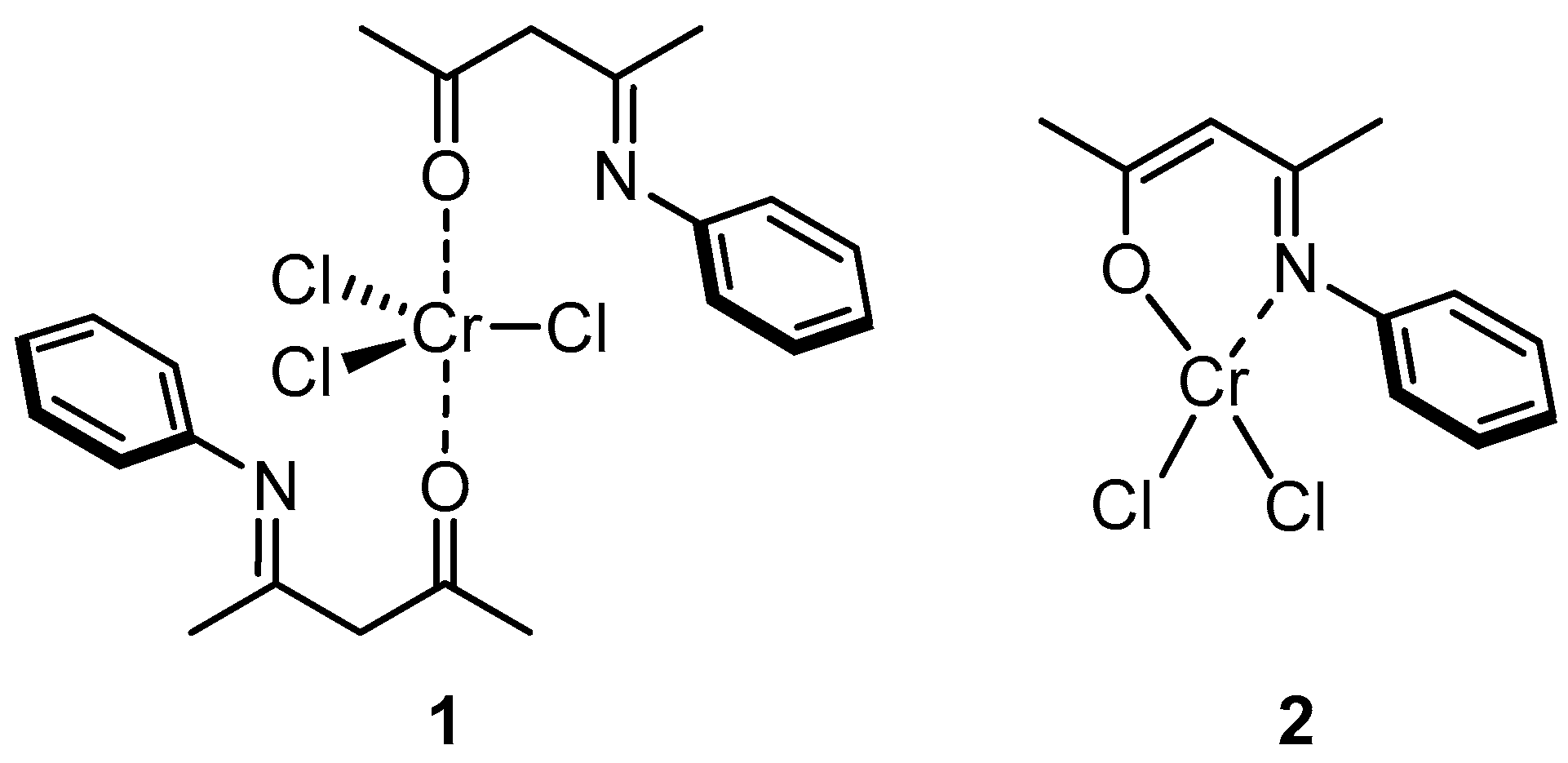
2.1. Synthesis of the Complexes
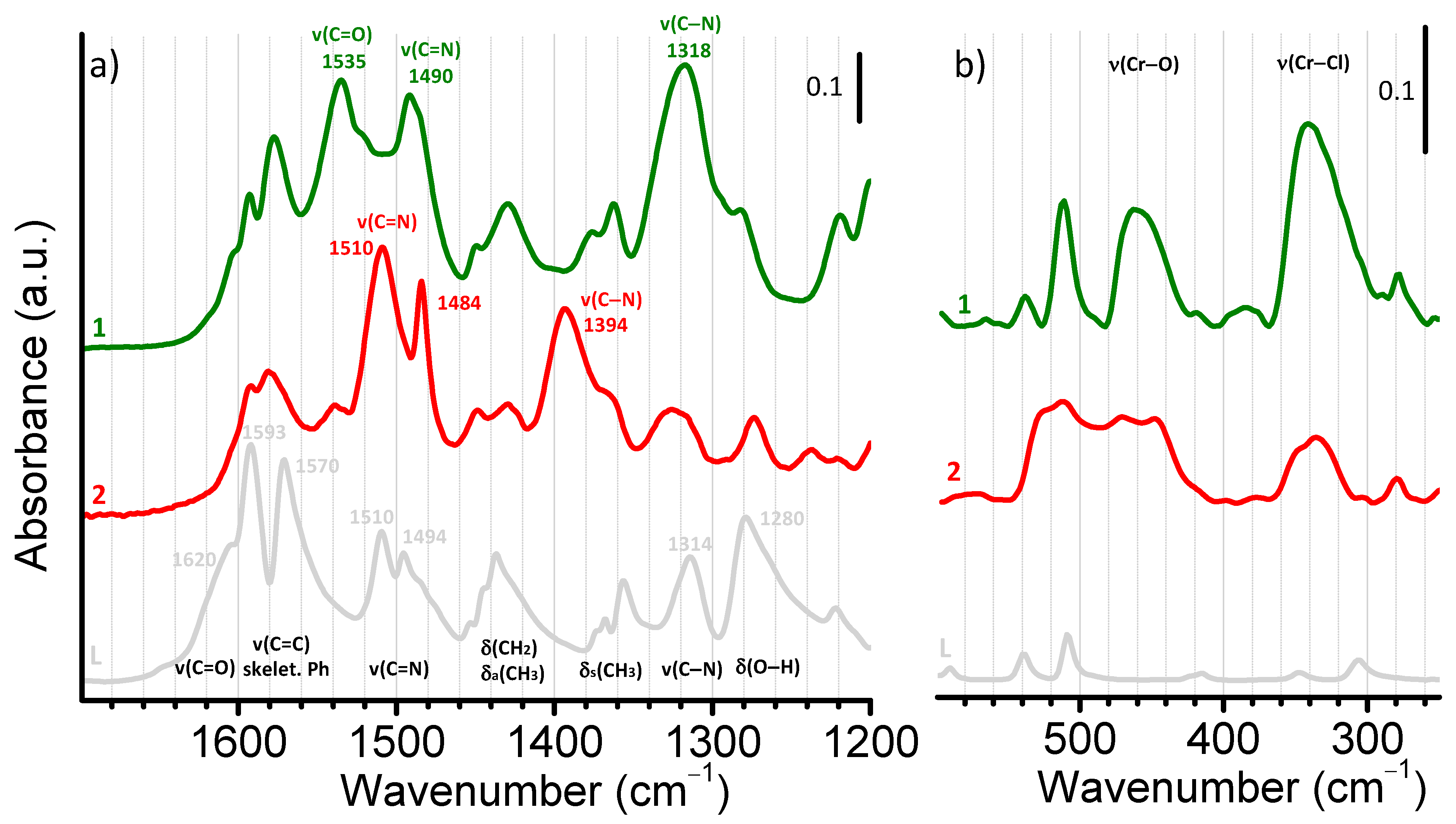
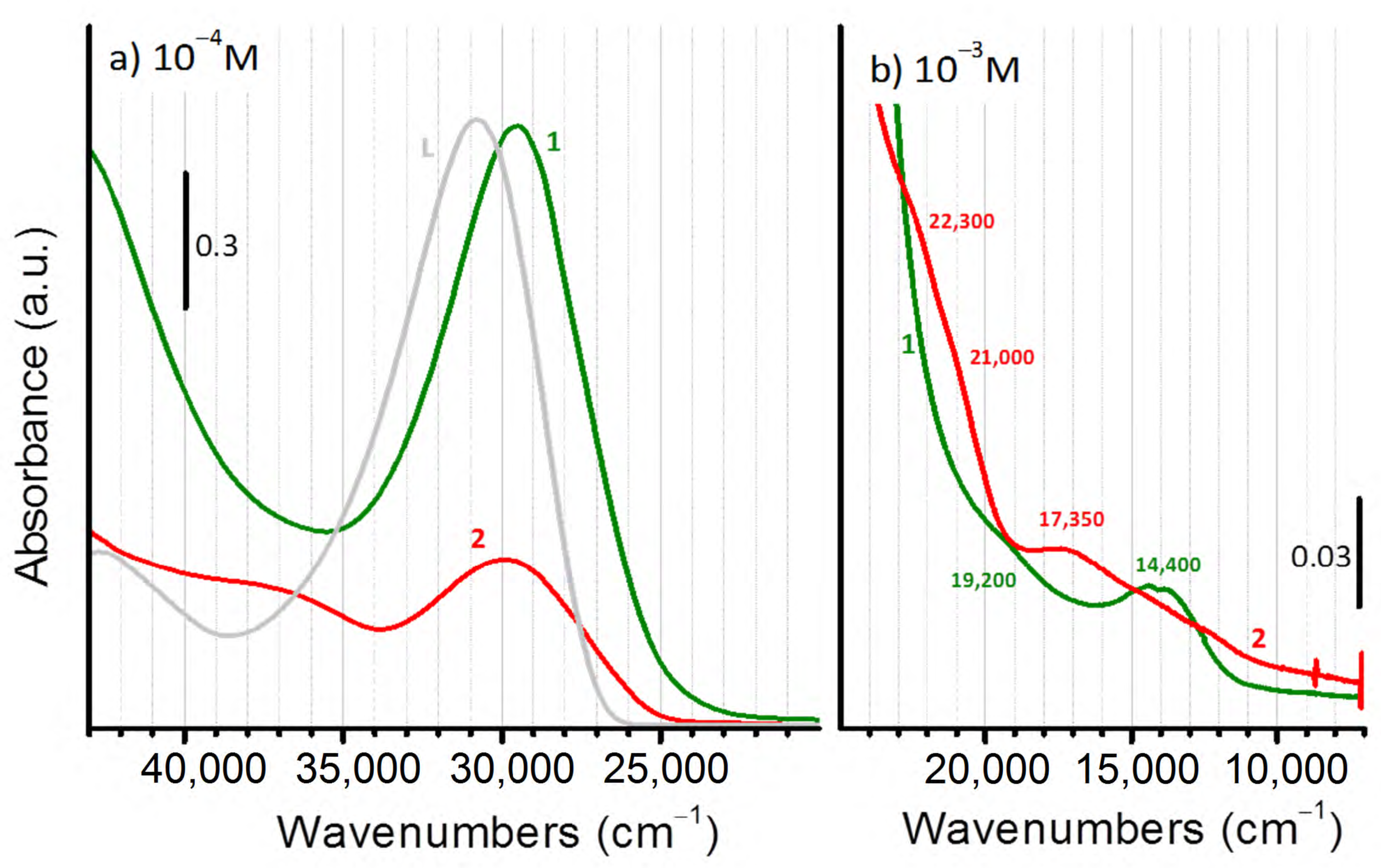
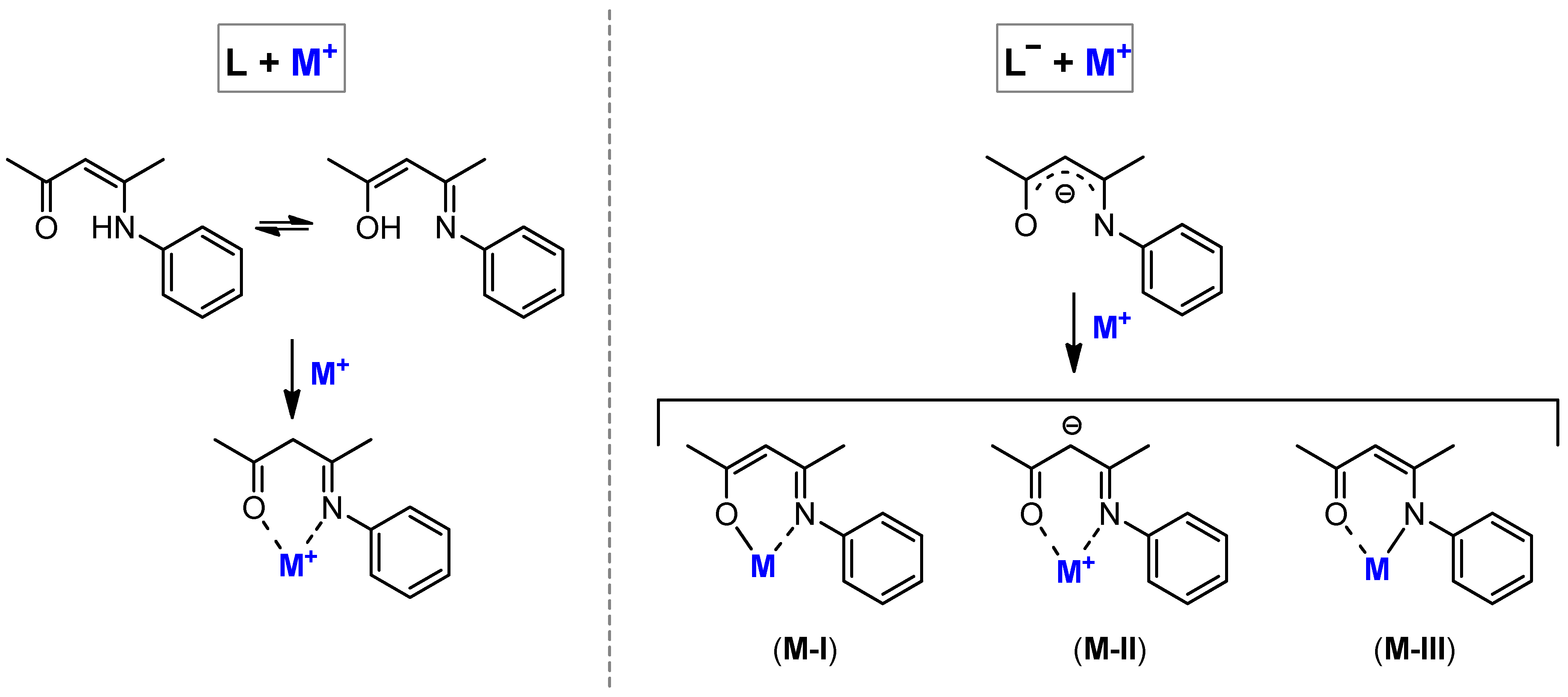
2.2. Characterization of the Bare Complexes
2.3. Polymerization Tests
2.3.1. Ethylene
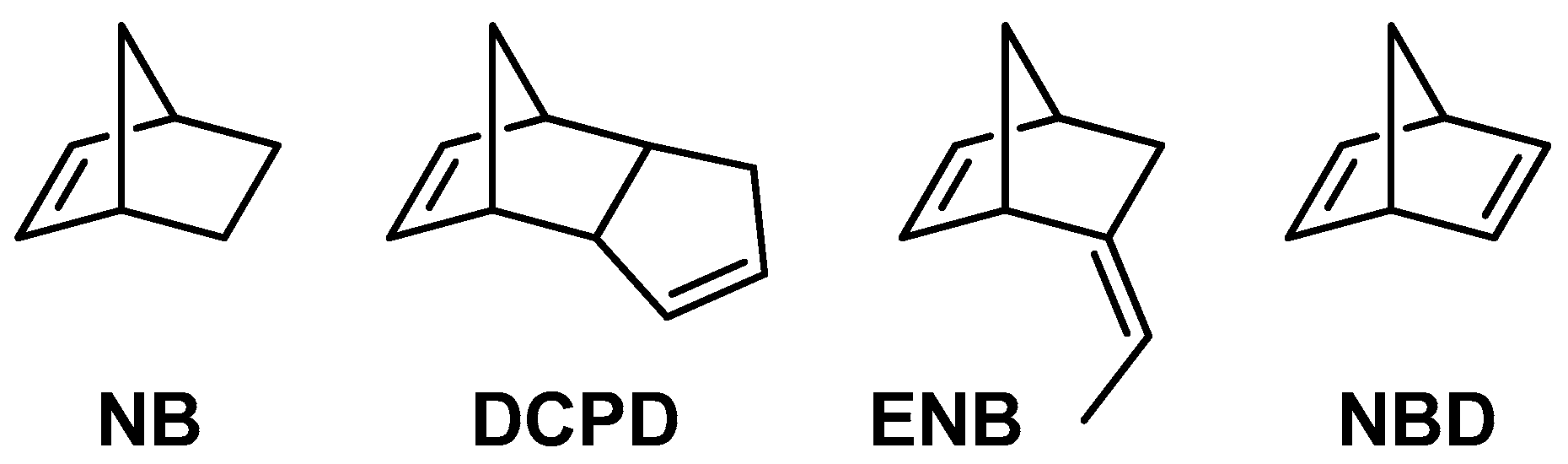
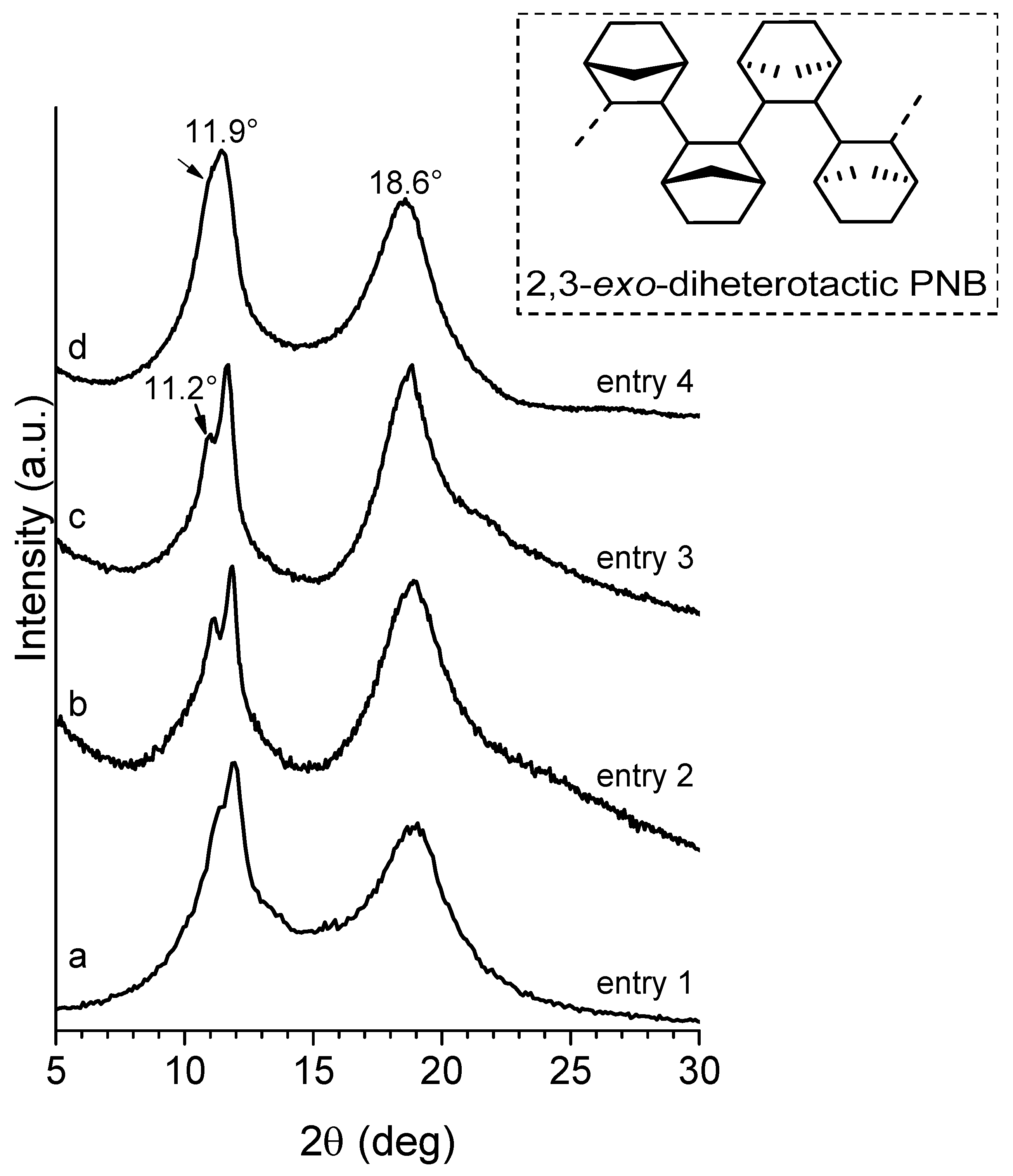
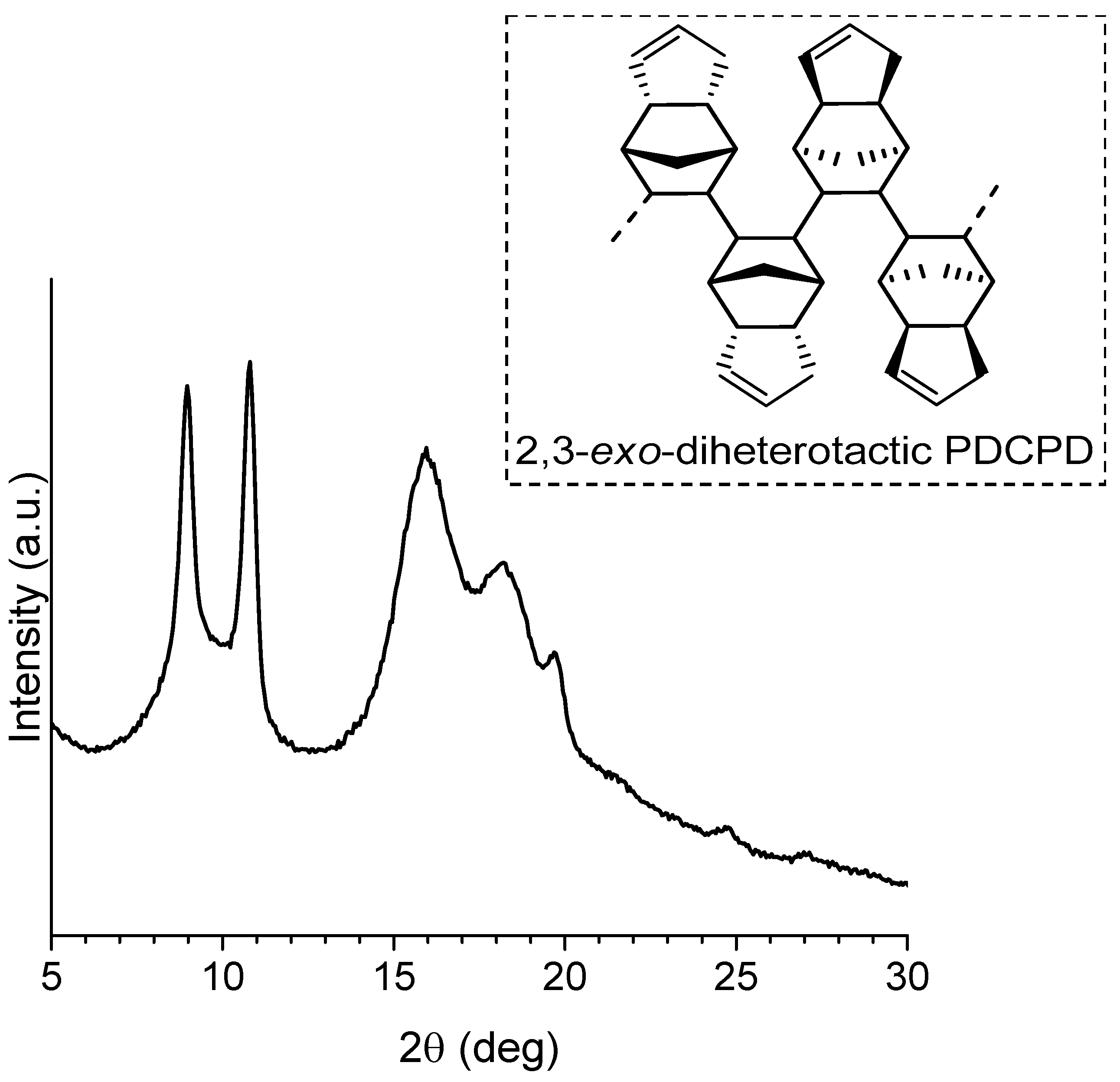
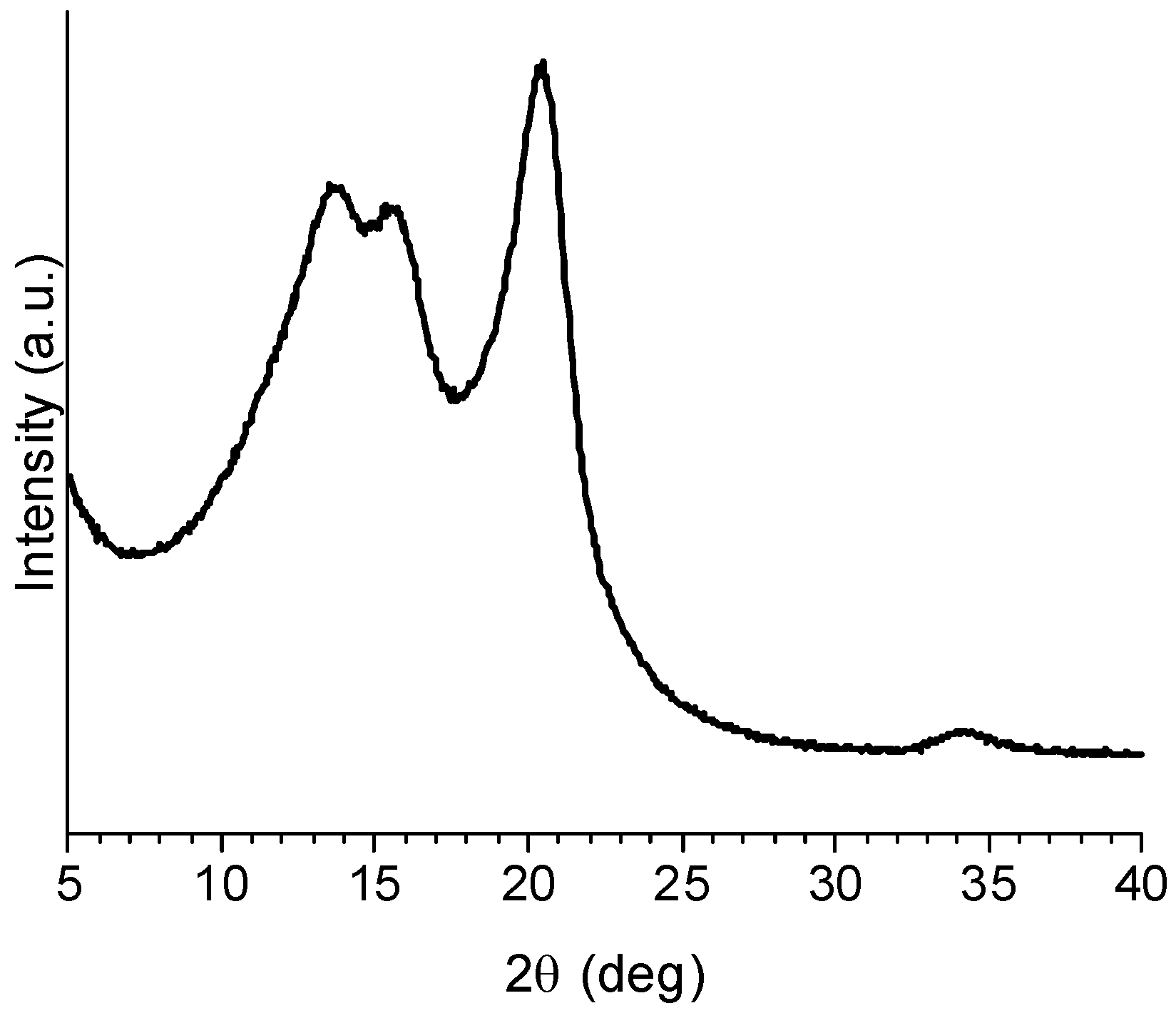
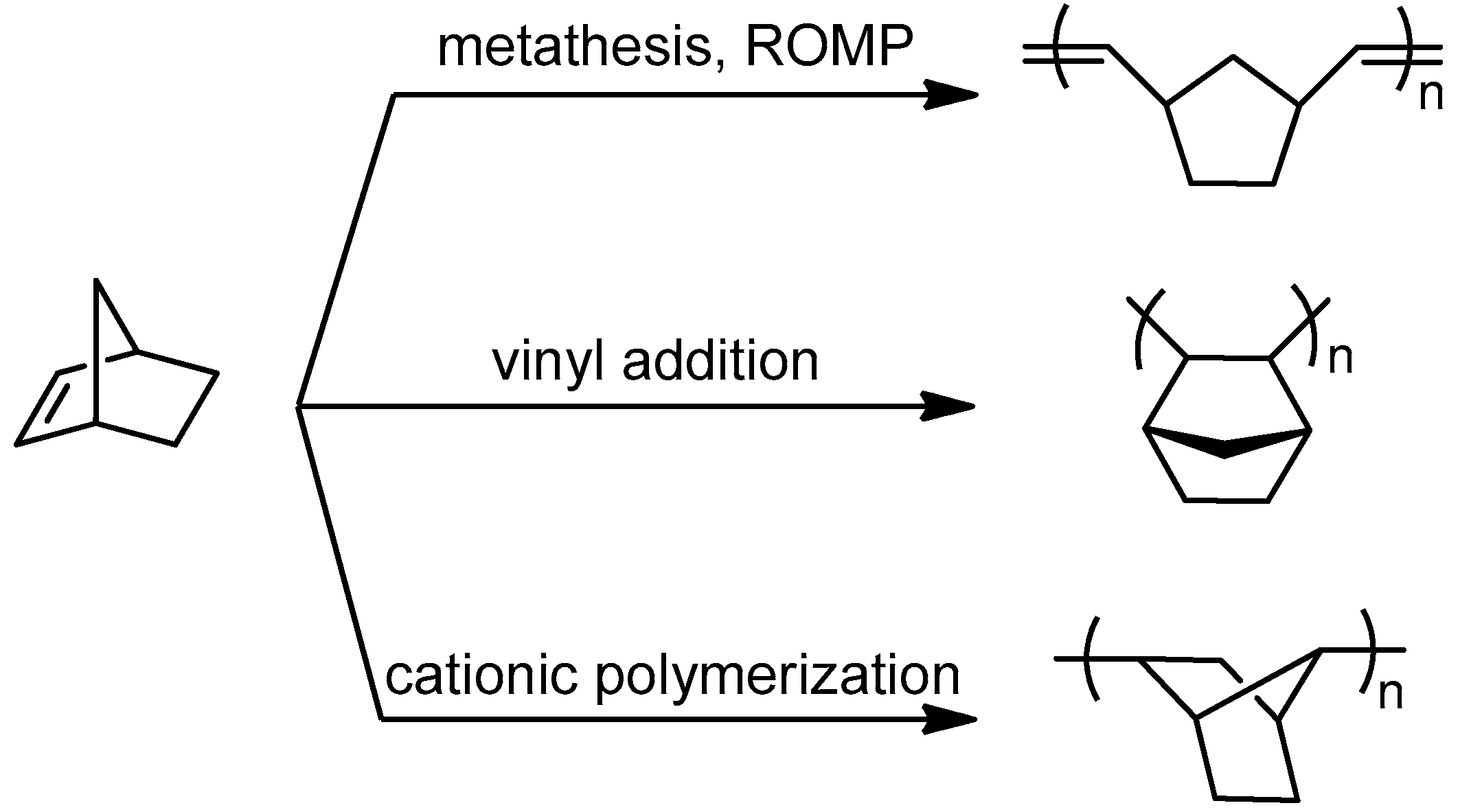
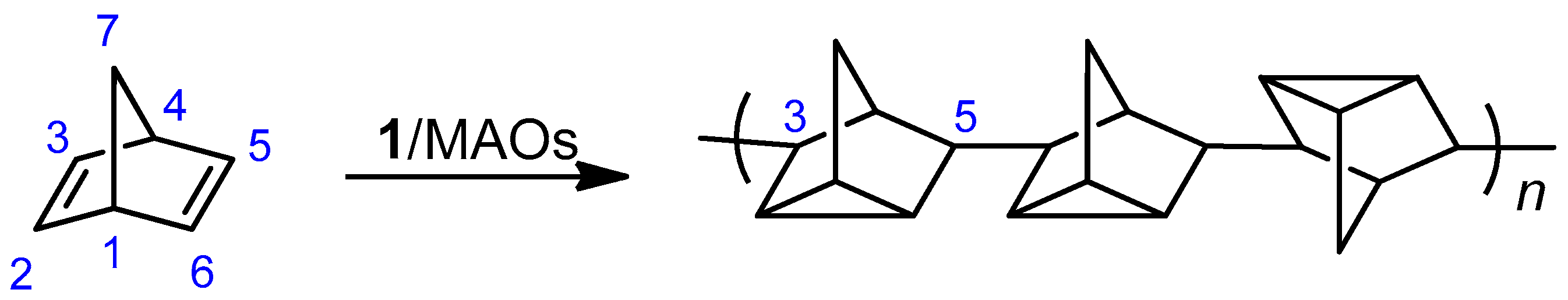
2.3.2. Cyclic Olefins
2.3.3. Cyclic Olefin Oligomers Characterization
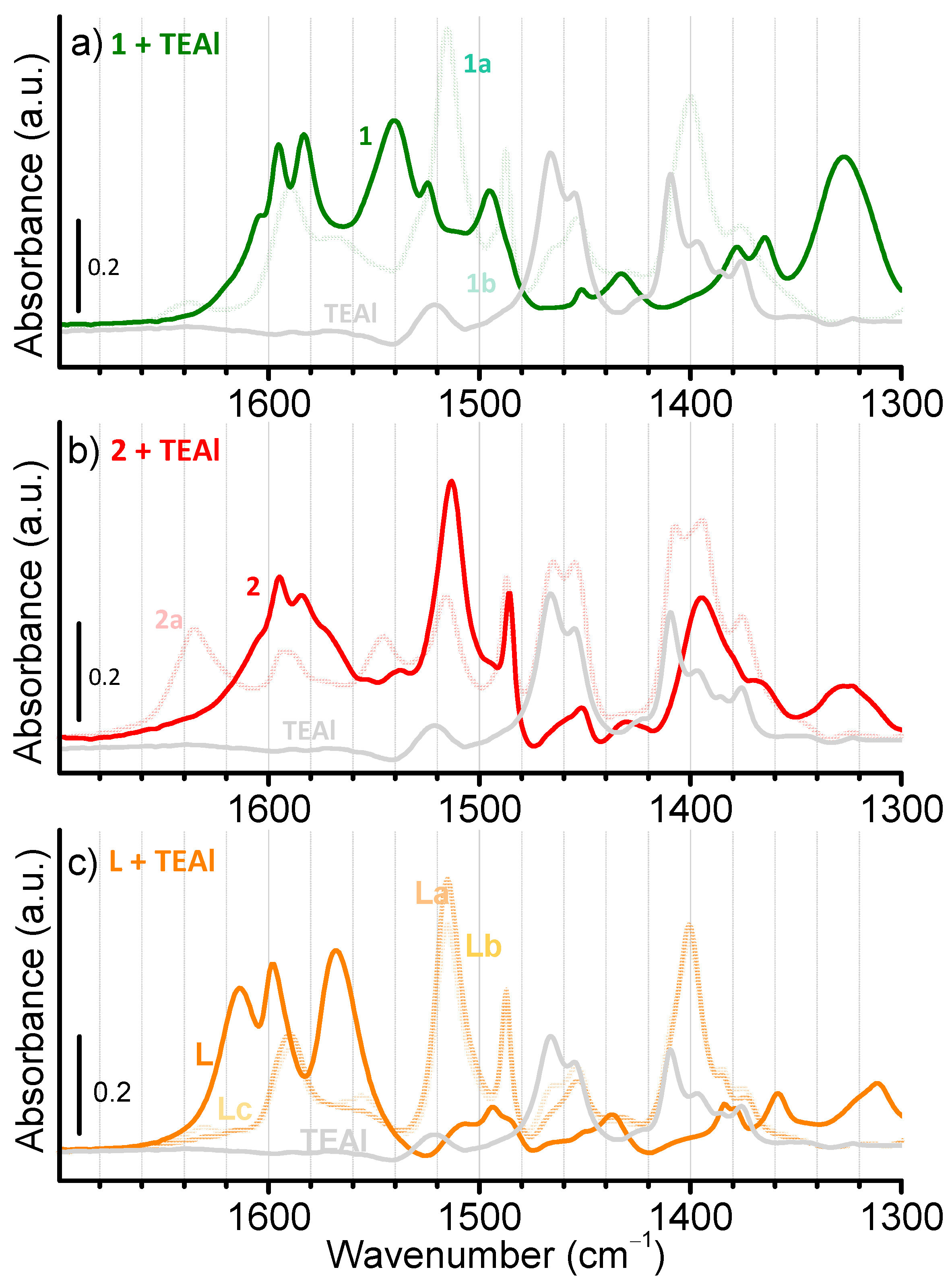
2.4. Characterization of the Activated Complexes
3. Materials and Methods
3.1. General Procedures and Material
3.2. Synthesis of the Ligand
3.3. Synthesis of the Cr complexes
3.3.1. Synthesis of Chromium Complex 1
3.3.2. Synthesis of Chromium Complex 2
3.4. Polymerization Procedure
3.5. Characterization Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holm, R.H.; Chakravorty, A.; Everett, G.W. Metal Complexes of Schiff Bases and β-Ketoamines. Prog. Inorg. Chem. 1966, 7, 83–214. [Google Scholar]
- Mehrotra, R.C.; Bohra, R.; Gaur, D.P. Metal β-Diketonates and Allied Derivatives; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef]
- Chamberlain, B.M.; Cheng, M.; Moore, D.R.; Ovitt, T.M.; Lobkovsky, E.B.; Coates, G.W. Polymerization of lactide with zinc and magnesium β-diiminate complexes: Stereocontrol and mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.G.; Piers, W.E.; McDonald, R. Cationic scandium methyl complexes supported by a β-diketiminato (“Nacnac”) ligand framework. J. Am. Chem. Soc. 2002, 124, 2132–2133. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Fevola, M.J.; Liable-Sands, L.M.; Rheingold, A.L.; Theopold, K.H. [(Ph)2nacnac]MCl2(THF)2 (M = Ti, V, Cr): A new class of homogeneous olefin polymerization catalysts featuring β-diiminate ligands. Organometallics 1998, 17, 4541–4543. [Google Scholar] [CrossRef]
- MacAdams, L.A.; Kim, W.K.; Liable-Sands, L.M.; Guzei, I.A.; Rheingold, A.L.; Theopold, K.H. The (Ph)2nacnac ligand in organochromium chemistry. Organometallics 2002, 21, 952–960. [Google Scholar] [CrossRef]
- Holland, P.L.; Tolman, W.B. Three-coordinate Cu(II) complexes: Structural models of trigonal-planar type 1 copper protein active sites. J. Am. Chem. Soc. 1999, 121, 7270–7271. [Google Scholar] [CrossRef]
- Basuli, F.; Bailey, B.C.; Tomaszewski, J.; Huffman, J.C.; Mindiola, D.J. A terminal and four-coordinate titanium alkylidene prepared by oxidatively induced α-hydrogen abstraction. J. Am. Chem. Soc. 2003, 125, 6052–6053. [Google Scholar] [CrossRef]
- Kogut, E.; Wiencko, H.L.; Zhang, L.; Cordeau, D.E.; Warren, T.H. A Terminal Ni(III)-Imide with Diverse Reactivity Pathways. J. Am. Chem. Soc. 2005, 127, 11248–11249. [Google Scholar] [CrossRef]
- MacLeod, K.C.; Vinyard, D.J.; Holland, P.L. A multi-iron system capable of rapid N2 formation and N2 cleavage. J. Am. Chem. Soc. 2014, 136, 10226–10229. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.; Chen, C.-H.; Pink, M.; Wu, G.; Mindiola, D.J. A Nitrido Salt Reagent of Titanium Scheme 1. Synthetic Protocol for the Parent Imido 2 and Nitride 3. J. Am. Chem. Soc. 2014, 136, 8197–8220. [Google Scholar] [CrossRef]
- Jang, E.S.; McMullin, C.L.; Käß, M.; Meyer, K.; Cundari, T.R.; Warren, T.H. Copper(II) anilides in sp3 C-H amination. J. Am. Chem. Soc. 2014, 136, 10930–10940. [Google Scholar] [CrossRef]
- Radwan, Y.K.; Maity, A.; Teets, T.S. Manipulating the Excited States of Cyclometalated Iridium Complexes with β-Ketoiminate and β-Diketiminate Ligands. Inorg. Chem. 2015, 54, 7122–7131. [Google Scholar] [CrossRef] [PubMed]
- Maya, R.A.; Maity, A.; Teets, T.S. Fluorination of Cyclometalated Iridium β-Ketoiminate and β-Diketiminate Complexes: Extreme Redox Tuning and Ligand-Centered Excited States. Organometallics 2016, 35, 2890–2899. [Google Scholar] [CrossRef]
- Lai, P.N.; Brysacz, C.H.; Alam, M.K.; Ayoub, N.A.; Gray, T.G.; Bao, J.; Teets, T.S. Highly Efficient Red-Emitting Bis-Cyclometalated Iridium Complexes. J. Am. Chem. Soc. 2018, 140, 10198–10207. [Google Scholar] [CrossRef] [PubMed]
- Yersin, H. (Hartmut) Highly Efficient OLEDs with Phosphorescent Materials; John Wiley & Sons: Hoboken, NJ, USA, 2007; p. 438. [Google Scholar]
- Mehrotra, R.C. Chemistry of metal β-diketonates. Pure Appl. Chem. 1988, 60, 1349–1356. [Google Scholar] [CrossRef]
- Gibson, D. Carbon-bonded beta-diketone complexes. Coord. Chem. Rev. 1969, 4, 225–240. [Google Scholar] [CrossRef]
- Arslan, E.; Lalancette, R.A.; Bernal, I. An historic and scientific study of the properties of metal(III) tris-acetylacetonates. Struct. Chem. 2016, 1, 201–212. [Google Scholar] [CrossRef]
- Latreche, S.; Schaper, F. Chromium(III) bis(diketiminate) complexes. Organometallics 2010, 29, 2180–2185. [Google Scholar] [CrossRef]
- Govil, N.; Jana, B. A review on aluminum, gallium and indium complexes of (Ph2-nacnac) ligand. Inorg. Chim. Acta 2021, 515, 120037. [Google Scholar] [CrossRef]
- Li, D.; Peng, Y.; Geng, C.; Liu, K.; Kong, D. Well-controlled ring-opening polymerization of cyclic esters initiated by dialkylaluminum β-diketiminates. Dalton Trans. 2013, 42, 11295–11303. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Dai, K.; Ye, W.P.; Pan, L.; Li, Y.S. New Titanium Complexes with Two β-Enaminoketonato Chelate Ligands: Syntheses, Structures, and Olefin Polymerization Activities. Organometallics 2004, 23, 1223–1230. [Google Scholar] [CrossRef]
- Altaf, C.T.; Wang, H.; Keram, M.; Yang, Y.; Ma, H. Aluminum methyl and isopropoxide complexes with ketiminate ligands: Synthesis, structural characterization and ring-opening polymerization of cyclic esters. Polyhedron 2014, 81, 11–20. [Google Scholar] [CrossRef]
- Jana, B.; Uhl, W. New aluminum and gallium complexes of β-diketiminato and β-ketiminato ligands. Inorg. Chim. Acta 2017, 455, 61–69. [Google Scholar] [CrossRef]
- Bera, S.K.; Panda, S.; Baksi, S.D.; Lahiri, G.K. Redox Non-Innocence and Isomer-Specific Oxidative Functionalization of Ruthenium-Coordinated β-Ketoiminate. Chem. Asian J. 2019, 14, 4236–4245. [Google Scholar] [CrossRef] [PubMed]
- Gibson, V.C.; Newton, C.; Redshaw, C.; Solan, G.A.; White, A.J.P.; Williams, D.J. Synthesis, structures and ethylene polymerisation behaviour of low valent β-diketiminato chromium complexes. Eur. J. Inorg. Chem. 2001, 7, 1895–1903. [Google Scholar] [CrossRef]
- Yoon, S.; Teets, T.S. Red to near-infrared phosphorescent Ir(iii) complexes with electron-rich chelating ligands. Chem. Commun. 2021, 57, 1975–1988. [Google Scholar] [CrossRef]
- Leone, G.; Groppo, E.; Zanchin, G.; Martino, G.A.; Piovano, A.; Bertini, F.; Martí-Rujas, J.; Parisini, E.; Ricci, G. Concerted Electron Transfer in Iminopyridine Chromium Complexes: Ligand Effects on the Polymerization of Various (Di)olefins. Organometallics 2018, 37, 4827–4840. [Google Scholar] [CrossRef]
- Zanchin, G.; Piovano, A.; Amodio, A.; De Stefano, F.; Di Girolamo, R.; Groppo, E.; Leone, G. NEt3-Triggered Synthesis of UHMWPE Using Chromium Complexes Bearing Non-Innocent Iminopyridine Ligands. Macromolecules 2021, 54, 1243–1253. [Google Scholar] [CrossRef]
- Bariashir, C.; Huang, C.; Solan, G.A.; Sun, W.H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization. Coord. Chem. Rev. 2019, 385, 208–229. [Google Scholar] [CrossRef] [Green Version]
- Cotton, F.A.; Ilsley, W.H.; Kaim, W. Chelate rings in a quadruply bonded dimetal compound: Preparation and structure of diacetatodi (4-phenylimino-2-pentanonato)dimolybdenum. Inorg. Chim. Acta 1979, 37, 267–272. [Google Scholar] [CrossRef]
- Ueno, K.; Martell, A.E. Infrared Study of Metal Chelates of Bisacetylacetone-Ethylidenediimine and Related Compounds. J. Phys. Chem. 1955, 59, 998–1004. [Google Scholar] [CrossRef]
- Camp, C.; Arnold, J. On the non-innocence of “Nacnacs”: Ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Martell, A.E.; Nakamoto, K. Infrared spectra of metal chelate compounds. VI. A normal coordinate treatment of oxalato metal complexes. J. Chem. Phys. 1962, 36, 324–331. [Google Scholar] [CrossRef]
- Ferraro, J.R. Low-Frequency Vibrations of Inorganic and Coordination Compounds; Springer: Boston, MA, USA, 1971. [Google Scholar]
- Chatt, J.; Hayter, R.G. Ligand Field Strengths of the Halide, Methyl, Phenyl, and Hydride Anions. J. Chem. Soc. 1961, 167, 772–774. [Google Scholar] [CrossRef]
- Figgis, B.N. Introduction to Ligand Fields; John Wiley & Sons, Ltd.: New York, NY, USA, 1966. [Google Scholar]
- Bermeshev, M.V.; Chapala, P.P. Addition polymerization of functionalized norbornenes as a powerful tool for assembling molecular moieties of new polymers with versatile properties. Prog. Polym. Sci. 2018, 84, 1–46. [Google Scholar] [CrossRef]
- Zanchin, G.; Leone, G. Polyolefin thermoplastic elastomers from polymerization catalysis: Advantages, pitfalls and future challenges. Prog. Polym. Sci. 2021, 113, 101342. [Google Scholar] [CrossRef]
- Rapallo, A.; Porzio, W.; Zanchin, G.; Ricci, G.; Leone, G. 2,3-exo-Diheterotactic Dicyclopentadiene Oligomers: An X-ray Powder Diffraction Study of a Challenging Multiphase Case. Chem. Mater. 2019, 31, 6650–6664. [Google Scholar] [CrossRef]
- Blank, F.; Janiak, C. Metal catalysts for the vinyl/addition polymerization of norbornene. Coord. Chem. Rev. 2009, 253, 827–861. [Google Scholar] [CrossRef]
- Ricci, G.; Boglia, A.; Boccia, A.C.; Zetta, L.; Famulari, A.; Meille, S.V. New stereoregularity in the stereospecific polymerization of bulky strained olefins: Diheterotactic polynorbornene. Macromolecules 2008, 41, 3109–3113. [Google Scholar] [CrossRef]
- Zanchin, G.; Leone, G.; Pierro, I.; Rapallo, A.; Porzio, W.; Bertini, F.; Ricci, G. Addition Oligomerization of Dicyclopentadiene: Reactivity of Endo and Exo Isomers and Postmodification. Macromol. Chem. Phys. 2017, 218, 1600602. [Google Scholar] [CrossRef]
- Arndt, M.; Engehausen, R.; Kaminsky, W.; Zoumis, K. Hydrooligomerization of cycloolefins -a view of the microstructure of polynorbornene. J. Mol. Catal. A Chem. 1995, 101, 171–178. [Google Scholar] [CrossRef]
- Roller, M.B.; Gillham, J.K.; Kennedy, J.P. Thermomechanical Behavior of a Polynorbornadiene. J. Appl. Polym. Sci. 1973, 17, 2223–2233. [Google Scholar] [CrossRef]
- De Rosa, C.; Malafronte, A.; Auriemma, F.; Scoti, M.; Di Girolamo, R.; D’Alterio, M.C.; Ricci, G.; Zanchin, G.; Leone, G. Synthesis, chain conformation and crystal structure of poly(norbornadiene) having repeating 3,5-enchained nortricyclene units. Polym. Chem. 2019, 10, 4593–4603. [Google Scholar] [CrossRef]
- Fabian, J.; Legrand, M.; Poirier, P. L’etude spectrographique infrarouge et Raman du groupe imine. Bull. Soc. Chim. Fr. 1956, 10, 1499–1509. [Google Scholar]
- Huang, Y.B.; Jin, G.X. Half-sandwich chromium(III) complexes bearing β-ketoiminato and β-diketiminate ligands as catalysts for ethylene polymerization. Dalton Trans. 2009, 5, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Piovano, A.; Zarupski, J.; Groppo, E. Disclosing the Interaction between Carbon Monoxide and Alkylated Ti3+ Species: A Direct Insight into Ziegler-Natta Catalysis. J. Phys. Chem. Lett. 2020, 11, 5632–5637. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
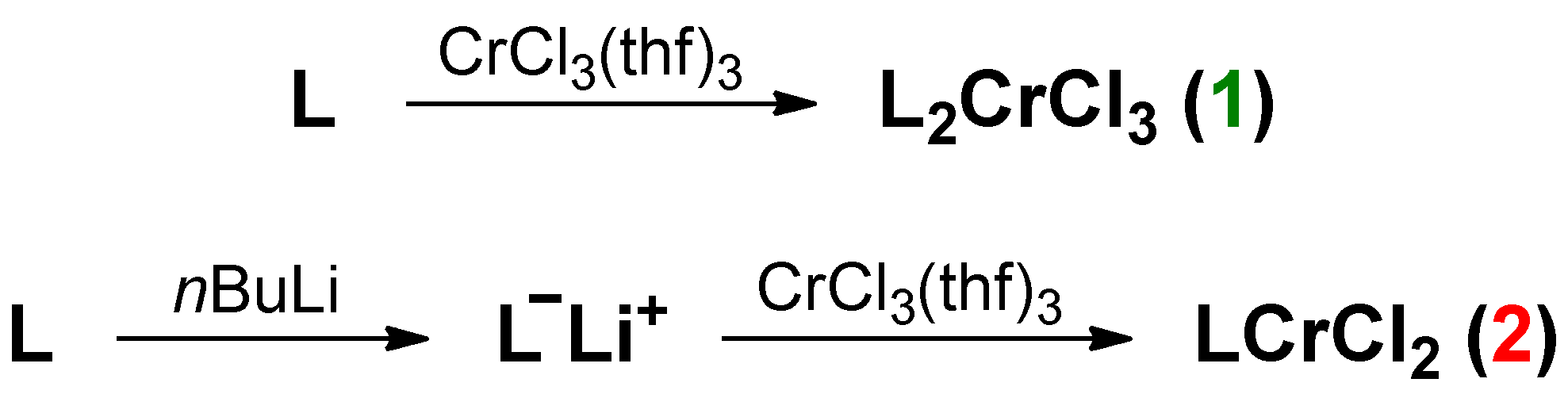{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | Complex | T | Yield | Mw b | Mn b | Mw/Mnb | |
|---|---|---|---|---|---|---|---|---|
| (°C) | (g) | (%) | (g mol−1) | (g mol−1) | ||||
| 1 | NB | 1 | 20 | 1.05 | 78 | 1790 | 1055 | 1.7 |
| 2 | NB | 2 | 20 | 1.00 | 74 | 1740 | 1210 | 1.4 |
| 3 | NB | 1 | 0 | 1.20 | 89 | 1990 | 1080 | 1.8 |
| 4 | NB | 2 | 0 | 1.20 | 89 | 2330 | 1385 | 1.7 |
| 5 | DCPD | 1 | 20 | 0.32 | 13 | 1140 | 880 | 1.3 |
| 6 | DCPD | 2 | 20 | 0.30 | 13 | 1140 | 855 | 1.3 |
| 7 | ENB | 1 | 20 | 1.98 | 92 | 2320 | 1420 | 1.6 |
| 8 c | NBD | 1 | 20 | 1.21 | 73 | insoluble | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amodio, A.; Zanchin, G.; De Stefano, F.; Piovano, A.; Palucci, B.; Guiotto, V.; Di Girolamo, R.; Leone, G.; Groppo, E. Cr(III) Complexes Bearing a β-Ketoimine Ligand for Olefin Polymerization: Are There Differences between Coordinative and Covalent Bonding? Catalysts 2022, 12, 119. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020119
Amodio A, Zanchin G, De Stefano F, Piovano A, Palucci B, Guiotto V, Di Girolamo R, Leone G, Groppo E. Cr(III) Complexes Bearing a β-Ketoimine Ligand for Olefin Polymerization: Are There Differences between Coordinative and Covalent Bonding? Catalysts. 2022; 12(2):119. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020119
Chicago/Turabian StyleAmodio, Alessia, Giorgia Zanchin, Fabio De Stefano, Alessandro Piovano, Benedetta Palucci, Virginia Guiotto, Rocco Di Girolamo, Giuseppe Leone, and Elena Groppo. 2022. "Cr(III) Complexes Bearing a β-Ketoimine Ligand for Olefin Polymerization: Are There Differences between Coordinative and Covalent Bonding?" Catalysts 12, no. 2: 119. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020119