
Organocatalysis for the Asymmetric Michael Addition of Aldehydes and α,β-Unsaturated Nitroalkenes


1
Department of Anatomy, Korea University College of Medicine, 46, Gaeunsa 2-gil, Seongbuk-gu, Seoul 02842, Korea
2
Department of Chemistry, Korea University, 145 Anam-ro Seongbuk-gu, Seoul 02841, Korea
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(2), 121; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020121
Submission received: 29 December 2021
/
Revised: 14 January 2022
/
Accepted: 17 January 2022
/
Published: 20 January 2022
(This article belongs to the Topic Catalysis for Sustainable Chemistry and Energy)
Abstract
:Michael addition is an important reaction because it can be used to synthesize a wide range of natural products or complex compounds that exhibit biological activities. In this study, a mirror image of an aldehyde and α,β-unsaturated nitroalkene were reacted in the presence of (R,R)-1,2-diphenylethylenediamine (DPEN). Herein, thiourea was introduced as an organic catalyst, and a selective Michael addition reaction was carried out. The primary amine moiety of DPEN reacts with aldehydes to form enamines, which is activated by the hydrogen bond formation between the nitro groups of α,β-unsaturated nitroalkenes and thiourea. Our aim was to obtain an asymmetric Michael product by adding 1,4-enamine to an alkene to form a new carbon–carbon bond. As a result, the primary amine of the chiral diamine was converted to an enamine. The reaction proceeded with a relatively high degree of enantioselectivity, which was achieved using double activation via hydrogen bonding of the nitro group and thiourea. Michael products with a high degree of enantioselectivity (97–99% synee) and diastereoselectivity (syn/anti = 9/1) were obtained in yields ranging from 94–99% depending on the aldehydes.
1. Introduction
In understanding the basic properties of a molecule, the spatial arrangement of atoms in a molecule has a significant meaning. Therefore, over the past few decades, organic chemists have put a lot of effort into developing stereoselective organic reactions. Organic reactions capable of efficiently controlling the stereochemistry of the products are not only desirable in academics but also in industries. Until recently, asymmetric synthesis involved a significant amount of metal-bonded catalysts. However, in the field of organocatalysis, many studies have focused on catalysts that do not contain metals [1,2,3,4,5,6,7,8,9,10,11,12]. Metals are often expensive, and metal ions contain air and moisture. Moreover, they are often unstable in the reaction environment. Furthermore, when a metal catalyst is used in a reaction, a small amount of it may remain in the product. To overcome these shortcomings, research on stereoselective synthesis using organic catalysts has attracted increasing attention [13,14,15,16,17,18,19,20,21,22].
In 2004, Barbas et al. reported a pyrrolidine-catalyzed Michael addition reaction involving an aldehyde, α,β-unsaturated nitroalkene, and trifluoroacetic acid [23]. In 2006, Tang et al. conducted the asymmetric Michael addition of isobutyraldehyde, pyrrolidine, and α,β-unsaturated nitroalkene using a bifunctional thiourea derivative as a catalyst [24]. In 2007, Connon et al. performed this reaction without using a solvent and obtained high yield and enantioselectivity by using a cinchona alkaloid derivative as a catalyst [25]. In addition, the case of Yan et al. was observed that the reaction proceeded only when a primary amine was present in the catalyst and that the reaction did not proceed in the absence of the primary amine [26]. In 2010, Chen et al. conducted this reaction only with a catalyst without additives and obtained high enantioselectivity; here, a pyrolidine derivative made from camphor was used as a catalyst. Consequently, a high diastereoselectivity was achieved [27].
As mentioned above, various reactions have been used to study organic catalysts [28,29,30]. However, to design an eco-friendly organocatalyzed reaction, many studies have focused on organic catalyst reactions that do not use solvents or use water. Therefore, we researched eco-friendly conditions for organic catalysts and applied thiourea catalysts derived from (R,R)-1,2-diphenylethylene diamine (DPEN) to various reactions.
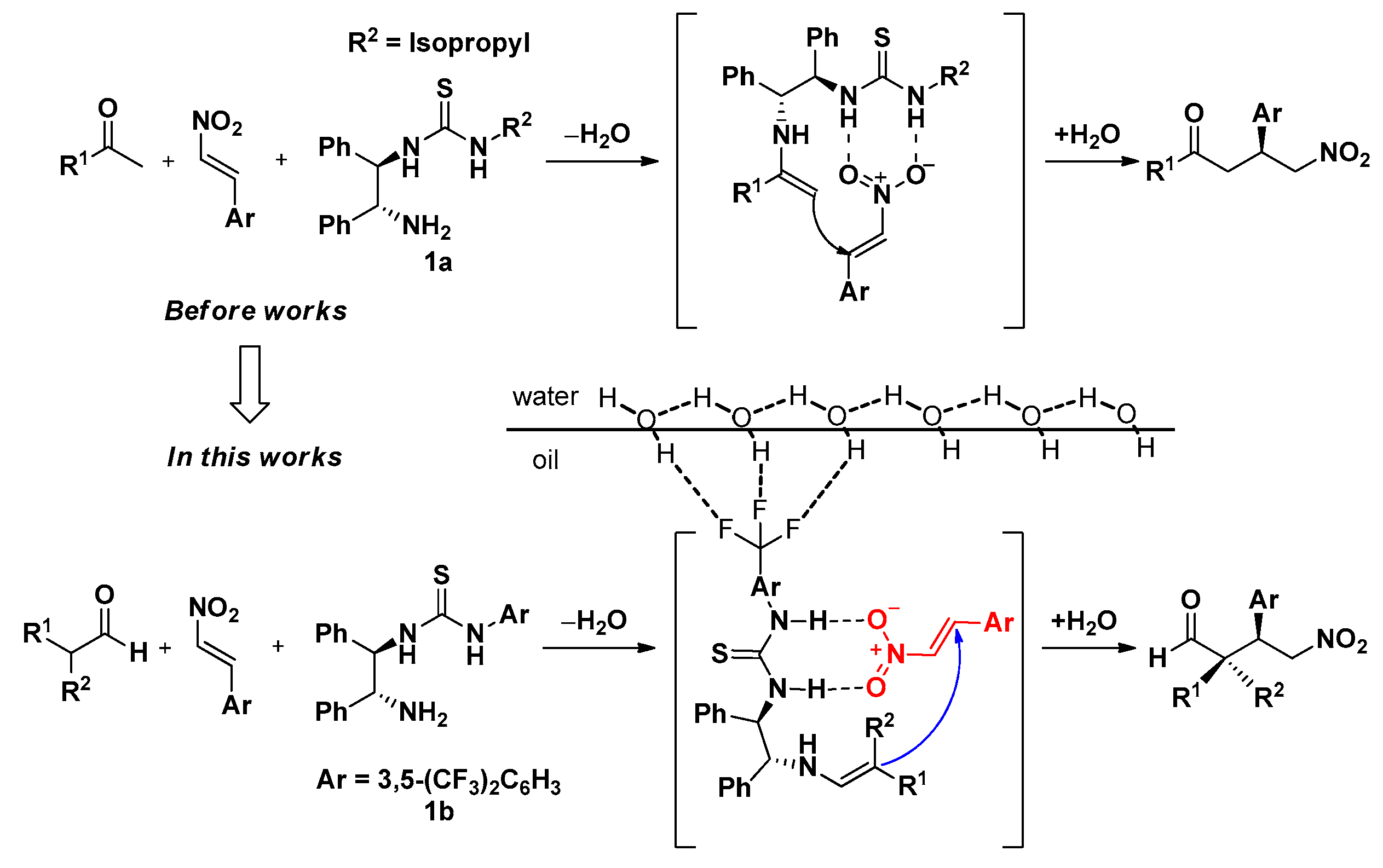
In this study, thiourea was introduced using (R,R)-1,2-diphenylethylenediamine, which was used as the basic skeleton of a chiral catalyst in previous studies [31,32,33,34]. Herein, a stereoselective Michael addition reaction was carried out using nitrostyrene, in which the nitro group acts as a strong electron-withdrawing electrophile, and the aldehyde acts as a nucleophile [6,30,35,36]. In addition, the reaction was carried out using water as a solvent for studying the eco-friendly organic catalysts (Figure 1).
In addition, the interfacial reaction mechanism of the DPEN-based catalyst and the factors that increase the catalytic reactivity were confirmed through quantum calculations under the reaction conditions without the acid additive used in the previous study. The study on the reaction mechanism according to the type of configurational diastereomers [37] and the reactivity of each solvent was verified through quantum calculation. In addition, the effect of the fluorine substituent on the organic catalyst 1b confirmed in the previous study was confirmed in more depth [33,34].
2. Results and Discussion
2.1. Asymmetric Michael Reaction of Various Aldehydes and α,β-Unsaturated Nitroalkenes Using a Thiourea Catalyst
2.1.1. Effect of the DPEN Catalyst, Temperature, and Equiv.
To investigate the effect of the catalyst on the enantioselective Michael addition of aldehydes and nitroalkynes, the reaction was performed using isobutyraldehyde and trans-beta-nitrostyrene. The basic skeleton of (R,R)-1,2-diphenylethylenediamine (DPEN) was used as the catalyst, and Michael addition was first performed using a catalyst in which one amine was substituted with 3-pentyl group and the other amine was substituted with thiourea. Subsequently, the reaction was performed using a thiourea catalyst that was not substituted with an amine (Figure 2).
To investigate the effect of the catalyst, first, a thiourea catalyst in which only one amine group was substituted with an alkyl group was used; toluene was used as the solvent, and the reaction was performed at ambient temperature (Table 1). The product of the addition reaction was not obtained, and it was observed that the catalyst (1h), in which the amine was substituted with an alkyl group, was not effective. When the reaction was performed using a thiourea catalyst without an alkyl group in the amine, it was confirmed that the reaction proceeded. Among all the catalysts, the thiourea catalyst substituted with the electron-donating para-methoxy group (1e) and that substituted with the electron-withdrawing para-fluorine (1f) group were evaluated. Subsequently, the yields and stereoselectivities of the two catalysts were compared, and the catalyst substituted with the electron-withdrawing para-fluorine (1f) group achieved higher yield and stereoselectivity. When the catalyst was substituted with an electron-withdrawing group, the hydrogen of thiourea involved in hydrogen bonding was more likely to participate in hydrogen bonding than that of the catalyst substituted with an electron-releasing group; thus, the stereoselectivity of the former was higher. The highest yield and stereoselectivity were achieved using the 3,5-bis(trifluoromethy- l)-substituted catalyst (1b). After confirming that this catalyst (1b) yielded the highest stereoselectivity, the reaction was performed at a lower temperature to further enhance the effect of the catalyst. It was confirmed that the reaction at low temperature exhibited a similar stereoselectivity to that of the reaction at room temperature; however, the yield was lower. Thus, it was confirmed that the 1b catalyst afforded the highest reactivity and stereoselectivity at ambient temperature (Table 1).
It was confirmed that the stereoselectivity did not depend on the equivalent weight of the aldehyde and the amount of the catalyst. However, these parameters affected the yield of the reaction. As the amount of aldehyde was decreased from 10 equivalents to 7 equivalents and 5 equivalents, there was no change in the stereoselectivity; however, the yield decreased. Subsequently, after fixing the aldehyde at 10 equivalents, when 5 mol% of the catalyst was added, both the yield and stereoselectivity were lower than when 10 mol% of the catalyst was used. The best results were obtained when 10 equivalents of aldehyde and 10 mol% of the catalyst were used. However, to reduce the amount of the aldehyde and catalyst, the solvent effect was confirmed by fixing the aldehyde to 5 equivalents and the amount of catalyst to 10 mol%.
2.1.2. Solvent Effect on the Reaction
From the previous experiment, it was observed that the highest enantioselectivity was achieved when the 3,5-bis(trifluoromethyl)-substituted catalyst (Figure 2, 1b) was used. Using this catalyst, it was confirmed that the reactivity and enantioselectivity depended on the solvent (Table 2). The reaction proceeded in all the solvents, and desirable yields were obtained with all the solvents except with hexane and tetrahydrofuran. In all the solvents, more than 96% stereoselectivity was obtained. In particular, when water was used as the solvent, the reaction proceeded for 4 h. Thus, water, which produced the highest yield and stereoselectivity and required the highest reaction time, was selected as the solvent.
2.1.3. Effects of the Types of α,β-Unsaturated Nitroalkenes on the Reaction
The reaction of isobutyraldehyde and various other α,β-unsaturated nitroalkenes was performed under the optimal conditions determined in the previous experiment. Here, a slightly lower yield than that achieved using unsubstituted nitrostyrene was obtained. Moreover, the reactions of the electron-withdrawing as well as the electron-donating groups exhibited desirable stereoselectivity and yield. Thus, it was confirmed that among the aromatic substituents, both the electron-withdrawing and electron-donating groups afforded desirable yields and stereoselectivity (Table 3).
2.1.4. Reaction Effect Depending on the Type of Aldehyde
In the previous experiment, the optimal conditions for the enantioselective Michael addition of isobutyraldehyde and nitroalkene were investigated. The reactions of various aldehydes and nitrostyrenes were conducted under the optimized conditions. In the case of propionaldehyde, the enantioselectivity was high; however, the diastereomeric selectivity was low. In subsequent experiments, entries 2 and 3 of Table 4 exhibited higher diastereoselectivity than that of propionaldehyde. Entry 4 exhibited desirable diastereomeric selectivity and mirror image selectivity. It was confirmed that the larger the alkyl group of the aldehyde, the higher the diastereomeric selectivity due to steric hindrance (Table 4).
2.1.5. Reaction Mechanism Inferred through Expected Transition States
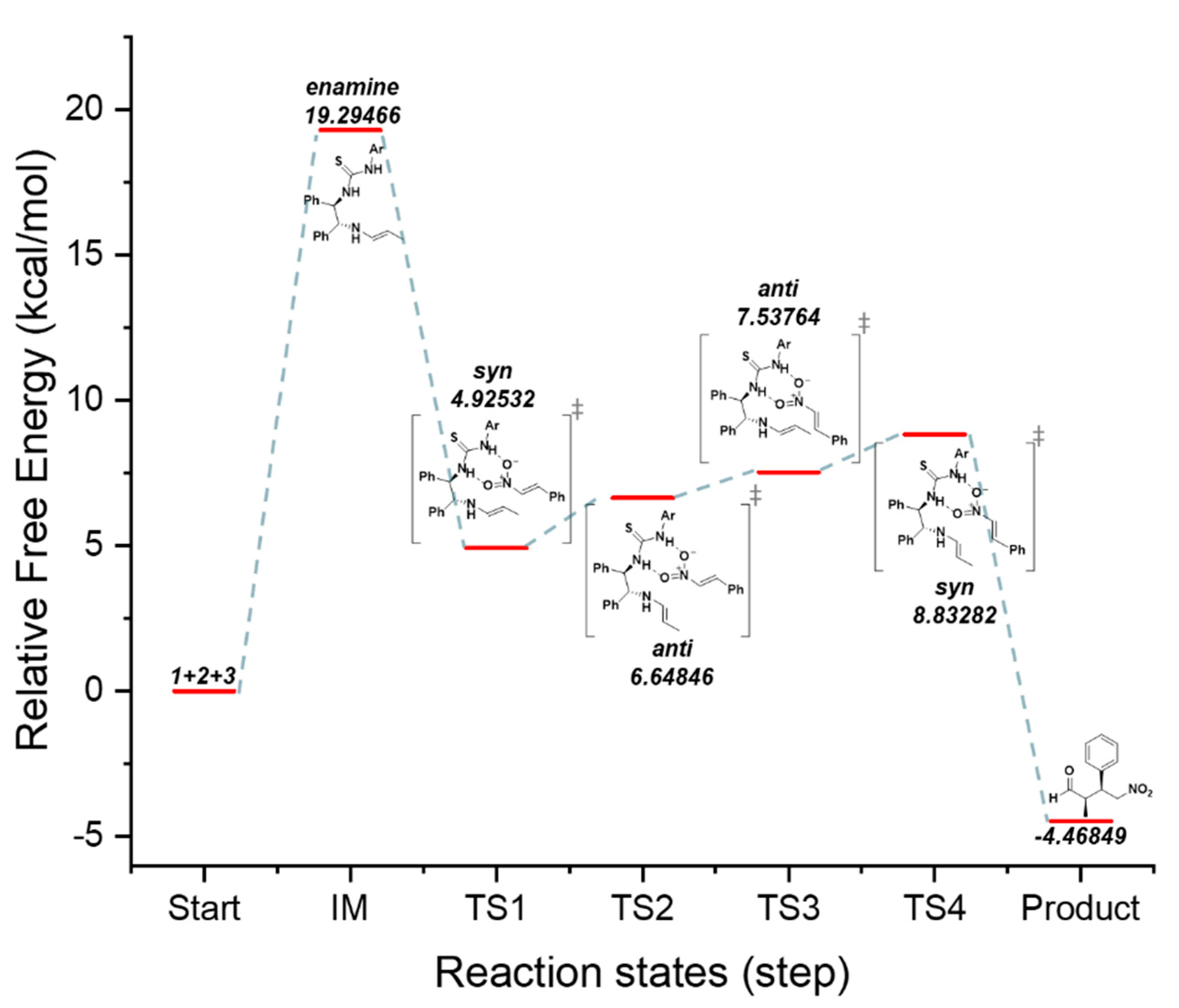
As shown in Figure 3, the expected energy of transition state 1 (TS1 syn major) was confirmed through DFT (Density Functional Theory) calculations. The calculated results show that, compared to TS2 (anti major), TS3 (anti minor), and TS4 (syn minor), the syn structure of TS 1 is the most stable based on its respective Gibbs free energy. In addition, minor TS 4 showed the highest Gibbs free energy than other TS structures. Moreover, as shown in Figure 4, the aldehyde reacts with the primary amine of the catalyst to pass through the imine form [38] to finally form the enamine [31]. TS 1 and TS 3 exhibit less steric hindrance because the double bond and thiourea are located on the same side. Furthermore, the hydrogen bonding between the nitro group of the nitroalkene and the thiourea of the catalyst causes the aromatic substituents on the alkene to be positioned like those in TS1 and TS2 with relatively lower steric hindrance. Thus, the nucleophilic enamine accesses the electrophile from the bottom, and the syn(2R, 3S) form is predicted to be predominantly produced. The above reaction focuses on the hydrogen bond between thiourea and water.
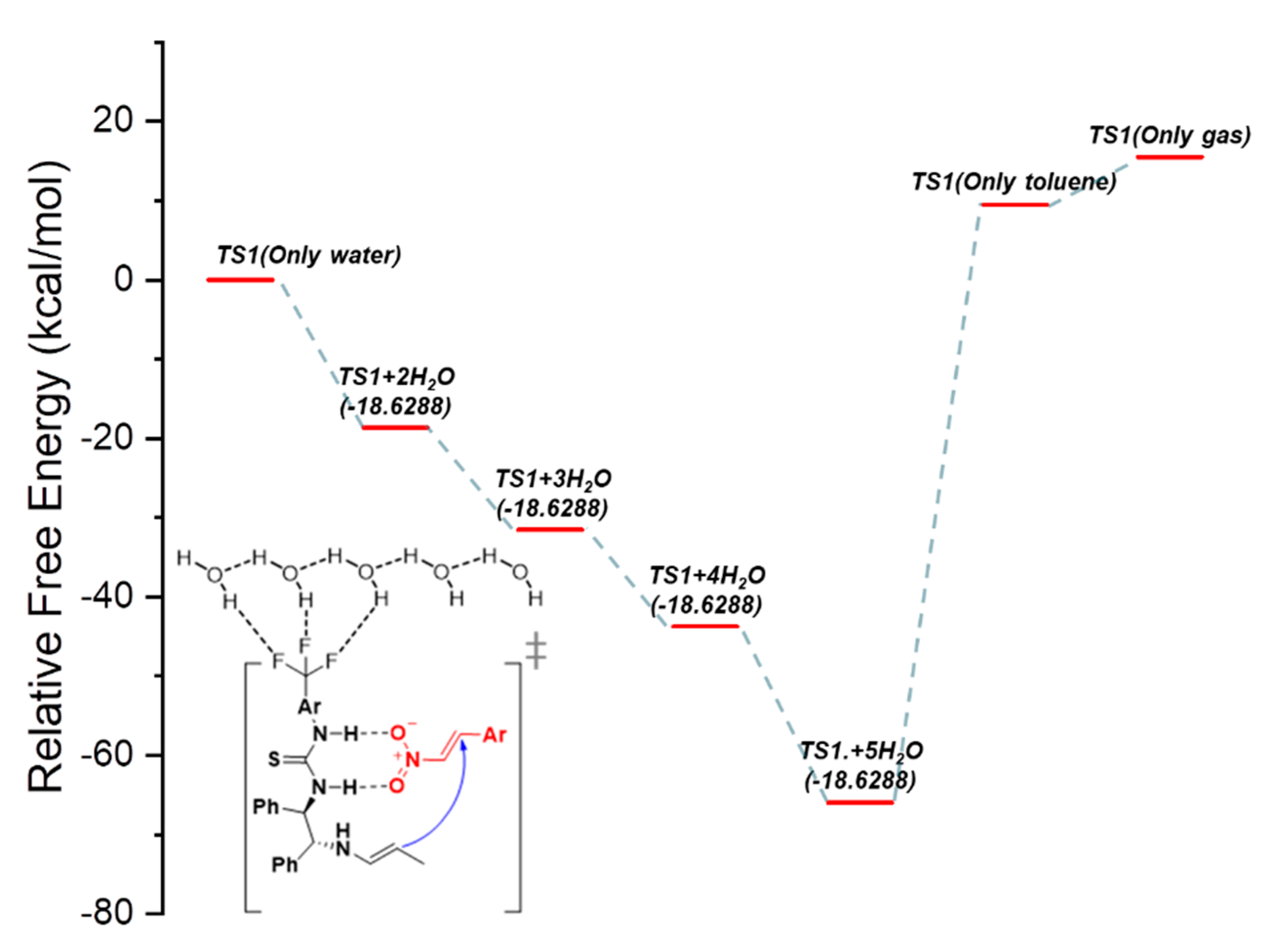
The relative free and thermal energies of the solvent effect for the Michael reaction steps are shown in Figure 5 through DFT calculations. We observed that the Michael addition reaction can be accelerated due to the hydrophobicity of thiourea-DPEN-based fluorine-substituted organic catalysts used in a previous study [32,33].
This reaction produces relatively high enantioselective and diastereoselective results. In addition, when the aromatic substituted nitroalkene is positioned in the thiourea direction, relatively little steric hindrance is observed. When water, a polar protic solvent, was used as the solvent, it was confirmed through an experiment that the reactivity was higher than that of the reactions using other solvents. In addition, it was confirmed through a quantum calculation that when water was used as a solvent, the reactivity was improved by stabilizing the transition state of the catalyst. Depending on the stabilized transition state, the aldehyde reacts with the amino group of the catalyst to form an enamine, and the thiourea moiety on the other side hydrogen bonds to the two oxygen atoms of the nitro group. The enamine nucleophile formed by the catalyst attacks the electrophile from below (Figure 5).
In order to predict the solvent effect of the catalyst, the relative free energy of the TS during the interfacial reaction between the hydrophobic substituent (CF3) and H2O of catalyst 1b were compared in an aqueous binary mixture (H2O + solvent), as shown in Figure 5. Consequently, it was confirmed that the relative free energy of the TS was the lowest when water was used as the solvent. When water is used as the solvent in a Michael addition reaction, the reactivity increases as the polarity of the catalyst increases. Therefore, it can be assumed that the reactivity increases owing to the stabilization of the relative energy and the hydrophobic effect of the hydration reaction. In addition, when the contact between the catalyst and water increased due to the hydrogen bonding, the degree of stabilization changed depending on the number of hydrogen bonds of the water molecules (Figure 5).
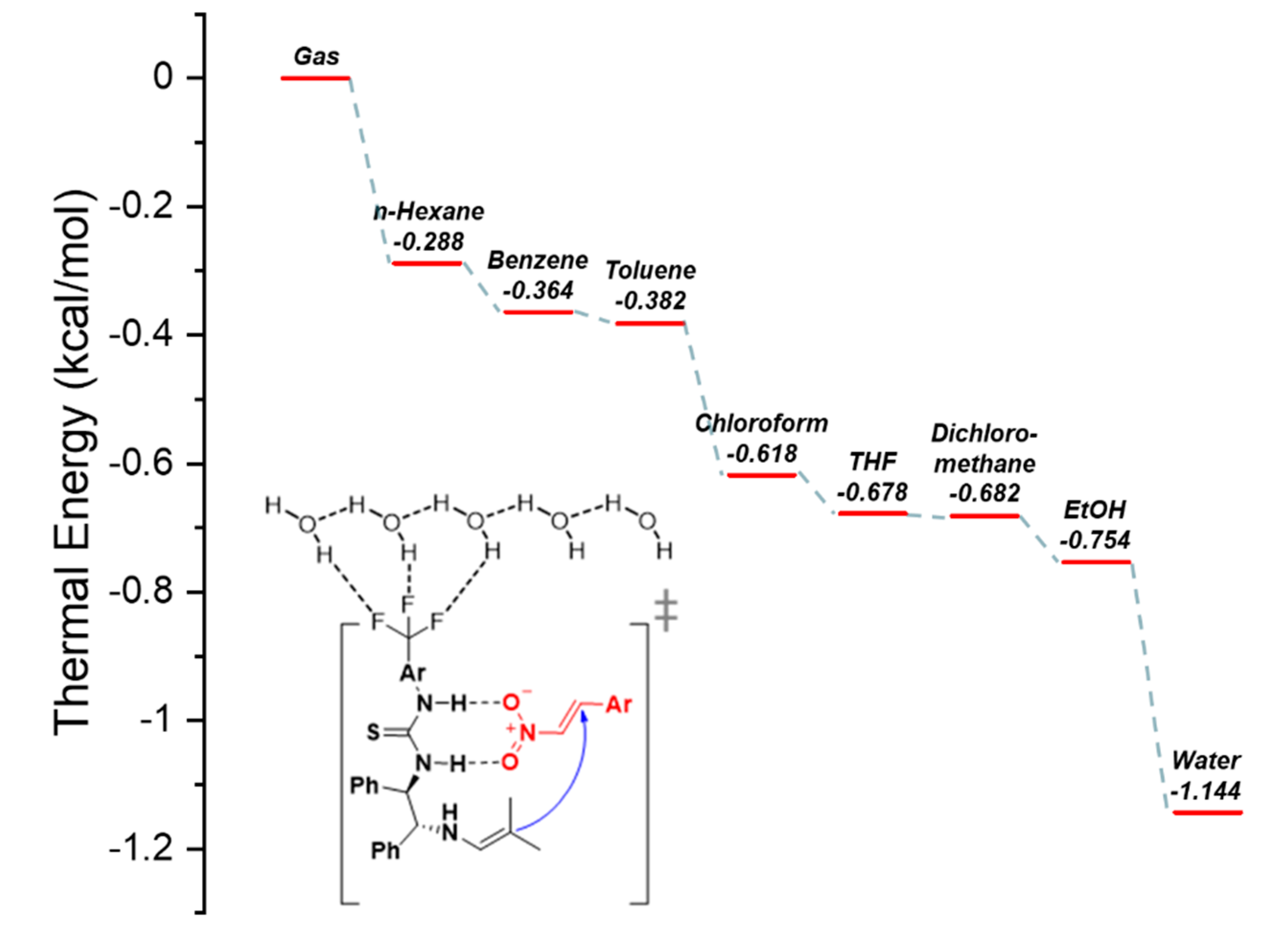
According to previous studies, hydrophobic, non-polar solvents such as n-hexane and toluene are known to favor the synthesis of Michael adducts [39]. However, as the solvent effect on the Michael reaction was confirmed in Table 2, a thermodynamic analysis was performed to determine the factors in which water influences the Michael addition reaction. To this end, quantum calculations were performed to predict the thermophysical data for the interfacial reaction system in the transition state of the catalyst. On comparing the actual reaction (Table 2) with the quantum calculation results, it was confirmed that the non-polar solvents, such as n-hexane and benzene exhibited the lowest reactivity. In addition, CHCl3, tetrahydrofuran, CH2Cl2, and EtOH showed similar reactivities in the calculated results. Among these solvents, it was confirmed that water exhibited the best reaction activity and stability (Figure 6).
Based on the mirror image selectivity obtained through the Michael reaction using isobutyraldehyde and α,β-unsaturated nitroalkenes, it was deduced that the catalytic reaction proceeded via the mechanism displayed in Figure 7.
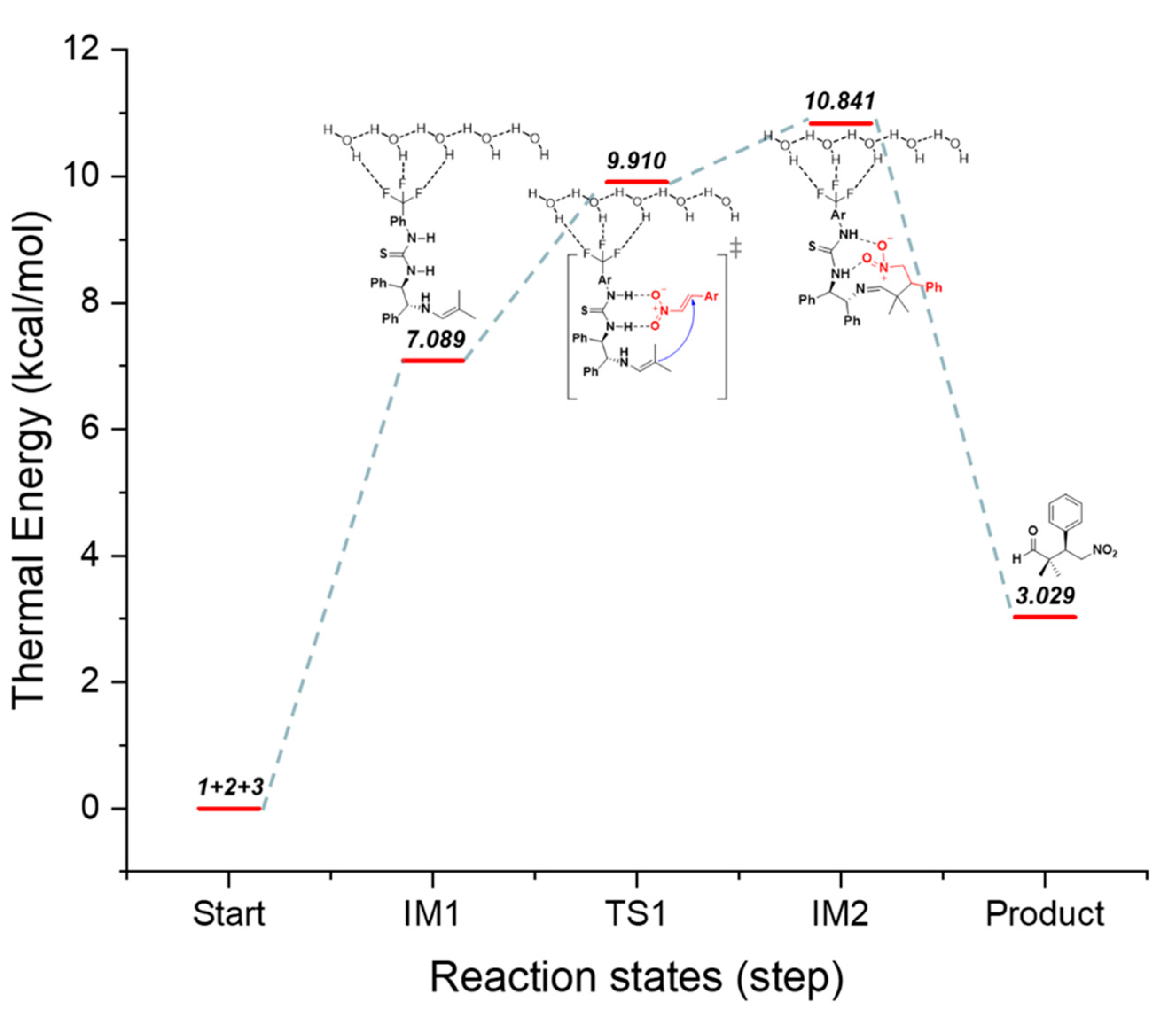
In the reaction, the hydrogen of the thiourea moiety formed a hydrogen bond with the oxygen in the nitroalkene. In addition, we predicted that the amine of the catalyst reacts with the aldehyde to produce the enamine, and the nucleophile approaches the rear side of the α,β-unsaturated nitroalkene to produce a compound with predominant stereoselectivity for the (R)-product. The optimization structures for DFT calculations mentioned in this article can be found on Supplementary Material pages 52–170.
3. Materials and Methods
3.1. Instruments and Reagents
IR spectrum was recorded using NICOLET 380 FT-IR spectrophotometer of Thermo electron corporation, and optical rotation was measured using an auto digital polarimeter. 1H-NMR and 13C-NMR spectrum were obtained using Varian Gemini 300 (300, 75 MHz), Varian Mercury 400 (400, 100 MHz) and Bruker Avance 500 (500, 125 MHz) using TMS as internal standards. HRMS spectra were obtained using a JEOL JMS-AX505WA mass spectrometer. Chiral HPLC analysis was performed using a Jasco LC-1500 Series HPLC system. All reactions were carried out under an argon environment in well-dried flasks in an oven. Toluene (CaH2), THF (Na, benzophenone), and CH2Cl2 (CaH2) reaction solvents were purified before use. The reagents used in this study were products such as Aldrich, Acros, Alfa, Sigma, Merck, Fluka, TCI, and Lancaster, and if necessary, purified or dried by a known method. Merck’s silica gel 60 (230–400 mech) was used as a stationary phase for column chromatography.
3.2. Experimental Method
3.2.1. Synthesis of N-mono-thiourea Catalyst
(R,R)-1.2-diphenylethylenediamine (200 mg, 0.942 mmol) was dissolved in toluene (1.00 mL), followed by the addition of isothiocyanate (0.140 mL, 0.942 mmol) at 0 °C for 1 h Stir. After completing the reaction with distilled water, extraction with dichloromethane (20 mL × 3 times), dehydration with MgSO4, filtration, concentration under reduced pressure, and column chromatography (SiO2, EtOAc:CH2Cl2 = 1:6) to isolate the product (Scheme 1).
3.2.2. Asymmetric Michael Reaction of Chitons and α,β-Unsaturated Nitroalkenes Using a Chiral Thiourea Catalyst
At room temperature, a thiourea catalyst (5 mol%), α,β-unsaturated nitroalkene (0.3 mmol) were put into a reaction vessel and then dissolved with water (1.0 mL) in air condition, followed by aldehyde (5 equiv.) was added and stirred for 4~12 h. After terminating the reaction with distilled water, extraction with dichloromethane (20 mL × 3 times), dehydration with MgSO4, filtration, concentration under reduced pressure, and column chromatography (SiO2, EtOAc: hexanes = 5:1) to isolate the product.
3.2.3. General Procedure of the Racemic Michael Addition
To α,β-unsaturated nitroalkene (0.3 mmol), aldehyde (10 equiv.), and 20 mol% of DL-Proline were added to toluene (0.1 M) and the reaction mixture was stirred at ambient temperature. The reaction conversion was checked by thin layer chromatography. After completion about 12 h, ethyl acetate (0.2 mL) was added to the reaction product. This solution was washed twice with water (2 × 1.0 mL), dried over magnesium sulfate (anhydrous), and concentrated to yield the desired product. The product was purified by chromatography on a silica gel column eluted with mixed solvent (hexanes/EA, 5/1).
3.3. Results of DFT Calculations and Discussion
Density functional theory (DFT) calculations were performed using Gaussian 16 and Gauss-View 6.0 programs. DFT calculations were performed to show the mechanisms of substrates and catalysts. The optimized geometry was described using the B3LYP/6-31G(d,p) level. After the shapes of reactants, intermediates (IM), transition states (TS), and products are fully optimized, zero-point energy (ZPE) is obtained through vibrational frequency calculation in the same level of theory, and the minimum or transition state of the potential energy surface (PES) is obtained. Enthalpy correction and entropy with temperature are calculated at 298 K and 1 atm pressure.
4. Conclusions
The Michael addition reaction of isobutyraldehyde and unsaturated nitroalkene was carried out using the N-monothiourea catalyst of (R,R)-1,2-diphenylethylene-diamine (DPEN). An enantiomeric excess of 97–99% ee was obtained. The catalyst substituted with the 3,5-bis-(trifluoromethyl) group (Figure 1, 1b) exhibited a simpler synthesis than the other organic catalysts. Michael 1,4-addition reaction using the 1b catalyst yielded relatively high enantioselectivity and diastereoselectivity. In addition, using water as a solvent is eco-friendly, does not use additives, and exhibits increased reactivity through hydration. The H2O molecule stabilizes the transition state of the catalyst through hydrogen bonding with fluorine of the catalyst and accelerates the reactivity through the hydrolysis of the enamine form of the product. However, this reaction requires a large amount of catalyst. Therefore, future research that focuses on optimizing these reaction conditions is essential. In addition, drug development of chiral compounds using this synthetic approaches is ongoing.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12020121/s1, Compound Characterization Data, Copy of NMR and MASS Spectra, Copy of HPLC Chromatograms, DFT Calculations for all Calculated Structures of the compounds mentioned in the text.
Author Contributions
Conceptualization, J.H.S. and D.-C.H.; Data curation, J.H.S. and S.H.C.; Funding acquisition, J.H.S.; Investigation, S.H.C. and D.-C.H.; Methodology, D.-C.H.; Project administration, J.H.S.; Resources, H.S.K.; Software, H.S.K.; Supervision, J.H.S.; Validation, S.H.C.; Writing—original draft, J.H.S.; Writing—review and editing, J.H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Research Foundation (NRF) and funded by the Korean government (MSIT) (2021R1A6A3A01087948). This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) and funded by the Korean government (MSIT) (2021M3A9G1097744). In addition, this study was supported by a Korea University grant.
Data Availability Statement
Not applicable.
Acknowledgments
We are also grateful for the financial support provided by Kim, K. H.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Pojarliev, P.; Martin, H.J. Efficient Proline-Catalyzed Michael Additions of Unmodified Ketones to Nitro Olefins. Org. Lett. 2001, 3, 2423–2425. [Google Scholar] [CrossRef]
- Mase, N.; Watanabe, K.; Yoda, H.; Takabe, K.; Tanaka, F.; BarbasIII, C.F. Organocatalytic Direct Michael Reaction of Ketones and Aldehydes with β-Nitrostyrene in Brine. J. Am. Chem. Soc. 2006, 128, 4966–4967. [Google Scholar] [CrossRef]
- Dalko, P.L.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Perrier, A.; Linhardt, R.; Travers, L.; Wittmann, S.; Caminade, A.-M.; Majoral, J.-P.; Reiser, O.; Ouali, A. Dendrimers or Nanoparticles as Supports for the Design of Efficient and Recoverable Organocatalysts? Adv. Synth. Catal. 2013, 355, 1748–1754. [Google Scholar] [CrossRef]
- De Simone, N.A.; Meninno, S.; Talotta, C.; Gaeta, C.; Neri, P.; Lattanzi, A. Solvent-Free Enantioselective Michael Reactions Catalyzed by a Calixarene-Based Primary Amine Thiourea. J. Org. Chem. 2018, 83, 10318–10325. [Google Scholar] [CrossRef] [PubMed]
- Seayad, J.; List, B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005, 3, 719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, S.; Wei, K.; Lin, J. Epiandrosterone-derived prolinamide as an efficient asymmetric catalyst for Michael addition reactions of aldehydes to nitroalkenes. RSC Adv. 2014, 4, 30850–30856. [Google Scholar] [CrossRef]
- Wang, Y.; Li, D.; Lin, J.; Wei, K. Organocatalytic asymmetric Michael addition of aldehydes and ketones to nitroalkenes catalyzed by adamantoyl l-prolinamide. RSC Adv. 2015, 5, 5863–5874. [Google Scholar] [CrossRef]
- Castán, A.; Badorrey, R.; Gálvez, J.A.; López-Ram-de-Víu, P.; Díaz-de-Villegas, M.D. Michael addition of carbonyl compounds to nitroolefins under the catalysis of new pyrrolidine-based bifunctional organocatalysts. Org. Biomol. Chem. 2018, 16, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorde, A.B.; Ramapanicker, R. d-Prolyl-2-(trifluoromethylsulfonamidopropyl)pyrrolidine: An Organocatalyst for Asymmetric Michael Addition of Aldehydes to β-Nitroalkenes at Ambient Conditions. J. Org. Chem. 2019, 84, 1523–1533. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 11, 1701–1716. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Wang, M.; He, P.; Lin, L.; Feng, X. Enantioselective Synthesis of 3,4-Dihydropyran Derivatives via Organocatalytic Michael Reaction of α,β-Unsaturated Enones. J. Org. Chem. 2012, 77, 4136–4142. [Google Scholar] [CrossRef]
- Kwiatkowski, P.; Lyzwa, D. Effect of High Pressure on the Organocatalytic Asymmetric Michael Reaction: Highly Enantioselective Synthesis of γ-Nitroketones with Quaternary Stereogenic Centers. Org. Lett. 2011, 13, 3624–3627. [Google Scholar] [CrossRef]
- Qing, G.; You, S.L. Desymmetrization of cyclohexadienones viacinchonine derived thiourea-catalyzed enantioselective aza-Michael reaction and total synthesis of (-)-Mesembrine. Chem. Sci. 2011, 2, 1519–1522. [Google Scholar]
- Li, H.Y.; Xu, L.W. A Total Synthesis of Paeoveitol. Org. Lett. 2011, 15, 3698–3701. [Google Scholar]
- Sigman, M.; Jacobsen, E.N. Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Uraguchi, D.; Terada, M. Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- McCooey, S.H.; McCabe, T.; Connon, S.J. Stereoselective Synthesis of Highly Functionalized Nitrocyclopropanes via Organocatalyic Conjugate Addition to Nitroalkenes. J. Org. Chem. 2006, 71, 7494–7497. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Torres, H.; Barbas, C.F., III. Rationally Designed Amide Donors for Organocatalytic Asymmetric Michael Reactions. Angew. Chem. Int. Ed. 2012, 51, 5381. [Google Scholar] [CrossRef] [PubMed]
- Curti, C.; Casiraghi, G. Bifunctional Cinchona Alkaloid/Thiourea Catalyzes Direct and Enantioselective Vinylogous Michael Addition of 3-Alkylidene Oxindoles to Nitroolefins. Angew. Chem. Int. Ed. 2012, 51, 6304–6308. [Google Scholar] [CrossRef]
- Mase, N.; Thayumanavan, R.; Tanaka, F.; Barbas, C.F. Direct Asymmetric Organocatalytic Michael Reactions of α,α-Disubstituted Aldehydes with β-Nitrostyrenes for the Synthesis of Quaternary Carbon-Containing Products. Org. Lett. 2004, 6, 2527–2530. [Google Scholar] [CrossRef]
- Cao, C.L.; Ye, M.C.; Sun, X.L.; Tang, Y. Pyrrolidine−Thiourea as a Bifunctional Organocatalyst: Highly Enantioselective Michael Addition of Cyclohexanone to Nitroolefins. Org. Lett. 2006, 8, 2901–2904. [Google Scholar] [CrossRef]
- McCooey, S.H.; Connon, S.J. Readily Accessible 9-epi-amino Cinchona Alkaloid Derivatives Promote Efficient, Highly Enantioselective Additions of Aldehydes and Ketones to Nitroolefins. Org. Lett. 2007, 9, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Liu, S.P.; Yan, M.; Chan, A.S.C. Highly enantioselective conjugate addition of aldehydes to nitroolefins catalyzed by chiral bifunctional sulfamides. Chem. Commun. 2009, 833–835. [Google Scholar] [CrossRef]
- Ting, Y.F.; Chang, C.; Reddy, R.J.; Magar, D.R.; Chen, K. Pyrrolidinyl–Camphor Derivatives as a New Class of Organocatalyst for Direct Asymmetric Michael Addition of Aldehydes and Ketones to β-Nitroalkenes. Chem. Eur. J. 2010, 16, 7030–7038. [Google Scholar] [CrossRef] [PubMed]
- Genc, H.N.; Ozgun, U.; Sirit, A. Design, synthesis and application of chiraltetraoxacalix[2]arene[2]triazine base dorganocatalysts in asymmetric Michael additionreactions. Chirality 2019, 31, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yu, S.; Ma, D. Highly Efficient Catalytic System for Enantioselective MichaelAddition of Aldehydes to Nitroalkenes in Water. Angew. Chem. Int. Ed. 2008, 47, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.C.; Shoaib, M.; Shoaib, A. Practical access to highly enantioenriched quaternary carbon Michael adducts using simple organocatalysts. Org. Biomol. Chem. 2011, 9, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Nam, S.H.; Kim, B.S.; Ha, D.C. Organocatalytic Asymmetric Michael Addition of Ketones to α,β-Unsaturated Nitro Compounds. Catalysts 2020, 10, 618. [Google Scholar] [CrossRef]
- Shim, J.H.; Park, S.J.; Ahn, B.K.; Lee, J.Y.; Kim, B.S.; Ha, D.C. Enantioselective Thiolysis and Aminolysis of Cyclic Anhydrides Using a Chiral Diamine-Derived Thiourea Catalyst. ACS Omega 2021, 6, 34501–34511. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Ahn, B.K.; Lee, J.Y..; Kim, H.S.; Ha, D.C. Organocatalysis for the Asymmetric Michael Addition of Cycloketones and α,β-Unsaturated Nitroalkenes. Catalysts 2021, 11, 1004. [Google Scholar] [CrossRef]
- Shim, J.H.; Hong, Y.; Kim, S.H.; Kim, J.H.; Ha, D.C. Organocatalytic Asymmetric Michael Addition in Aqueous Media by a Hydrogen-Bonding Catalyst and Application for Inhibitors of GABAB Receptor. Catalysts 2021, 11, 1134. [Google Scholar] [CrossRef]
- Gorde, A.B.; Ramapanicker, R. Enantioselective Michael Addition of Aldehydes to β-Nitrostyrenes Catalyzed by (S)-N-(D-Prolyl)-1-triflicamido-3-phenylpropan-2-amine. Eur. J. Org. Chem. 2019, 26, 4745–4751. [Google Scholar] [CrossRef]
- Ying, A.; Liu, S.; Li, Z.; Chen, G.; Yang, J.; Yan, H.; Xu, S. Magnetic Nanoparticles-Supported Chiral Catalyst with an Imidazolium Ionic Moiety: An Efficient and Recyclable Catalyst for Asymmetric Michael and Aldol Reactions. Adv. Synth. Catal. 2016, 358, 2116–2125. [Google Scholar] [CrossRef]
- Shim, J.H.; Kim, M.J.; Lee, J.Y.; Kim, K.H.; Ha, D.C. Organocatalytic Asymmetric Aldol Reaction Using Protonated Chiral 1,2-Diamines. Tetrahedron Lett. 2020, 61, 152295. [Google Scholar] [CrossRef]
- Luis, Á.S.; Eusebio, J.; Alexa, B.A.C.; Jorge, P.F.; Fanny, A.C.R.; Jaime, E. Efficient Solvent-Free Preparation of Imines, and Their Subsequent Oxidation with m-CPBA to Afford Oxaziridines. Green Sustain. Chem. 2019, 9, 143–154. [Google Scholar]
- José, D.R.R.; Jaime, E.; Agustín, L.M.; Alain, M.; Edmundo, C. Thermodynamically controlled chemoselectivity in lipase-catalyzed aza-Michael additions. J. Mol. Catal. B Enzym. 2015, 112, 76–82. [Google Scholar]
Figure 1.
Mechanism of the Michael reaction of aldehydes and nitroalkenes using a thiourea-based catalyst.
Figure 1.
Mechanism of the Michael reaction of aldehydes and nitroalkenes using a thiourea-based catalyst.

Figure 2.
DPEN and thiourea based catalysts.

Figure 3.
Possible stereochemical transition state models and expected syn and anti products. This figure shows the reaction mechanism proposed for the Michael reaction presented below.
Figure 3.
Possible stereochemical transition state models and expected syn and anti products. This figure shows the reaction mechanism proposed for the Michael reaction presented below.

Figure 4.
Proposed catalytic mechanism based on B3LYP/6-31G(d,p) calculations and the free energy diagram of the (R,R)-1,2-diphenylethylenediamine(DPEN)-thiourea-catalyzed enantioselective Michael reaction.
Figure 4.
Proposed catalytic mechanism based on B3LYP/6-31G(d,p) calculations and the free energy diagram of the (R,R)-1,2-diphenylethylenediamine(DPEN)-thiourea-catalyzed enantioselective Michael reaction.

Figure 5.
Proposed catalytic mechanism based on the B3LYP/6-31G(d,p) method. Here, the calculations were performed using water, toluene, and solvent + H2O conditions. The relative free energy diagram of the (R,R)-1,2-diphenylethylen-ediamine(DPEN)-thiourea catalyzed enantioselective Michael reaction.
Figure 5.
Proposed catalytic mechanism based on the B3LYP/6-31G(d,p) method. Here, the calculations were performed using water, toluene, and solvent + H2O conditions. The relative free energy diagram of the (R,R)-1,2-diphenylethylen-ediamine(DPEN)-thiourea catalyzed enantioselective Michael reaction.

Figure 6.
Proposed catalytic mechanism based on B3LYP/6-31G(d,p), on water solvent + H2O condition calculations and the thermal energy diagram of the (R,R)-1,2-diphenylethylenediamine(DPE- N)-thiourea-catalyzed enantioselective Michael reaction.
Figure 6.
Proposed catalytic mechanism based on B3LYP/6-31G(d,p), on water solvent + H2O condition calculations and the thermal energy diagram of the (R,R)-1,2-diphenylethylenediamine(DPE- N)-thiourea-catalyzed enantioselective Michael reaction.

Figure 7.
Proposed catalytic mechanism based on the B3LYP/6-31G(d,p) method and the water solvent + H2O conditions. The thermal energy diagram of the (R,R)-1,2-diphenylethylene diamine(DPEN)-thiourea-catalyzed enantioselective Michael reaction.
Figure 7.
Proposed catalytic mechanism based on the B3LYP/6-31G(d,p) method and the water solvent + H2O conditions. The thermal energy diagram of the (R,R)-1,2-diphenylethylene diamine(DPEN)-thiourea-catalyzed enantioselective Michael reaction.

Scheme 1.
Synthesis of N-mono-thiourea catalyst.


{kind=link}
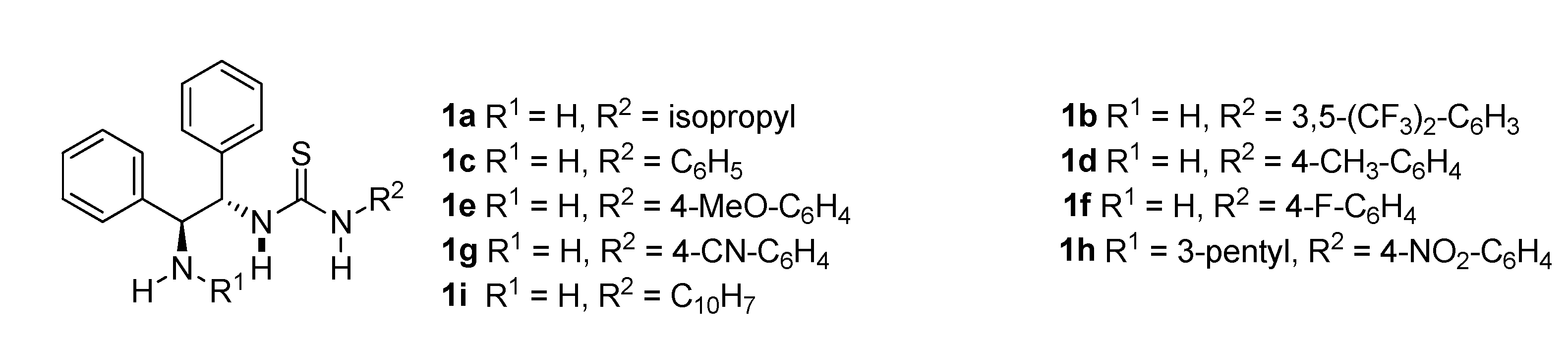{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
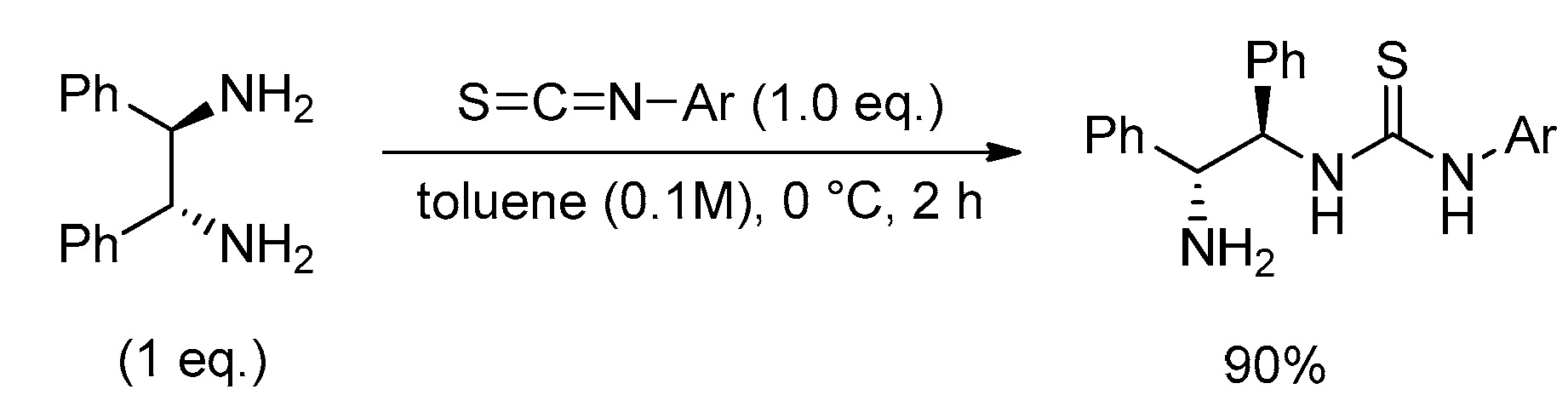{kind=link}
Table 1.
Catalyst optimization.
 | ||||||
| Entry | Catalyst | Temp (°C) | Equiv. | mol% | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|---|
| 1 | 1a | rt | 10 | 10 | 50 | 92 |
| 2 | 1b | rt | 10 | 10 | 91 | 97 |
| 3 | 1c | rt | 10 | 10 | 67 | 93 |
| 4 | 1d | rt | 10 | 10 | 70 | 93 |
| 5 | 1e | rt | 10 | 10 | 50 | 92 |
| 6 | 1f | rt | 10 | 10 | 68 | 97 |
| 7 | 1g | rt | 10 | 10 | 86 | 94 |
| 8 | 1h | rt | 10 | 10 | N.R | - |
| 9 | 1i | rt | 10 | 10 | 45 | 94 |
| 10 | 1b | 10 | 10 | 74 | 97 | |
| 11 | 1b | rt | 7 | 10 | 86 | 97 |
| 12 | 1b | rt | 5 | 10 | 85 | 97 |
| 13 | 1b | rt | 10 | 5 | 83 | 96 |
a Isolated yield. b The ee values were determined by chiral phase HPLC using OD-H column.
Table 2.
Solvent optimization.
 | |||
| Entry | Solvent | Yield (%) a | ee (%) b |
|---|---|---|---|
| 1 | n-hexane | 52 | 97 |
| 2 | CHCl3 | 80 | 97 |
| 3 | THF | 60 | 96 |
| 4 | benzene | 87 | 97 |
| 5 | EtOH | 58 | 97 |
| 6 | toluene | 85 | 97 |
| 7 c | water | 99 | 99 |
| 8 | CH2Cl2 | 85 | 97 |
a Isolated yield. b The ee values were determined by HPLC using the OD-H column. c The reactions were run with catalyst of 5 mol%, 12 h.
Table 3.
Asymmetric Michael additions of aldehyde to nitro-olefins.
 | |||
| Entry | Ar | Yield (%) a | ee (%) b |
|---|---|---|---|
| 1 | Ph | 99 | 99 |
| 2 | 4-Cl-Ph | 94 | 99 |
| 3 | 4-Br-Ph | 94 | 98 |
| 4 | 4-Me-Ph | 94 | 99 |
| 5 | 2-Furyl | 96 | 99 |
| 6 | 4-MeO-Ph | 97 | 99 |
| 7 | 2-MeO-Ph | 96 | 99 |
| 8 | 4-OH-Ph | 96 | 99 |
a Isolated yield. b The ee values were determined by HPLC using OD-H column.
Table 4.
Asymmetric Michael reaction of various aldehyde and unsaturated nitroalkenes.
 | |||||
| Entry | R1 | R2 | Yield (%) a | drb (syn:anti) | ee (%) c |
|---|---|---|---|---|---|
| 1 | Me | H | 95 | 67:33 | 99 |
| 2 | Et | H | 94 | 83:17 | 99 |
| 3 | n-Pr | H | 94 | 83:17 | 98 |
| 4 | i-Pr | H | 93 | 93:07 | 99 |
a Isolated yield. b Determined by 1H-NMR analysis. c The ee values were determined by HPLC. using the OD-H and AD-H columns.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shim, J.H.; Cheun, S.H.; Kim, H.S.; Ha, D.-C. Organocatalysis for the Asymmetric Michael Addition of Aldehydes and α,β-Unsaturated Nitroalkenes. Catalysts 2022, 12, 121. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020121
AMA Style
Shim JH, Cheun SH, Kim HS, Ha D-C. Organocatalysis for the Asymmetric Michael Addition of Aldehydes and α,β-Unsaturated Nitroalkenes. Catalysts. 2022; 12(2):121. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020121
Chicago/Turabian StyleShim, Jae Ho, Seok Hyun Cheun, Hyeon Soo Kim, and Deok-Chan Ha. 2022. "Organocatalysis for the Asymmetric Michael Addition of Aldehydes and α,β-Unsaturated Nitroalkenes" Catalysts 12, no. 2: 121. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020121
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.