
En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches

 , and
, and
Abstract
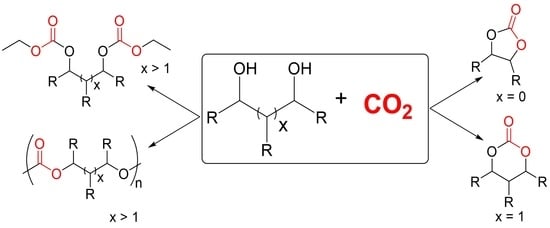
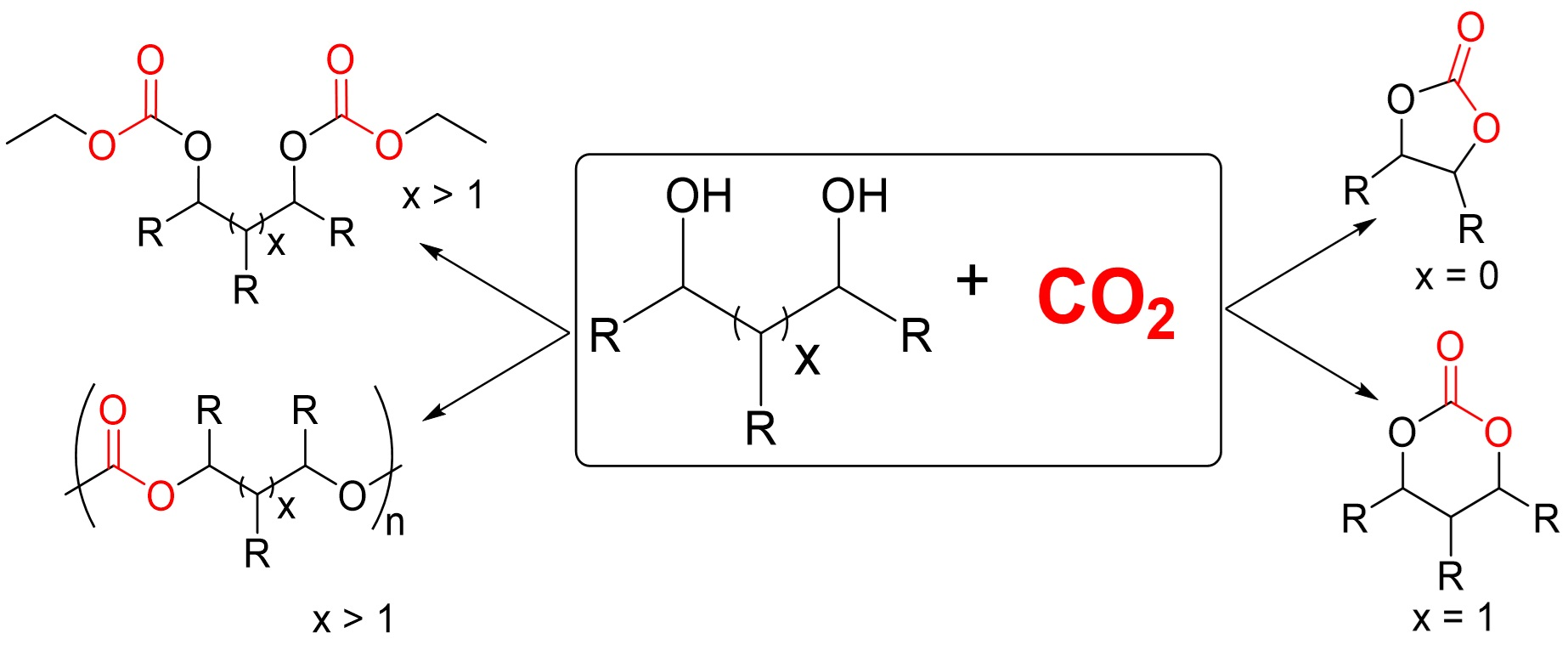
:
{kind=link}
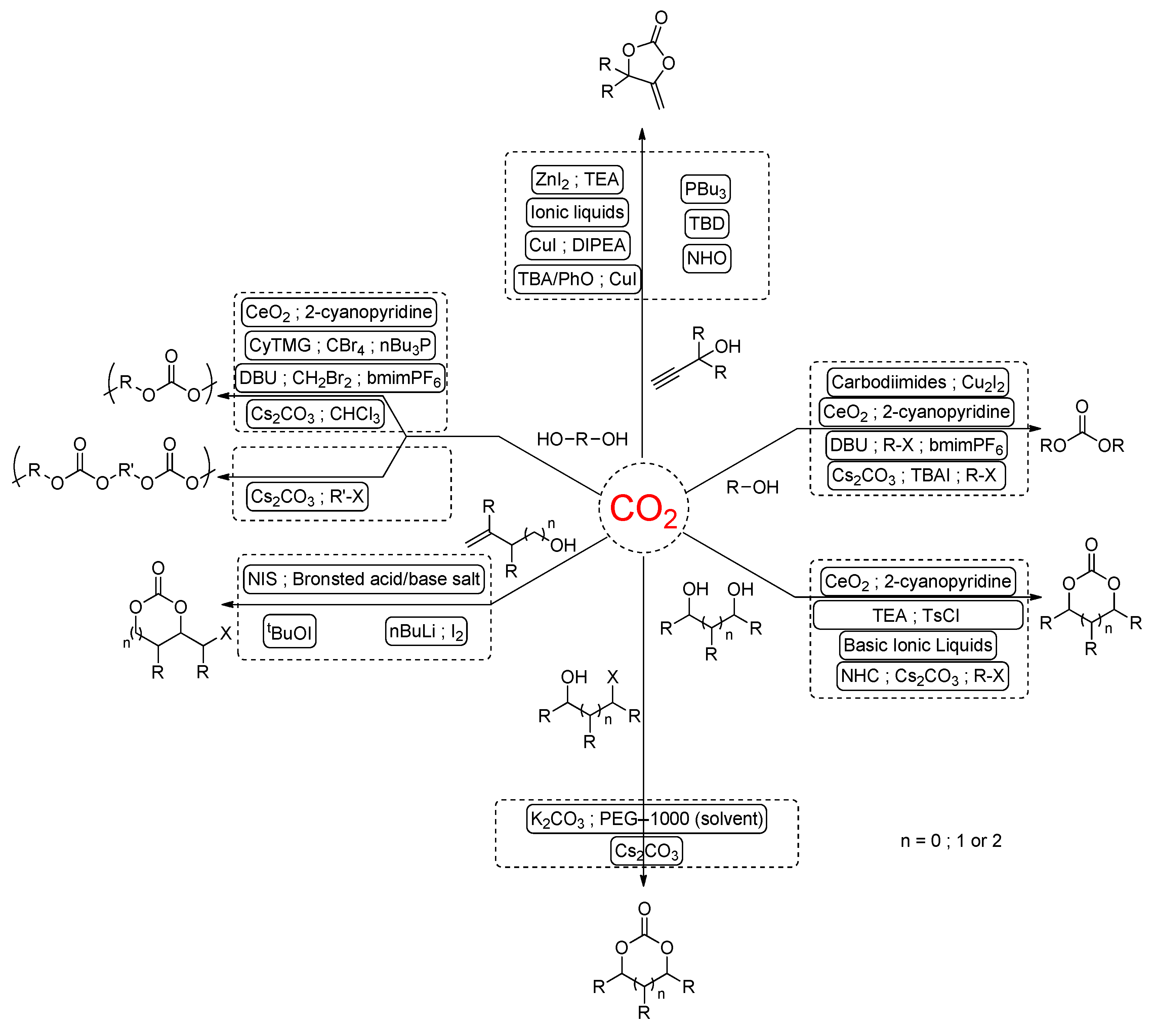{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
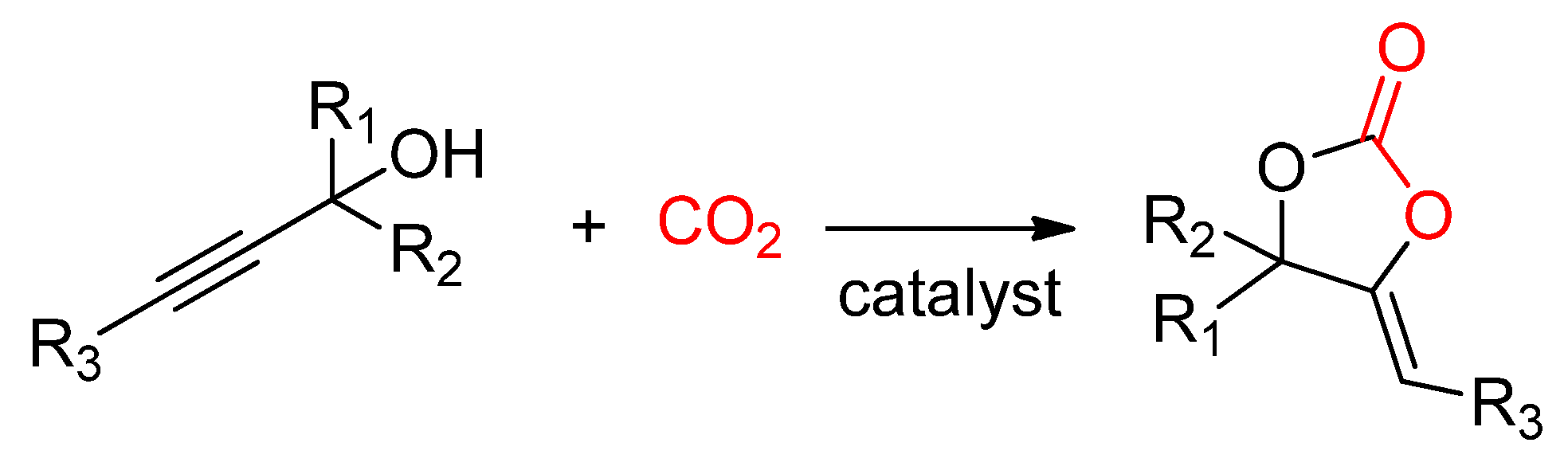{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
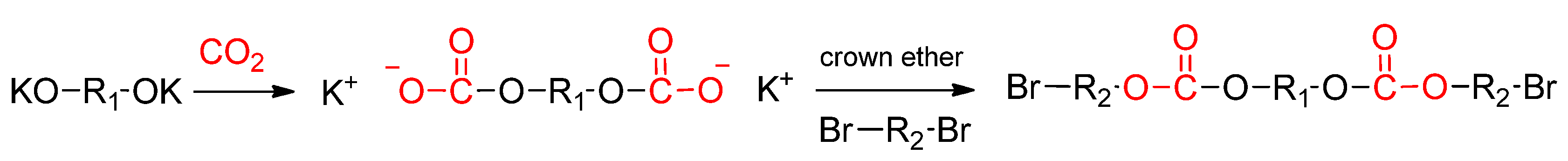{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
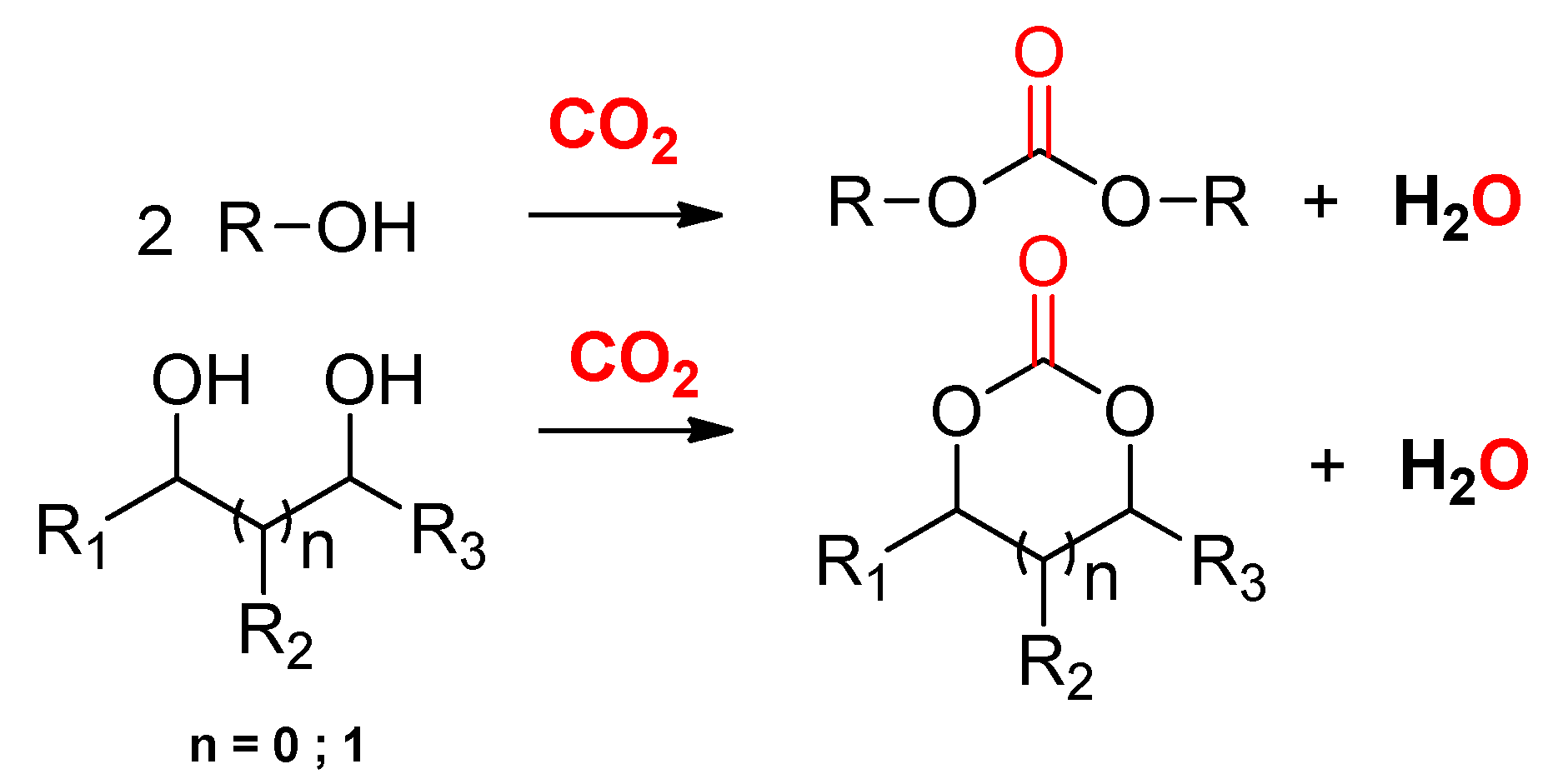
2. CO2-Based Synthons: Organic Carbonate Synthesis from the Carbonation of Alcohols
- (i)
- The “dehydrative condensation” approach, utilizing the synergy between a catalyst, which facilitates the CO2 fixation onto the alcohol moiety, with physical or reactive dehydrating agents to trap or consume water;
- (ii)
- The “alkylation” strategy, involving the activation of CO2 under the form of a hemi-carbonate ion in the presence of an excess of base prior to further reaction with an alkyl halide via nucleophilic substitution. As a variant, the “leaving group” approach proposes the in situ formation of a highly reactive carbonate intermediate from the ter-reaction between CO2, an alcohol and a dihalide, which undergoes facile transcarbonation into an acyclic carbonate;
- (iii)
- The “protected alcohols” route, which generates in situ alcohols from ketals, trimethyl phosphates, or orthoesters via water consumption prior to reaction with CO2
2.1. Synthesis of Acyclic Organic Carbonates
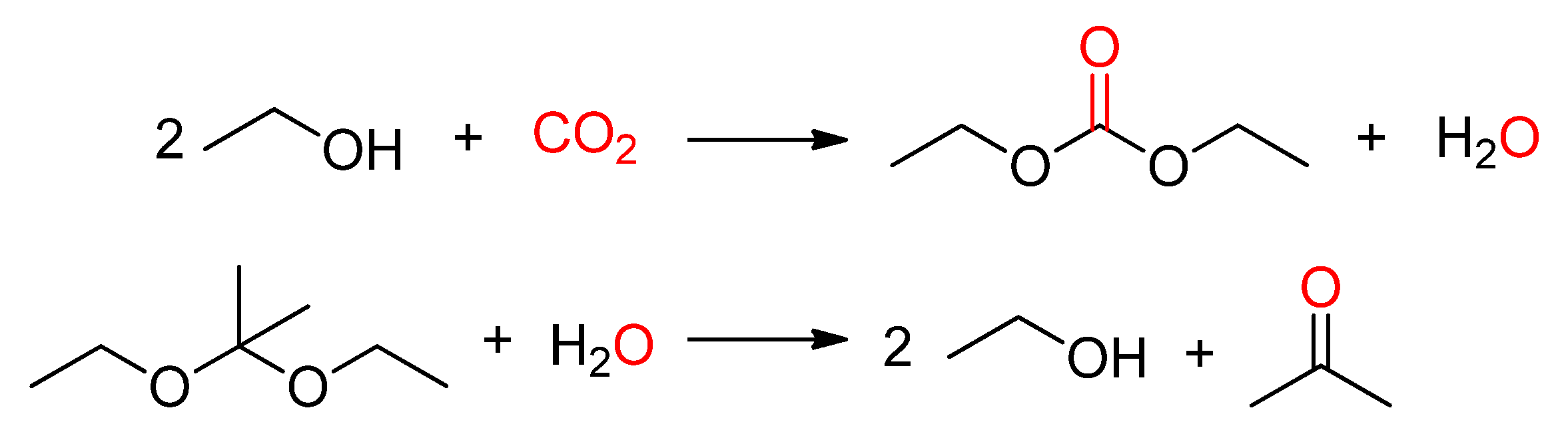
2.1.1. DMC Synthesis by Dehydrative Condensation
- Physical drying systems;
- Reactive dehydrative systems;
2.1.2. Synthesis of Acyclic Carbonates via the Alkylation Route
2.1.3. Synthesis of Acyclic Carbonates Using the “Leaving Group” Strategy
2.1.4. Synthesis of Acyclic Carbonates via the Carbodiimide Route
2.1.5. Synthesis of Acyclic Carbonates from the “Protected Alcohols” Route
2.2. Synthesis of Cyclic Organic Carbonates
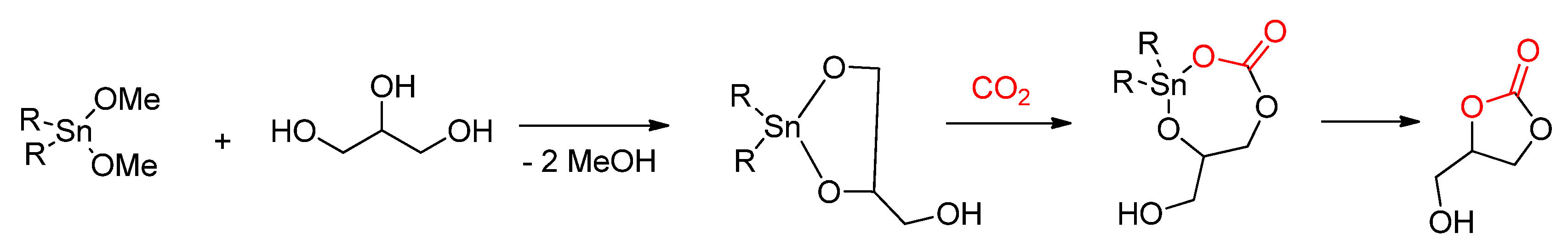
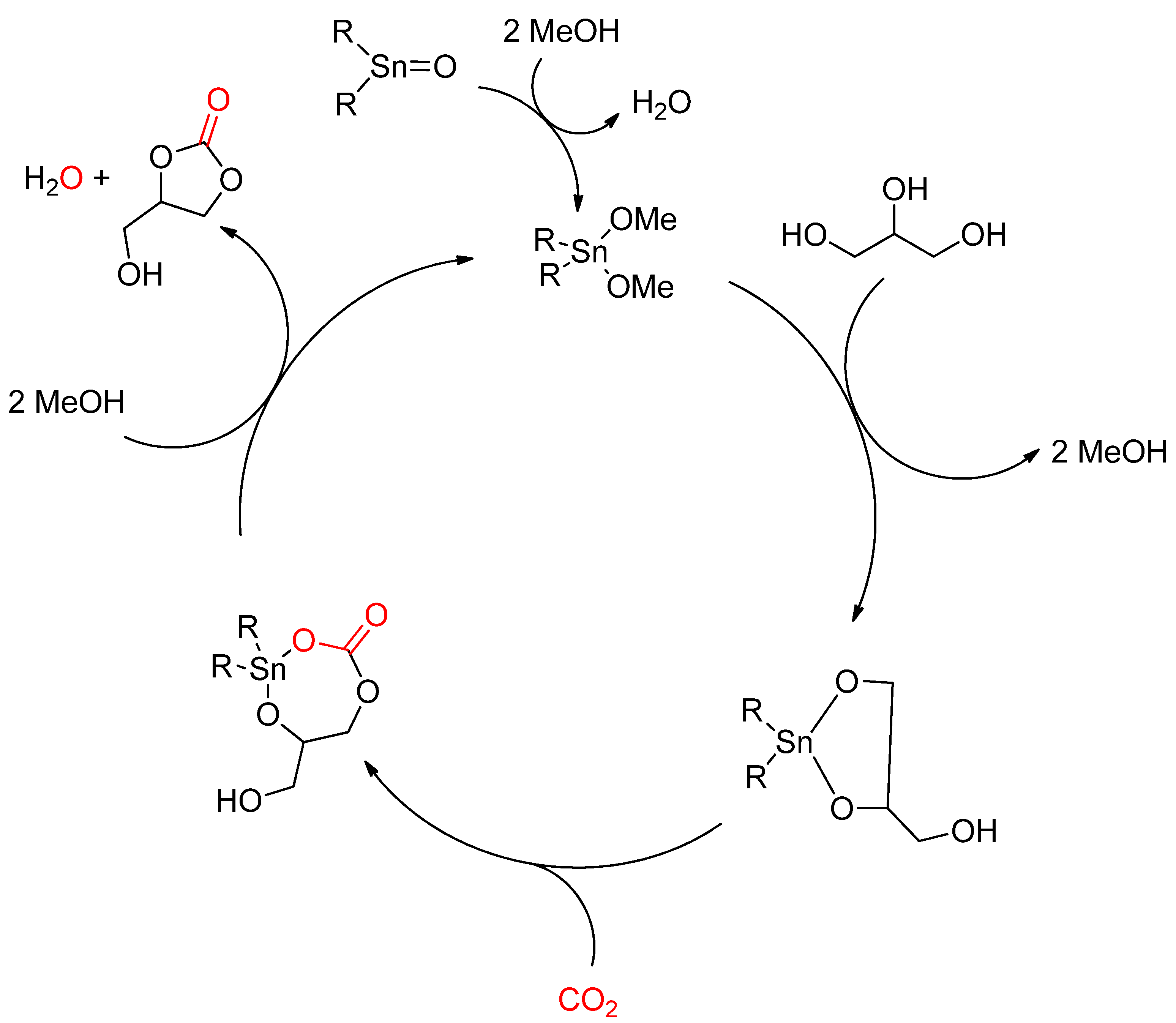
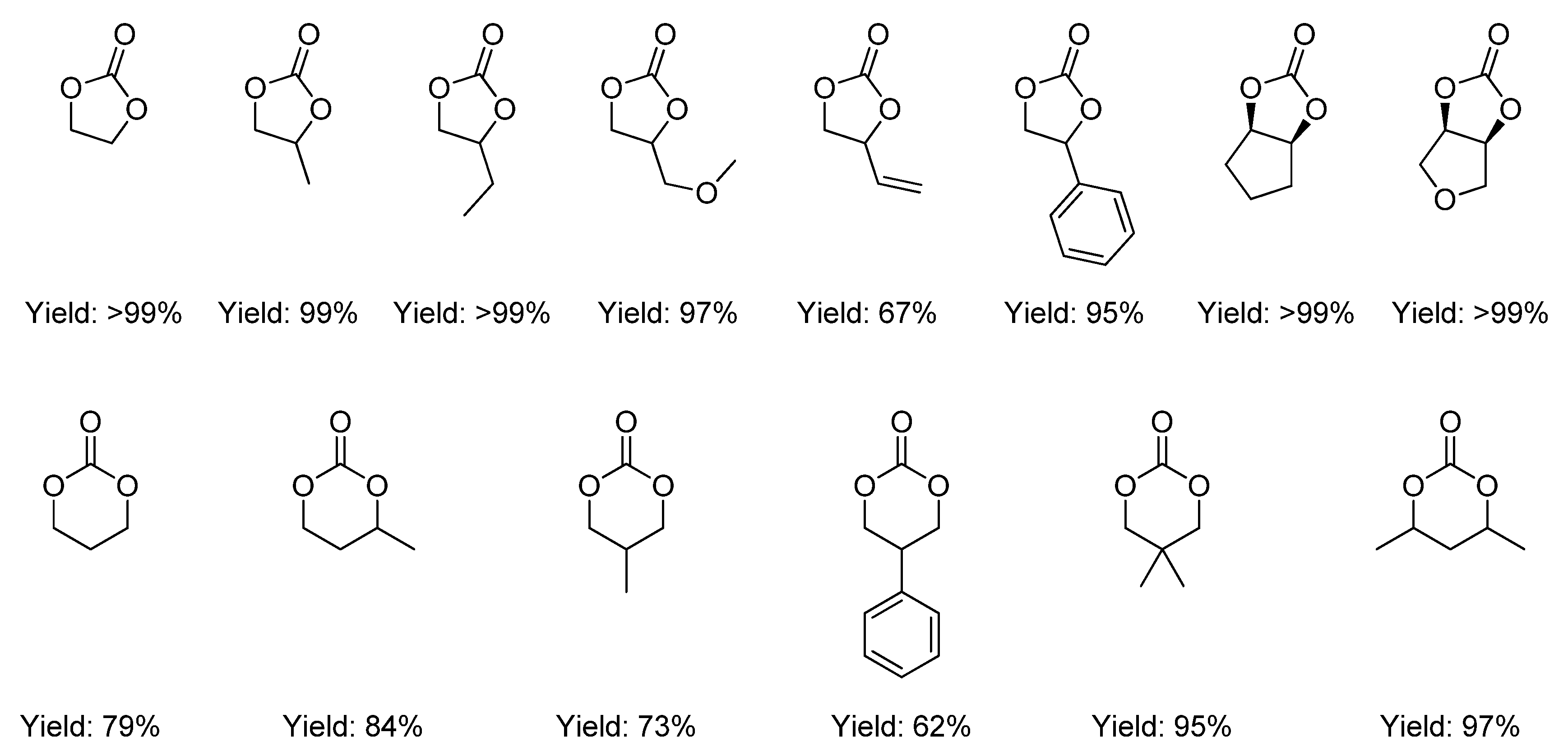
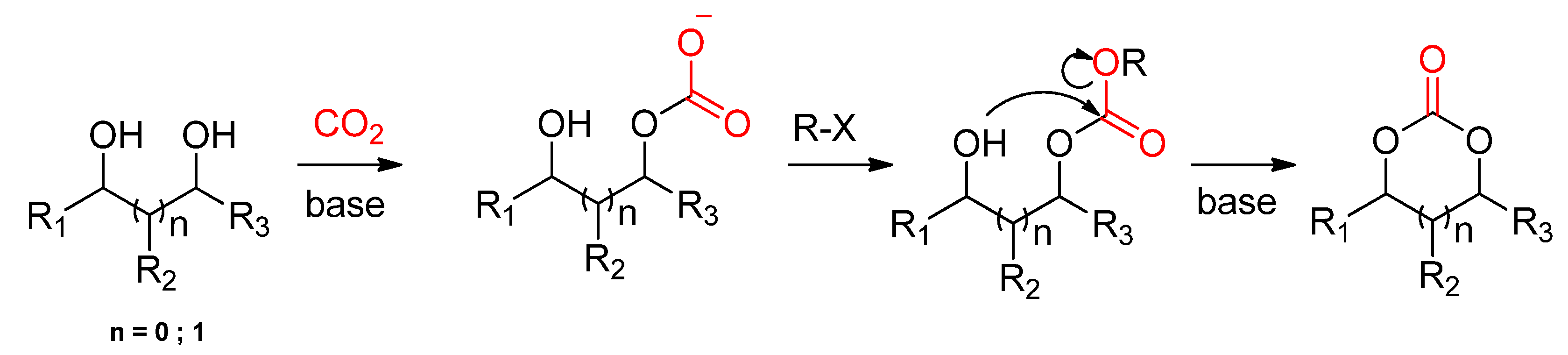
2.2.1. Synthesis of Cyclic Carbonates from Diols
2.2.1.1. Dehydrative Condensation
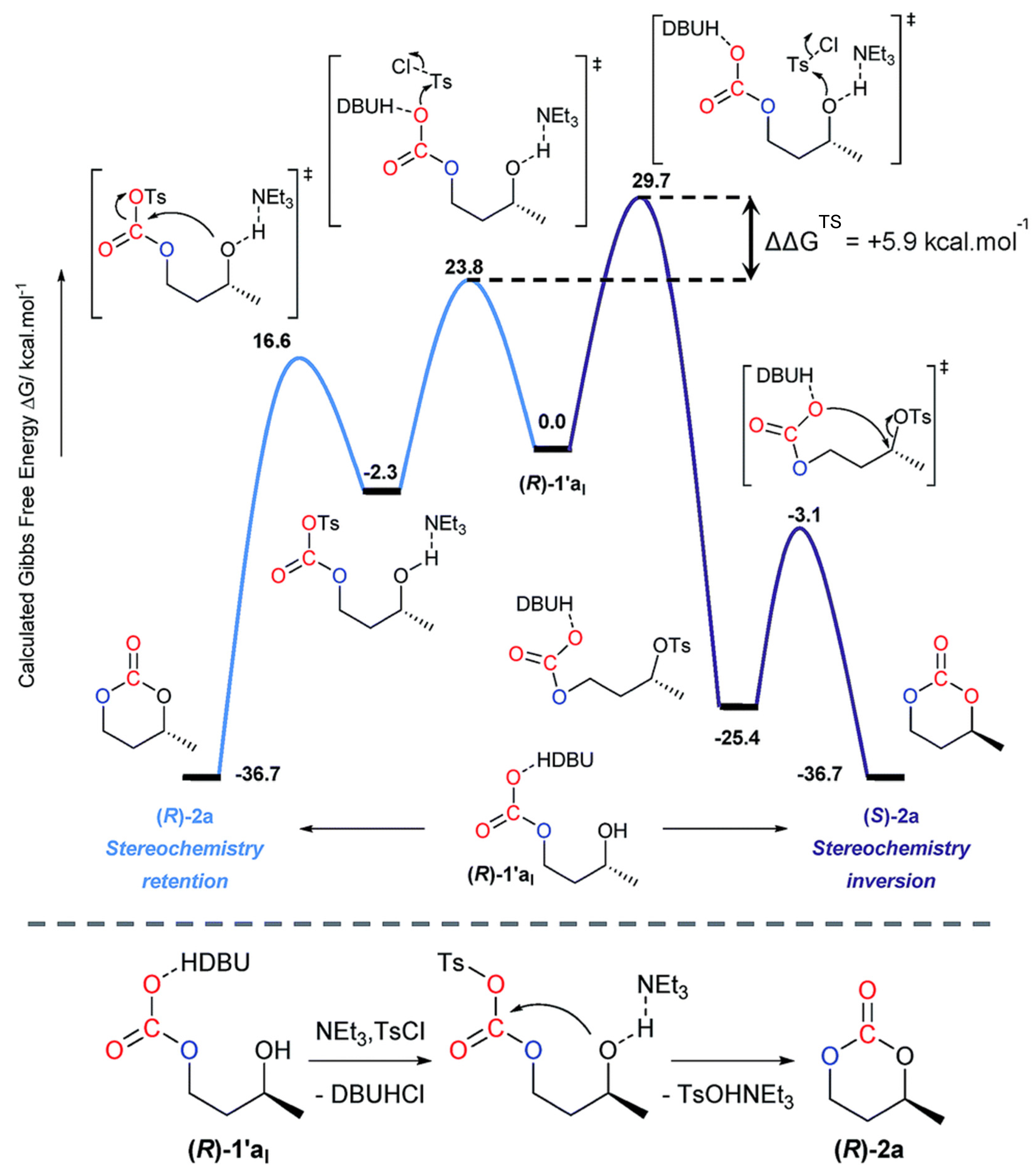
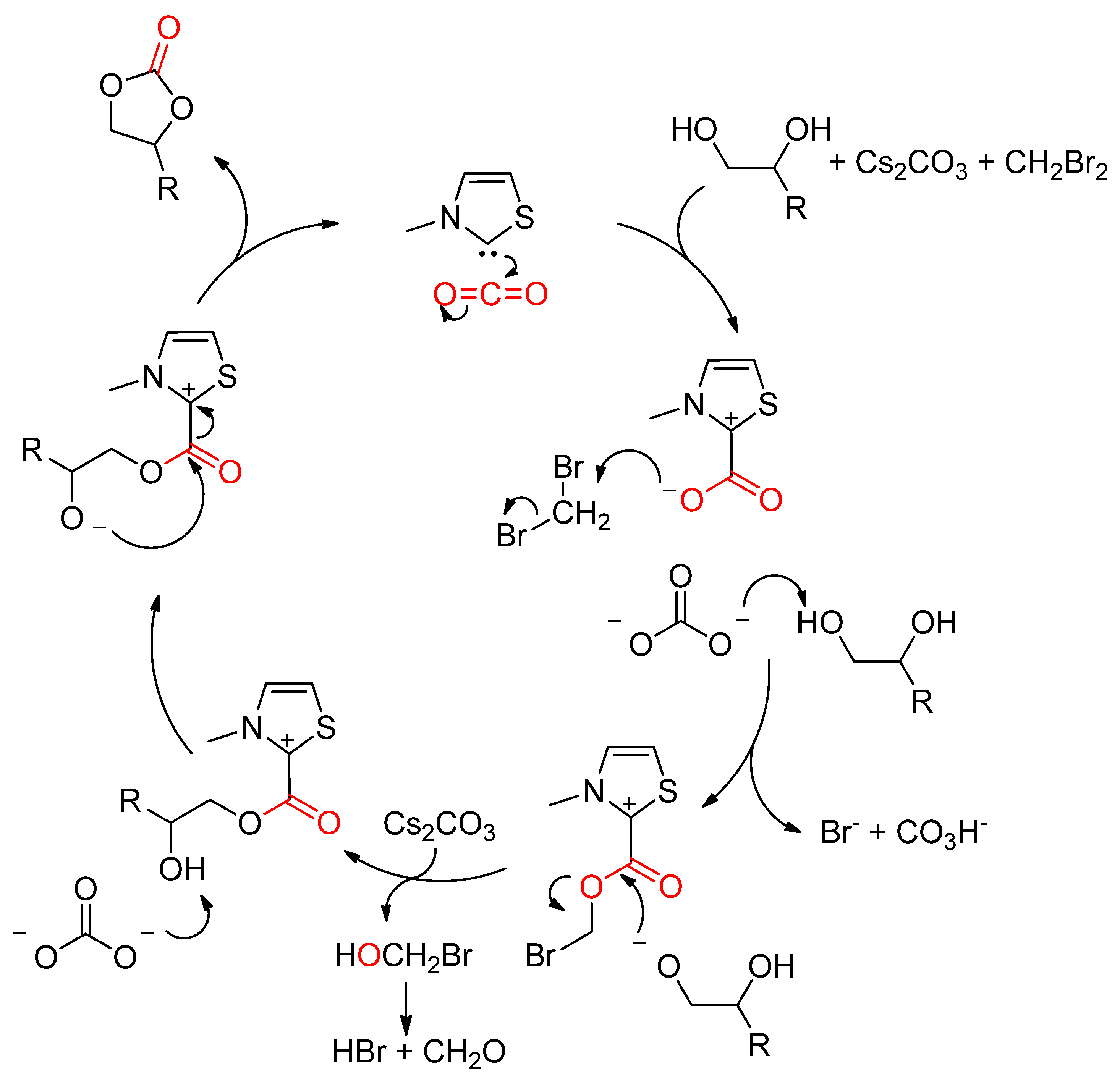
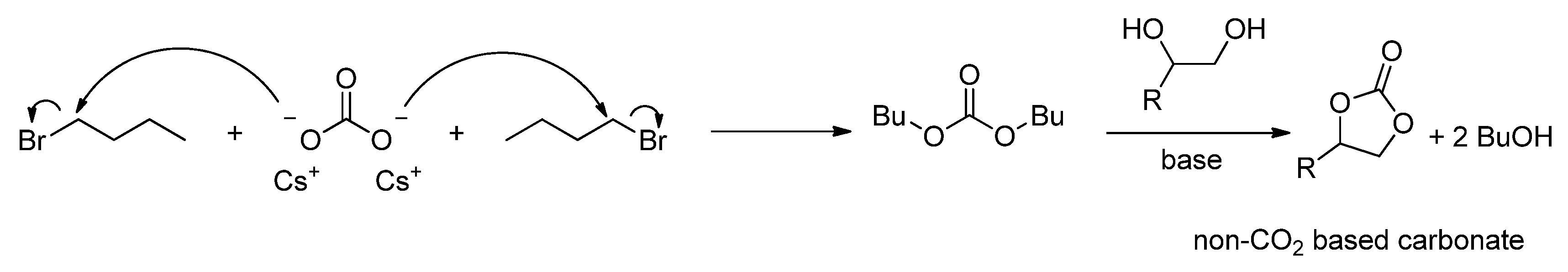
2.2.1.2. Cyclic Carbonates Synthesis via the Leaving Group Strategy or Alkylation Route
2.2.2. Synthesis of Cyclic Carbonates from Allyl Alcohols
2.2.3. Synthesis of Cyclic Carbonates from Halohydrin
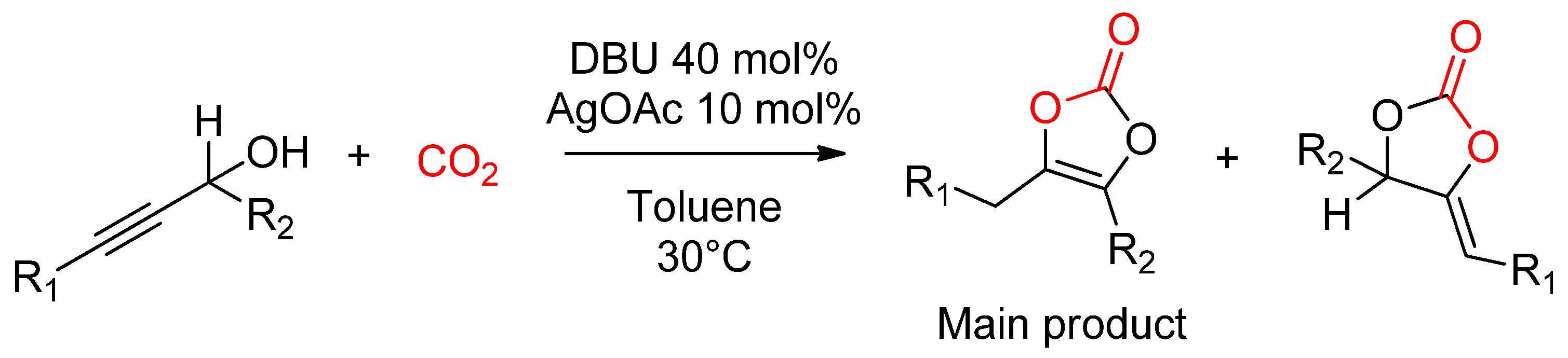
2.2.4. Synthesis of Cyclic Carbonates from Propargylic Alcohols
3. Strategies to Afford CO2-Based Polycarbonates
3.1. Direct Polymerization of CO2 with Diols
3.1.1. Direct Copolymerization by Dehydrative Polycondensation
3.1.2. Direct Copolymerization via Alkylation
3.1.3. Terpolymerization Methods
3.2. Ring-Opening Polymerization of Cyclic Carbonates
3.2.1. 5-Membered Cyclic Carbonates
3.2.2. 6-Membered Cyclic Carbonates
3.3. Polycondensation of Alcohols with (a)Cyclic Carbonates
3.3.1. DMC and DPC as Carbonylating Agents
Aliphatic PC
Aromatic and Cycloaliphatic PCs
3.3.2. Polycondensation between Bis(α-Alkylidene Cyclic Carbonates) and Diols
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stocker, T.F. Intergovernmental Panel on Climate Change 2013: Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Friedlingstein, P.; O’sullivan, M.; Jones, M.W.; Andrew, R.M.; Hauck, J.; Olsen, A.; Peters, G.P.; Peters, W.; Pongratz, J.; Sitch, S.; et al. Global Carbon Budget 2020. Earth Syst. Sci. Data 2020, 12, 3269–3340. [Google Scholar] [CrossRef]
- Jackson, R.B.; Le Quéré, C.; Andrew, R.M.; Canadell, J.G.; Peters, G.P.; Roy, J.; Wu, L. Warning signs for stabilizing global CO2 emissions. Environ. Res. Lett. 2017, 12, 110202. [Google Scholar] [CrossRef] [Green Version]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Maeda, C.; Miyazaki, Y.; Ema, T. Recent progress in catalytic conversions of carbon dioxide. Catal. Sci. Technol. 2014, 4, 1482–1497. [Google Scholar] [CrossRef] [Green Version]
- Tappe, N.A.; Reich, R.M.; D’elia, V.; Kühn, F.E. Current advances in the catalytic conversion of carbon dioxide by molecular catalysts: An update. Dalton Trans. 2018, 47, 13281–13313. [Google Scholar] [CrossRef]
- Ruscic, B.; Feller, D.; Peterson, K.A. Active Thermochemical Tables: Dissociation energies of several homonuclear first-row diatomics and related thermochemical values. Theor. Chem. Acc. 2013, 133, 1415. [Google Scholar] [CrossRef] [Green Version]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Boyaval, A.; Méreau, R.; De Winter, J.; Gerbaux, P.; Detrembleur, C.; Tassaing, T.; Jérôme, C. Organocatalytic Coupling of CO2 with Oxetane. ChemSusChem 2017, 10, 1128–1138. [Google Scholar] [CrossRef]
- Rintjema, J.; Guo, W.; Martin, E.; Escudero-Adan, E.C.; Kleij, A.W. Highly Chemoselective Catalytic Coupling of Substituted Oxetanes and Carbon Dioxide. Chem. Eur. J. 2015, 21, 10754–10762. [Google Scholar] [CrossRef]
- Katie, J.L.; Ian, D.V.I.; Michael, N.; Mani, S. Valorization of Carbon Dioxide into Oxazolidinones by Reaction with Aziridines. Curr. Green Chem. 2019, 6, 32–43. [Google Scholar]
- Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Carbon dioxide utilization with C–N bond formation: Carbon dioxide capture and subsequent conversion. Energy Environ. Sci. 2012, 5, 6602. [Google Scholar] [CrossRef]
- Harder, A.; Escher, B.I.; Landini, P.; Tobler, N.B.; Schwarzenbach, R.P. Evaluation of Bioanalytical Assays for Toxicity Assessment and Mode of Toxic Action Classification of Reactive Chemicals. Environ. Sci. Technol. 2003, 37, 4962–4970. [Google Scholar] [CrossRef]
- Kostal, J.; Voutchkova-Kostal, A.; Weeks, B.; Zimmerman, J.B.; Anastas, P.T. A Free Energy Approach to the Prediction of Olefin and Epoxide Mutagenicity and Carcinogenicity. Chem. Res. Toxicol. 2012, 25, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
- Santos, B.A.V.; Silva, V.M.T.M.; Loureiro, J.M.; Barbosa, D.; Rodrigues, A.E. Modeling of physical and chemical equilibrium for the direct synthesis of dimethyl carbonate at high pressure conditions. Fluid Phase Equilib. 2012, 336, 41–51. [Google Scholar] [CrossRef]
- Santos, B.a.V.; Pereira, C.S.M.; Silva, V.M.T.M.; Loureiro, J.M.; Rodrigues, A.E. Kinetic study for the direct synthesis of dimethyl carbonate from methanol and CO2 over CeO2 at high pressure conditions. Appl. Catal. Gen. 2013, 455, 219–226. [Google Scholar] [CrossRef]
- Müller, K.; Mokrushina, L.; Arlt, W. Thermodynamic Constraints for the Utilization of CO2. Chem. Ing. Technol. 2014, 86, 497–503. [Google Scholar] [CrossRef]
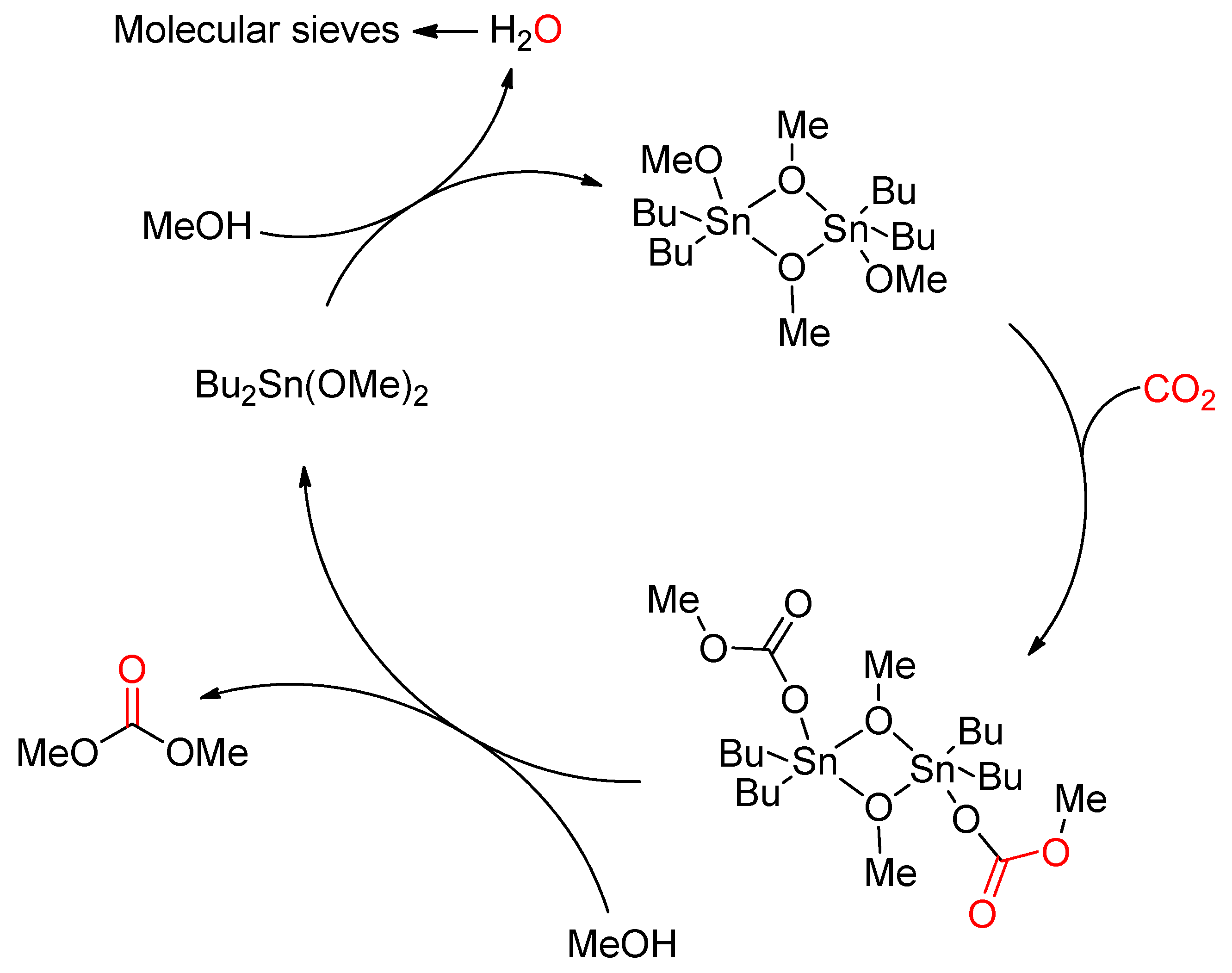
- Kizlink, J.; Pastucha, I. Preparation of Dimethyl Carbonate from Methanol and Carbon Dioxide in the Presence of Organotin Compounds. Collect. Czech. Chem. Commun. 1994, 59, 2116–2118. [Google Scholar] [CrossRef]
- Kizlink, J. Synthesis of Dimethyl Carbonate from Carbon Dioxide and Methanol in the Presence of Organotin Compounds. Collect. Czech. Chem. Commun. 1993, 58, 1399–1402. [Google Scholar] [CrossRef]
- Kizlink, J.; Pastucha, I. Preparation of Dimethyl Carbonate from Methanol and Carbon Dioxide in the Presence of Sn(IV) and Ti(IV) Alkoxides and Metal Acetates. Collect. Czech. Chem. Commun. 1995, 60, 687–692. [Google Scholar] [CrossRef]
- Zhao, T.; Han, Y.; Sun, Y. Novel reaction route for dimethyl carbonate synthesis from CO2 and methanol. Fuel Process. Technol. 2000, 62, 187–194. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Pastore, C.; Pápai, I.; Schubert, G. Reaction mechanism of the direct carboxylation of methanol to dimethylcarbonate: Experimental and theoretical studies. Top. Catal. 2006, 40, 71–81. [Google Scholar] [CrossRef]
- Du, Y.; He, L.-N.; Kong, D.-L. Magnesium-catalyzed synthesis of organic carbonate from 1,2-diol/alcohol and carbon dioxide. Catal. Commun. 2008, 9, 1754–1758. [Google Scholar] [CrossRef]
- Tomishige, K.; Sakaihori, T.; Ikeda, Y.; Fujimoto, K. A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia. Catal. Lett. 1999, 58, 225–229. [Google Scholar] [CrossRef]
- Tomishige, K.; Ikeda, Y.; Sakaihori, T.; Fujimoto, K. Catalytic properties and structure of zirconia catalysts for direct synthesis of dimethyl carbonate from methanol and carbon dioxide. J. Catal. 2000, 192, 355–362. [Google Scholar] [CrossRef]
- Ballivet-Tkatchenko, D.; Dos Santos, J.H.Z.; Philippot, K.; Vasireddy, S. Carbon dioxide conversion to dimethyl carbonate: The effect of silica as support for SnO2 and ZrO2 catalysts. C. R. Chim. 2011, 14, 780–785. [Google Scholar] [CrossRef]
- Ikeda, Y.; Asadullah, M.; Fujimoto, K.; Tomishige, K. Structure of the Active Sites on H3PO4/ZrO2 Catalysts for Dimethyl Carbonate Synthesis from Methanol and Carbon Dioxide. J. Phys. Chem. 2001, 105, 10653–10658. [Google Scholar] [CrossRef]
- Ikeda, Y.; Sakaihori, T.; Tomishige, K.; Fujimoto, K. Promoting effect of phosphoric acid on zirconia catalysts in selective synthesis of dimethyl carbonate from methanol and carbon dioxide. Catal. Lett. 2000, 66, 59–62. [Google Scholar] [CrossRef]
- Yoshida, Y.; Arai, Y.; Kado, S.; Kunimori, K.; Tomishige, K. Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts. Catal. Today 2006, 115, 95–101. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, L.; Wang, W.; Zhao, Y.; Zhang, G.; Ma, X.; Gong, J. Morphology control of ceria nanocrystals for catalytic conversion of CO2 with methanol. Nanoscale 2013, 5, 5582–5588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. DMC Formation over Ce0.5Zr0.5O2 Prepared by Complex-decomposition Method. Catal. Lett. 2009, 129, 428–436. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, S.; Song, I.K.; Jung, J.C. Direct Synthesis of Dimethyl Carbonate from Methanol and Carbon Dioxide over Ga2O3/Ce0.6Zr0.4O2 Catalysts: Effect of Acidity and Basicity of the Catalysts. Catal. Lett. 2011, 141, 531–537. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, S.; Jung, J.C.; Song, I.K. Direct synthesis of dimethyl carbonate from methanol and carbon dioxide over H3PW12O40/CeXZr1−XO2 catalysts: Effect of acidity of the catalysts. Korean J. Chem. Eng. 2011, 28, 1518–1522. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Xiao, M.; Han, D.; Lu, Y.; Meng, Y. Novel Cu–Fe bimetal catalyst for the formation of dimethyl carbonate from carbon dioxide and methanol. RSC Adv. 2012, 2, 6831–6837. [Google Scholar] [CrossRef]
- Bian, J.; Xiao, M.; Wang, S.J.; Lu, Y.X.; Meng, Y.Z. Graphite oxide as a novel host material of catalytically active Cu–Ni bimetallic nanoparticles. Catal. Commun. 2009, 10, 1529–1533. [Google Scholar] [CrossRef]
- Bian, J.; Wei, X.W.; Wang, L.; Guan, Z.P. Graphene nanosheet as support of catalytically active metal particles in DMC synthesis. Chin. Chem. Lett. 2011, 22, 57–60. [Google Scholar] [CrossRef]
- La, K.W.; Youn, M.H.; Chung, J.S.; Baeck, S.H.; Song, I.K. Synthesis of Dimethyl Carbonate from Methanol and Carbon Dioxide by Heteropolyacid/Metal Oxide Catalysts. Solid State Phenom. 2007, 119, 287–290. [Google Scholar] [CrossRef]
- Li, C.-F.; Zhong, S.-H. Study on application of membrane reactor in direct synthesis DMC from CO2 and CH3OH over Cu–KF/MgSiO catalyst. Catal. Today 2003, 82, 83–90. [Google Scholar] [CrossRef]
- Choi, J.-C.; He, L.-N.; Yasuda, H.; Sakakura, T. Selective and high yield synthesis of dimethyl carbonate directly from carbon dioxide and methanol. Green Chem. 2002, 4, 230–234. [Google Scholar] [CrossRef]
- George, J.; Patel, Y.; Pillai, S.M.; Munshi, P. Methanol assisted selective formation of 1,2-glycerol carbonate from glycerol and carbon dioxide using nBu2SnO as a catalyst. J. Mol. Catal. Chem. 2009, 304, 1–7. [Google Scholar] [CrossRef]
- Iwakabe, K.; Nakaiwa, M.; Sakakura, T.; Choi, J.-C.; Yasuda, H.; Takahashi, T.; Ooshima, Y. Reaction Rate of the Production of Dimethyl Carbonate Directly from the Supercritical CO2 and Methanol. J. Chem. Eng. Jpn. 2005, 38, 1020–1024. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, L.; Yin, S.; Yang, G.; Han, B. Driving dimethyl carbonate synthesis from CO2 and methanol and production of acetylene simultaneously using CaC2. Chem. Commun. 2018, 54, 4410–4412. [Google Scholar] [CrossRef]
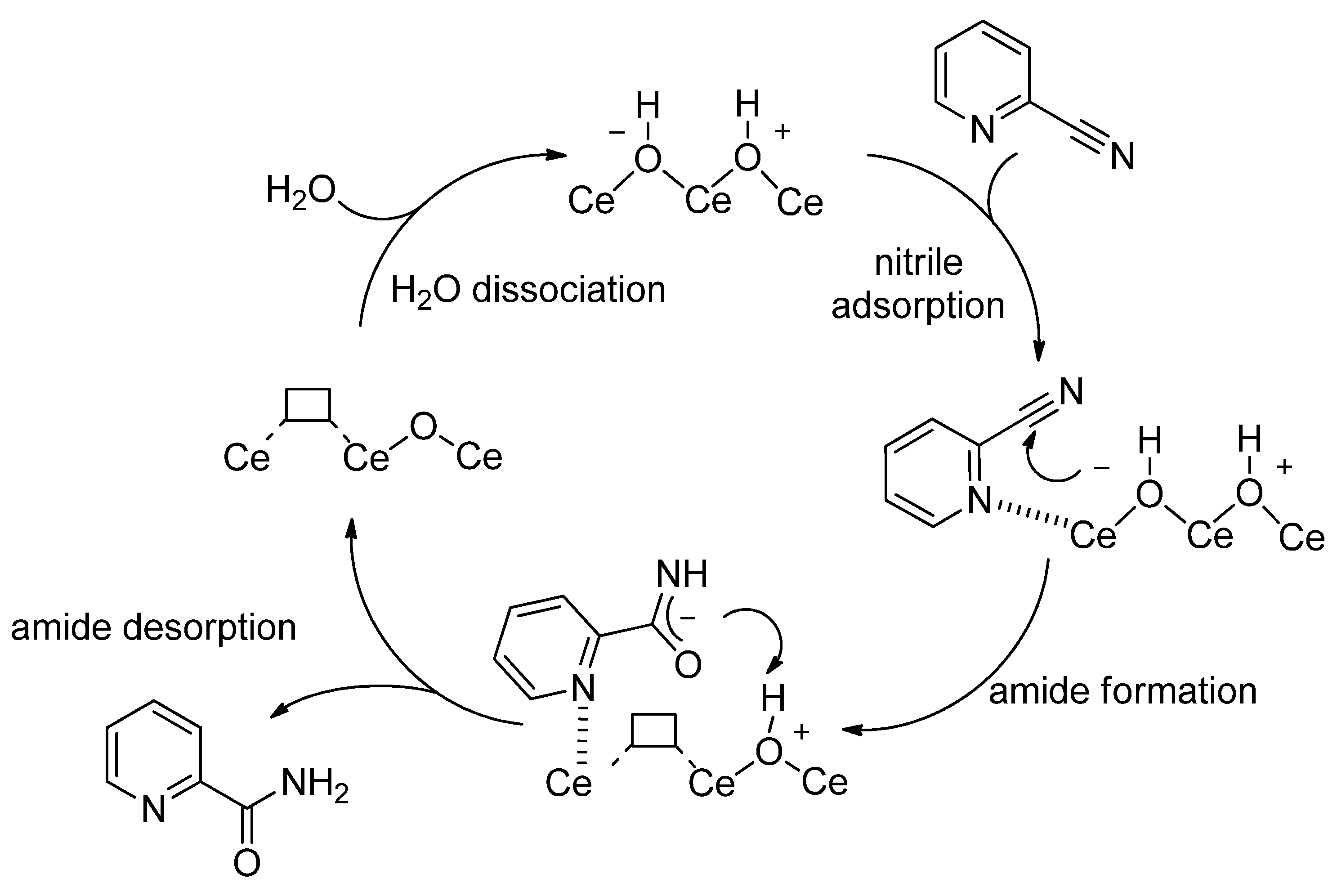
- Tamura, M.; Satsuma, A.; Shimizu, K.-I. CeO2-catalyzed nitrile hydration to amide: Reaction mechanism and active sites. Catal. Sci. Technol. 2013, 3, 1386–1393. [Google Scholar] [CrossRef]
- Tomishige, K.; Furusawa, Y.; Ikeda, Y.; Asadullah, M.; Fujimoto, K. CeO2–ZrO2 Solid Solution Catalyst for Selective Synthesis of Dimethyl Carbonate from Methanol and Carbon Dioxide. Catal. Lett. 2001, 76, 71–74. [Google Scholar] [CrossRef]
- Lamotte, J.; Morávek, V.; Bensitel, M.; Lavalley, J.C. FT-IR study of the structure and reactivity of methoxy species on ThO2 and CeO2. React. Kinet. Catal. Lett. 1988, 36, 113–118. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. Use of methanol as an IR molecular probe to study the surface of polycrystalline ceria. J. Chem. Soc. Faraday Trans. 1997, 93, 1159–1168. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Honda, M.; Kuno, S.; Begum, N.; Fujimoto, K.-I.; Suzuki, K.; Nakagawa, Y.; Tomishige, K. Catalytic synthesis of dialkyl carbonate from low pressure CO2 and alcohols combined with acetonitrile hydration catalyzed by CeO2. Appl. Catal. Gen. 2010, 384, 165–170. [Google Scholar] [CrossRef]
- Honda, M.; Suzuki, A.; Noorjahan, B.; Fujimoto, K.-I.; Suzuki, K.; Tomishige, K. Low pressure CO2 to dimethyl carbonate by the reaction with methanol promoted by acetonitrile hydration. Chem. Commun. 2009, 30, 4596–4598. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Kuno, S.; Sonehara, S.; Fujimoto, K.-I.; Suzuki, K.; Nakagawa, Y.; Tomishige, K. Tandem Carboxylation-Hydration Reaction System from Methanol, CO2 and Benzonitrile to Dimethyl Carbonate and Benzamide Catalyzed by CeO2. ChemCatChem 2011, 3, 365–370. [Google Scholar] [CrossRef]
- Tamura, M.; Shimizu, K.-I.; Satsuma, A. Comprehensive IR study on acid/base properties of metal oxides. Appl. Catal. Gen. 2012, 4339, 135–145. [Google Scholar] [CrossRef]
- Honda, M.; Tamura, M.; Nakagawa, Y.; Sonehara, S.; Suzuki, K.; Fujimoto, K.-I.; Tomishige, K. Ceria-Catalyzed Conversion of Carbon Dioxide into Dimethyl Carbonate with 2-Cyanopyridine. ChemSusChem 2013, 6, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Tamura, M.; Nakagawa, Y.; Nakao, K.; Suzuki, K.; Tomishige, K. Organic carbonate synthesis from CO2 and alcohol over CeO2 with 2-cyanopyridine: Scope and mechanistic studies. J. Catal. 2014, 318, 95–107. [Google Scholar] [CrossRef]
- Salvatore, R.N.; Chu, F.; Nagle, A.S.; Kapxhiu, E.A.; Cross, R.M.; Jung, K.W. Efficient Cs2CO3-promoted solution and solid phase synthesis of carbonates and carbamates in the presence of TBAI. Tetrahedron 2002, 58, 3329–3347. [Google Scholar] [CrossRef]
- Shi, M.; Shen, Y.-M. Synthesis of Mixed Carbonates via a Three-Component Coupling of Alcohols, CO2, and Alkyl Halides in the Presence of K2CO3 and Tetrabutylammonium Iodide. Molecules 2002, 7, 386–393. [Google Scholar] [CrossRef]
- Lethesh, K.C.; Shah, S.N.; Mutalib, M.I.A. Synthesis, Characterization, and Thermophysical Properties of 1,8-Diazobicyclo[5.4.0]undec-7-ene Based Thiocyanate Ionic Liquids. J. Chem. Eng. Data 2014, 59, 1788–1795. [Google Scholar] [CrossRef]
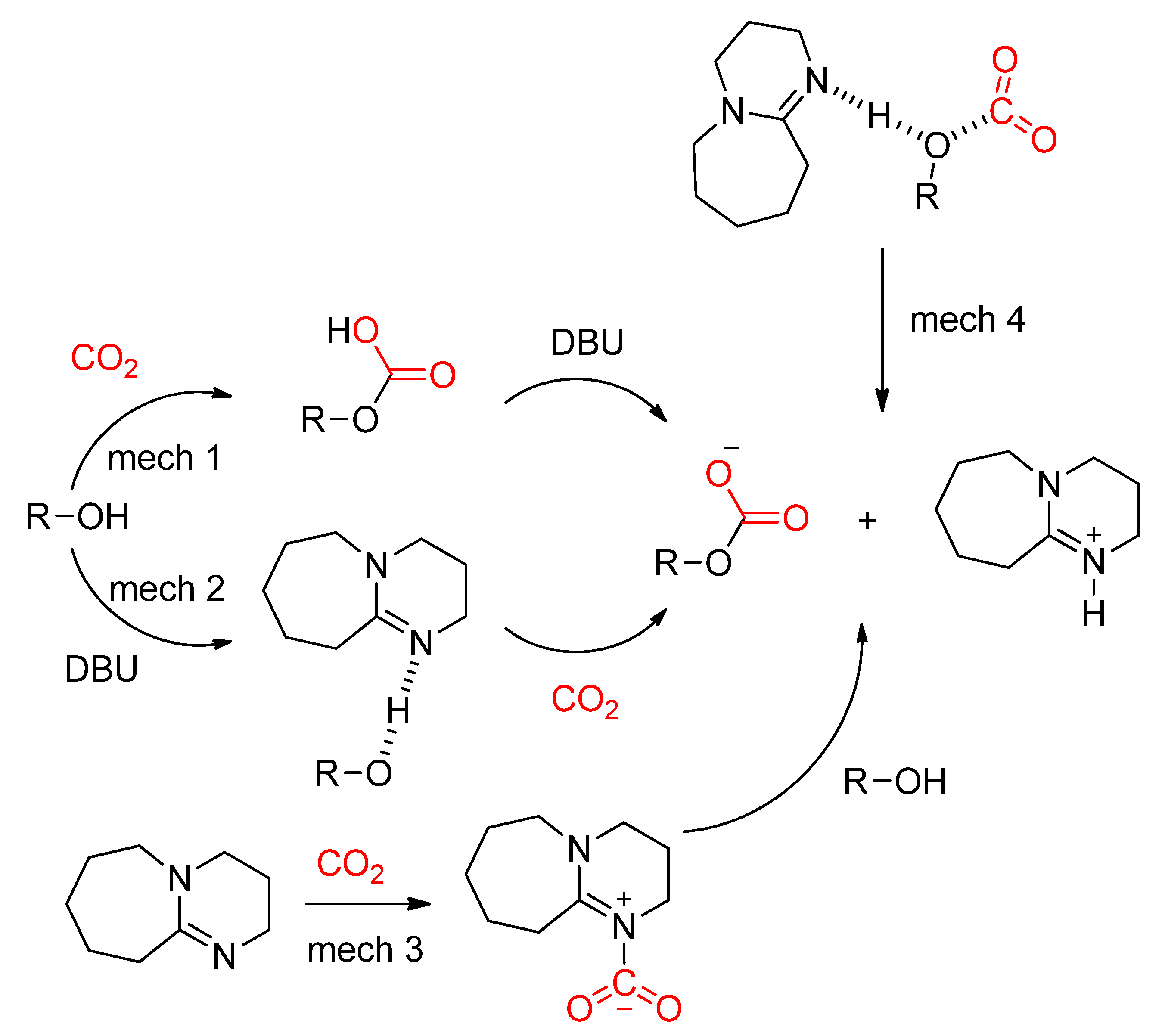
- Wang, Y.; Han, Q.; Wen, H. Theoretical discussion on the mechanism of binding CO2by DBU and alcohol. Mol. Simul. 2013, 39, 822–827. [Google Scholar] [CrossRef]
- Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. An Isolated CO2 Adduct of a Nitrogen Base: Crystal and Electronic Structures. Angew. Chem. Int. Ed. 2010, 49, 3465–3468. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, X.; Zhao, N.; Al-Arifi, A.S.N.; Aouak, T.; Al-Othman, Z.A.; Xiao, F.; Wei, W.; Sun, Y. Theoretical study of TBD-catalyzed carboxylation of propylene glycol with CO2. J. Mol. Catal. Chem. 2010, 315, 76–81. [Google Scholar] [CrossRef]
- Goodrich, P.; Gunaratne, H.Q.N.; Jin, L.; Lei, Y.; Seddon, K.R. Carbon Dioxide Utilisation for the Synthesis of Unsymmetrical Dialkyl and Cyclic Carbonates Promoted by Basic Ionic Liquids. Aust. J. Chem. 2018, 71, 181–185. [Google Scholar] [CrossRef]
- Lim, Y.N.; Lee, C.; Jang, H.-Y. Metal-Free Synthesis of Cyclic and Acyclic Carbonates from CO2and Alcohols. Eur. J. Org. Chem. 2014, 2014, 1823–1826. [Google Scholar] [CrossRef]
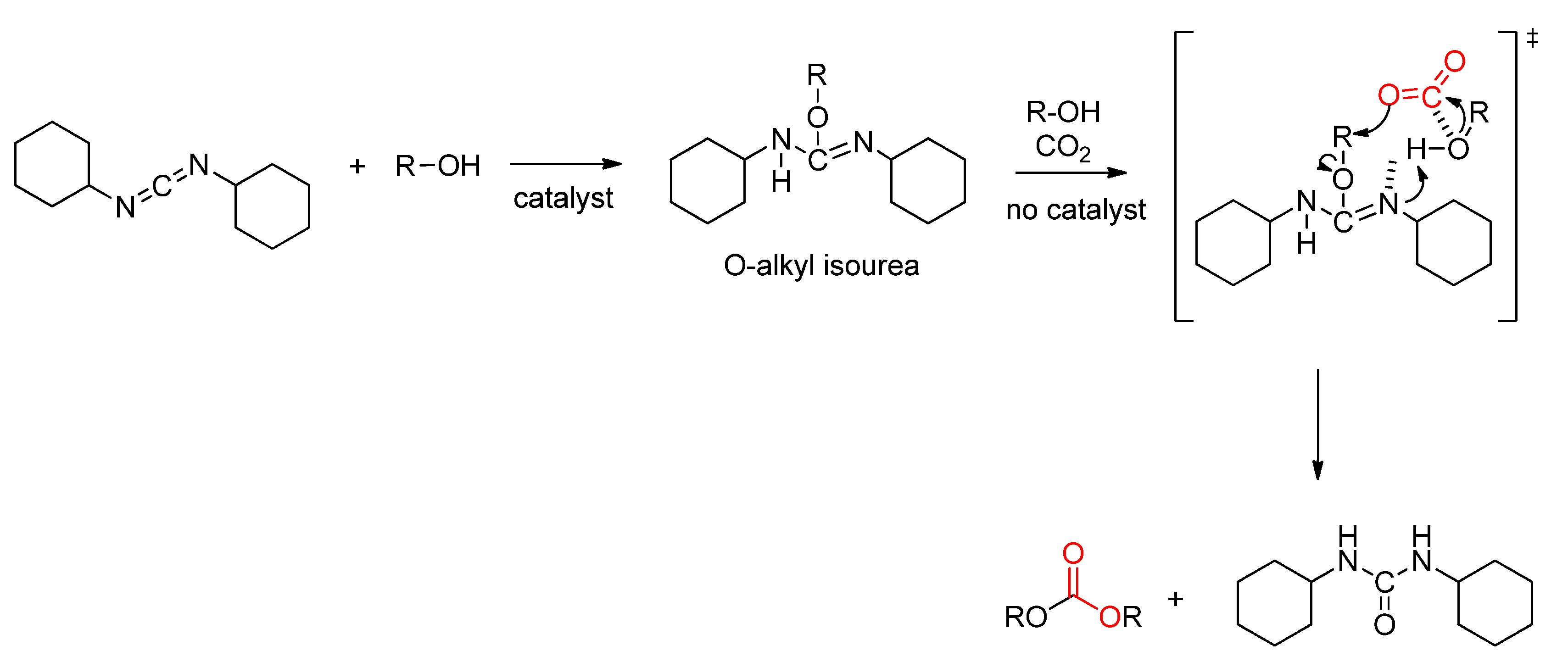
- Aresta, M.; Dibenedetto, A.; Fracchiolla, E.; Giannoccaro, P.; Pastore, C.; Pápai, I.; Schubert, G. Mechanism of Formation of Organic Carbonates from Aliphatic Alcohols and Carbon Dioxide under Mild Conditions Promoted by Carbodiimides. DFT Calculation and Experimental Study. J. Org. Chem. 2005, 70, 6177–6186. [Google Scholar] [PubMed]
- Däbritz, E. Syntheses and Reactions of O,N,N′-Trisubstituted Isoureas. Angew. Chem. Int. Ed. Engl. 1966, 5, 470–477. [Google Scholar] [CrossRef]
- Imberdis, A.; Lefèvre, G.; Thuéry, P.; Cantat, T. Metal-Free and Alkali-Metal-Catalyzed Synthesis of Isoureas from Alcohols and Carbodiimides. Angew. Chem. 2018, 130, 3138–3142. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhou, J.; Zhao, S.; Zhao, Y.; Ma, X. Enhancement of Dimethyl Carbonate Synthesis with In Situ Hydrolysis of 2,2-Dimethoxy Propane. Chem. Eng. Technol. 2016, 39, 723–729. [Google Scholar] [CrossRef]
- Tomishige, K.; Kunimori, K. Catalytic and direct synthesis of dimethyl carbonate starting from carbon dioxide using CeO2-ZrO2 solid solution heterogeneous catalyst: Effect of H2O removal from the reaction system. Appl. Catal. Gen. 2002, 237, 103–109. [Google Scholar] [CrossRef]
- Chang, T.; Tamura, M.; Nakagawa, Y.; Fukaya, N.; Choi, J.-C.; Mishima, T.; Matsumoto, S.; Hamura, S.; Tomishige, K. An effective combination catalyst of CeO2 and zeolite for the direct synthesis of diethyl carbonate from CO2 and ethanol with 2,2-diethoxypropane as a dehydrating agent. Green Chem. 2020, 22, 7321–7327. [Google Scholar] [CrossRef]
- Sakakura, T.; Saito, Y.; Okano, M.; Choi, J.-C.; Sako, T. Selective Conversion of Carbon Dioxide to Dimethyl Carbonate by Molecular Catalysis. J. Org. Chem. 1998, 63, 7095–7096. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Liu, Z.-W.; Lu, J.; Liu, Z.-T. Synthesis of Dimethyl Carbonate from Carbon Dioxide and Methanol over CexZr1-xO2 and [EMIM]Br/Ce0.5Zr0.5O2. Ind. Eng. Chem. Res. 2011, 50, 1981–1988. [Google Scholar] [CrossRef]
- Putro, W.S.; Ikeda, A.; Shigeyasu, S.; Hamura, S.; Matsumoto, S.; Lee, V.Y.; Choi, J.-C.; Fukaya, N. Sustainable Catalytic Synthesis of Diethyl Carbonate. ChemSusChem 2020, 14, 842–846. [Google Scholar] [CrossRef]
- Guidi, S.; Calmanti, R.; Noè, M.; Perosa, A.; Selva, M. Thermal (Catalyst-Free) Transesterification of Diols and Glycerol with Dimethyl Carbonate: A Flexible Reaction for Batch and Continuous-Flow Applications. ACS Sustain. Chem. Eng. 2016, 4, 6144–6151. [Google Scholar] [CrossRef]
- Baral, E.R.; Lee, J.H.; Kim, J.G. Diphenyl Carbonate: A Highly Reactive and Green Carbonyl Source for the Synthesis of Cyclic Carbonates. J. Org. Chem. 2018, 83, 11768–11776. [Google Scholar] [CrossRef]
- Ochoa-Gómez, J.R.; Gómez-Jiménez-Aberasturi, O.; Ramírez-López, C.; Maestro-Madurga, B. Synthesis of glycerol 1,2-carbonate by transesterification of glycerol with dimethyl carbonate using triethylamine as a facile separable homogeneous catalyst. Green Chem. 2012, 14, 3368. [Google Scholar] [CrossRef]
- Schutyser, W.; Van Den Bosch, S.; Renders, T.; De Boe, T.; Koelewijn, S.F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of bio-based solvents on the catalytic reductive fractionation of birch wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Koelewijn, S.F.; Van Den Bosch, S.; Renders, T.; Schutyser, W.; Lagrain, B.; Smet, M.; Thomas, J.; Dehaen, W.; Van Puyvelde, P.; Witters, H.; et al. Sustainable bisphenols from renewable softwood lignin feedstock for polycarbonates and cyanate ester resins. Green Chem. 2017, 19, 2561–2570. [Google Scholar] [CrossRef]
- Ma, J.; Liu, J.; Zhang, Z.; Han, B. Mechanisms of ethylene glycol carbonylation with carbon dioxide. Comput. Theor. Chem. 2012, 992, 103–109. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C. A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: The role of the catalyst, solvent and reaction conditions. J. Mol. Catal. Chem. 2006, 257, 149–153. [Google Scholar] [CrossRef]
- Tamura, M.; Honda, M.; Nakagawa, Y.; Tomishige, K. Direct conversion of CO2 with diols, aminoalcohols and diamines to cyclic carbonates, cyclic carbamates and cyclic ureas using heterogeneous catalysts. J. Chem. Technol. Biotechnol. 2014, 89, 19–33. [Google Scholar] [CrossRef]
- Su, X.; Lin, W.; Cheng, H.; Zhang, C.; Wang, Y.; Yu, X.; Wu, Z.; Zhao, F. Metal-free catalytic conversion of CO2and glycerol to glycerol carbonate. Green Chem. 2017, 19, 1775–1781. [Google Scholar] [CrossRef]
- Honda, M.; Tamura, M.; Nakao, K.; Suzuki, K.; Nakagawa, Y.; Tomishige, K. Direct Cyclic Carbonate Synthesis from CO2 and Diol over Carboxylation/Hydration Cascade Catalyst of CeO2 with 2-Cyanopyridine. ACS Catal. 2014, 4, 1893–1896. [Google Scholar] [CrossRef]
- Huang, S.; Ma, J.; Li, J.; Zhao, N.; Wei, W.; Sun, Y. Efficient propylene carbonate synthesis from propylene glycol and carbon dioxide via organic bases. Catal. Commun. 2008, 9, 276–280. [Google Scholar] [CrossRef]
- Kitamura, T.; Inoue, Y.; Maeda, T.; Oyamada, J. Convenient synthesis of ethylene carbonates from carbon dioxide and 1,2-diols at atmospheric pressure of carbon dioxide. Synth. Commun. 2015, 46, 39–45. [Google Scholar] [CrossRef]
- Gregory, G.L.; Ulmann, M.; Buchard, A. Synthesis of 6-membered cyclic carbonates from 1,3-diols and low CO2 pressure: A novel mild strategy to replace phosgene reagents. RSC Adv. 2015, 5, 39404–39408. [Google Scholar] [CrossRef] [Green Version]
- Reithofer, M.R.; Sum, Y.N.; Zhang, Y. Synthesis of cyclic carbonates with carbon dioxide and cesium carbonate. Green Chem. 2013, 15, 2086–2090. [Google Scholar] [CrossRef]
- Mcguire, T.M.; López-Vidal, E.M.; Gregory, G.L.; Buchard, A. Synthesis of 5- to 8-membered cyclic carbonates from diols and CO2: A one-step, atmospheric pressure and ambient temperature procedure. J. CO2 Util. 2018, 27, 283–288. [Google Scholar] [CrossRef]
- Brege, A.; Méreau, R.; Mcgehee, K.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. The coupling of CO2 with diols promoted by organic dual systems: Towards products divergence via benchmarking of the performance metrics. J. CO2 Util. 2020, 38, 88–98. [Google Scholar] [CrossRef]
- Voutchkova, A.M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R.H. Imidazolium Carboxylates as Versatile and Selective N-Heterocyclic Carbene Transfer Agents: Synthesis, Mechanism, and Applications. J. Am. Chem. Soc. 2007, 129, 12834–12846. [Google Scholar] [CrossRef]
- Riduan, S.N.; Zhang, Y.; Ying, J.Y. Conversion of Carbon Dioxide into Methanol with Silanes over N-Heterocyclic Carbene Catalysts. Angew. Chem. Int. Ed. 2009, 48, 3322–3325. [Google Scholar] [CrossRef] [PubMed]
- Bobbink, F.D.; Gruszka, W.; Hulla, M.; Das, S.; Dyson, P.J. Synthesis of cyclic carbonates from diols and CO2 catalyzed by carbenes. Chem. Commun. 2016, 52, 10787–10790. [Google Scholar] [CrossRef] [Green Version]
- Cardillo, G.; Orena, M.; Porzi, G.; Sandri, S. A new regio- and stereo-selective functionalization of allylic and homoallylic alcohols. J. Chem. Soc. Chem. Commun. 1981, 10, 465–466. [Google Scholar] [CrossRef]
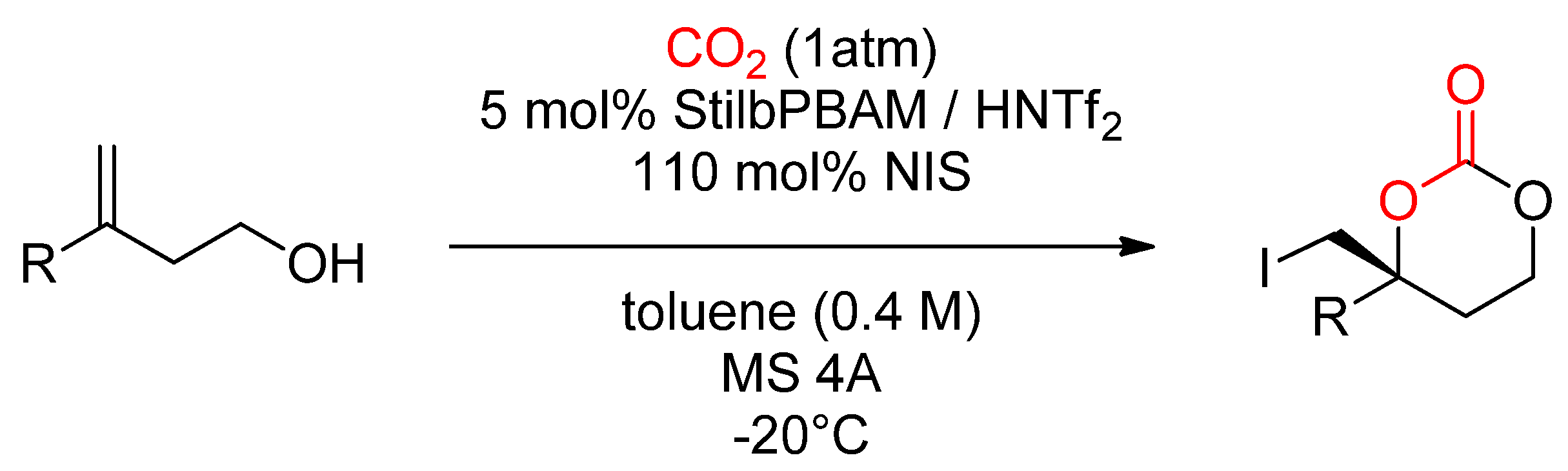
- Vara, B.A.; Struble, T.J.; Wang, W.; Dobish, M.C.; Johnston, J.N. Enantioselective Small Molecule Synthesis by Carbon Dioxide Fixation using a Dual Brønsted Acid/Base Organocatalyst. J. Am. Chem. Soc. 2015, 137, 7302–7305. [Google Scholar] [CrossRef] [Green Version]
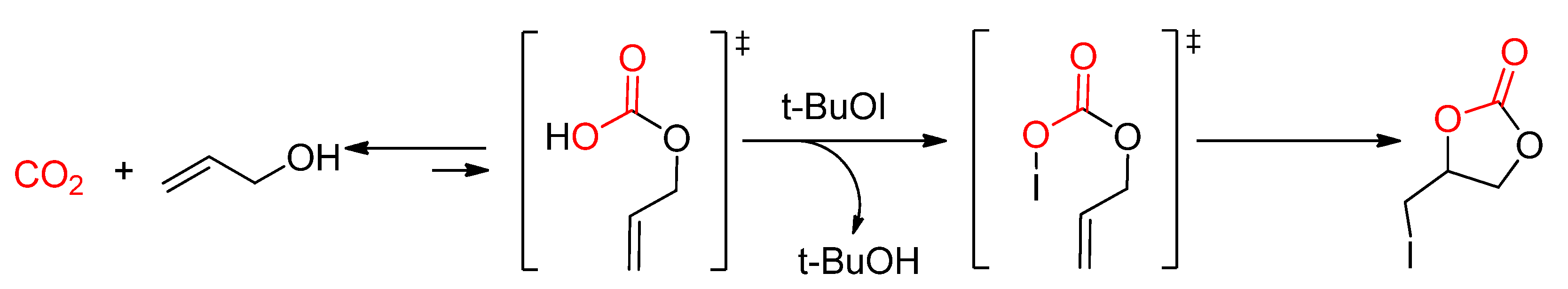
- Minakata, S.; Sasaki, I.; Ide, T. Atmospheric CO2 Fixation by Unsaturated Alcohols Using tBuOI under Neutral Conditions. Angew. Chem. Int. Ed. 2010, 49, 1309–1311. [Google Scholar] [CrossRef]
- Wang, J.-L.; He, L.-N.; Dou, X.-Y.; Wu, F. Poly(ethylene glycol): An Alternative Solvent for the Synthesis of Cyclic Carbonate from Vicinal Halohydrin and Carbon Dioxide. Aust. J. Chem. 2009, 62, 917–920. [Google Scholar] [CrossRef]
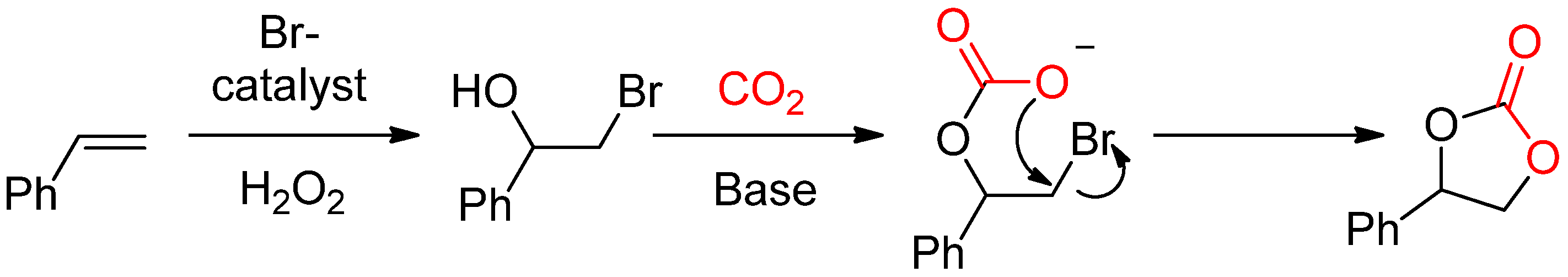
- Eghbali, N.; Li, C.-J. Conversion of carbon dioxide and olefins into cyclic carbonates in water. Green Chem. 2007, 9, 213–215. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. (Eds.) March’s Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 5th; Wiley Interscience: New York, NY, USA, 2001; ISBN 0-471-58589-0. [Google Scholar]
- Lang, X.D.; He, L.N. Green Catalytic Process for Cyclic Carbonate Synthesis from Carbon Dioxide under Mild Conditions. Chem. Rec. 2016, 16, 1337–1352. [Google Scholar] [CrossRef]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. Stereoselective Formation of α-Alkylidene Cyclic Carbonates via Carboxylative Cyclization of Propargyl Alcohols in Supercritical Carbon Dioxide. J. Org. Chem. 2007, 72, 647–649. [Google Scholar] [CrossRef]
- Sun, Y.-L.; Wei, Y.; Shi, M. Phosphine-catalyzed fixation of CO2 with γ-hydroxyl alkynone under ambient temperature and pressure: Kinetic resolution and further conversion. Org. Chem. Front. 2019, 6, 2420–2429. [Google Scholar] [CrossRef]
- Matthews, C.N.; Driscoll, J.S.; Birum, G.H. Mesomeric phosphonium inner salts. Chem. Commun. 1966, 20, 736–737. [Google Scholar] [CrossRef]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. Int. Ed. 2009, 48, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, N.; Lyu, Y. Theoretical Insights into the Catalytic Mechanism of N-Heterocyclic Olefins in Carboxylative Cyclization of Propargyl Alcohol with CO2. J. Org. Chem. 2016, 81, 5303–5313. [Google Scholar] [CrossRef]
- Yan, Z.-E.; Huo, R.-P.; Guo, L.-H.; Zhang, X. The mechanisms for N-heterocyclic olefin-catalyzed formation of cyclic carbonate from CO2 and propargylic alcohols. J. Mol. Model. 2016, 22, 94. [Google Scholar] [CrossRef]
- Li, W.; Huang, D.; Lyu, Y. A comparative computational study of N-heterocyclic olefin and N-heterocyclic carbene mediated carboxylative cyclization of propargyl alcohols with CO2. Org. Biomol. Chem. 2016, 14, 10875–10885. [Google Scholar] [CrossRef] [PubMed]
- Boyaval, A.; Méreau, R.; Grignard, B.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic Coupling of CO2 with a Propargylic Alcohol: A Comprehensive Mechanistic Study. ChemSusChem 2017, 10, 1241–1248. [Google Scholar] [CrossRef]
- Méreau, R.; Grignard, B.; Boyaval, A.; Detrembleur, C.; Jerome, C.; Tassaing, T. Tetrabutylammonium Salts: Cheap Catalysts for the Facile and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Alkynols. ChemCatChem 2018, 10, 956–960. [Google Scholar] [CrossRef]
- Grignard, B.; Ngassamtounzoua, C.; Gennen, S.; Gilbert, B.; Méreau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Boosting the Catalytic Performance of Organic Salts for the Fast and Selective Synthesis of α-Alkylidene Cyclic Carbonates from Carbon Dioxide and Propargylic Alcohols. ChemCatChem 2018, 10, 2584–2592. [Google Scholar] [CrossRef]
- Gennen, S.; Grignard, B.; Tassaing, T.; Jerome, C.; Detrembleur, C. CO2-Sourced alpha-Alkylidene Cyclic Carbonates: A Step Forward in the Quest for Functional Regioregular Poly(urethane)s and Poly(carbonate)s. Angew. Chem. Int. Ed. Engl. 2017, 56, 10394–10398. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhao, Y.; Li, Z.; Wang, H.; Fan, M.; Wang, J. Efficient Ionic-Liquid-Promoted Chemical Fixation of CO2 into α-Alkylidene Cyclic Carbonates. ChemSusChem 2017, 10, 1120–1127. [Google Scholar] [CrossRef]
- Chen, K.; Shi, G.; Dao, R.; Mei, K.; Zhou, X.; Li, H.; Wang, C. Tuning the basicity of ionic liquids for efficient synthesis of alkylidene carbonates from CO2 at atmospheric pressure. Chem. Commun. 2016, 52, 7830–7833. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Song, J.; Xie, C.; Wu, H.; Jiang, T.; Yang, G.; Han, B. Transformation of CO2 into α-Alkylidene Cyclic Carbonates at Room Temperature Cocatalyzed by CuI and Ionic Liquid with Biomass-Derived Levulinate Anion. ACS Sustain. Chem. Eng. 2019, 7, 5614–5619. [Google Scholar] [CrossRef]
- Ouyang, L.; Tang, X.; He, H.; Qi, C.; Xiong, W.; Ren, Y.; Jiang, H. Copper-Promoted Coupling of Carbon Dioxide and Propargylic Alcohols: Expansion of Substrate Scope and Trapping of Vinyl Copper Intermediate. Adv. Synth. Catal. 2015, 357, 2556–2565. [Google Scholar] [CrossRef]
- Satoshi, K.; Shunsuke, Y.; Yuudai, S.; Wataru, Y.; Hau-Man, C.; Kosuke, F.; Kohei, S.; Izumi, I.; Taketo, I.; Tohru, Y. Silver-Catalyzed Carbon Dioxide Incorporation and Rearrangement on Propargylic Derivatives. Bull. Chem. Soc. Jpn. 2011, 84, 698–717. [Google Scholar]
- Yamada, T.; Ugajin, R.; Kikuchi, S. Silver-Catalyzed Efficient Synthesis of Vinylene Carbonate Derivatives from Carbon Dioxide. Synlett 2014, 25, 1178–1180. [Google Scholar] [CrossRef]
- Yuan, Y.; Xie, Y.; Zeng, C.; Song, D.; Chaemchuen, S.; Chen, C.; Verpoort, F. A recyclable AgI/OAc− catalytic system for the efficient synthesis of α-alkylidene cyclic carbonates: Carbon dioxide conversion at atmospheric pressure. Green Chem. 2017, 19, 2936–2940. [Google Scholar] [CrossRef]
- Li, J.-Y.; Han, L.-H.; Xu, Q.-C.; Song, Q.-W.; Liu, P.; Zhang, K. Cascade Strategy for Atmospheric Pressure CO2 Fixation to Cyclic Carbonates via Silver Sulfadiazine and Et4NBr Synergistic Catalysis. ACS Sustain. Chem. Eng. 2019, 7, 3378–3388. [Google Scholar] [CrossRef]
- Yuan, Y.; Xie, Y.; Song, D.; Zeng, C.; Chaemchuen, S.; Chen, C.; Verpoort, F. One-pot carboxylative cyclization of propargylic alcohols and CO2 catalysed by N-heterocyclic carbene/Ag systems. Appl. Organomet. Chem. 2017, 31, e3867. [Google Scholar] [CrossRef]
- Cui, M.; Qian, Q.; He, Z.; Ma, J.; Kang, X.; Hu, J.; Liu, Z.; Han, B. Synthesizing Ag Nanoparticles of Small Size on a Hierarchical Porosity Support for the Carboxylative Cyclization of Propargyl Alcohols with CO2 under Ambient Conditions. Chem.–A Eur. J. 2015, 21, 15924–15928. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Chen, Y.; Ma, Z.; Ji, G.; Gao, X.; Han, B.; Liu, Z. Metalated Mesoporous Poly(triphenylphosphine) with Azo Functionality: Efficient Catalysts for CO2 Conversion. ACS Catal. 2016, 6, 1268–1273. [Google Scholar] [CrossRef]
- Zhou, Z.; He, C.; Yang, L.; Wang, Y.; Liu, T.; Duan, C. Alkyne Activation by a Porous Silver Coordination Polymer for Heterogeneous Catalysis of Carbon Dioxide Cycloaddition. ACS Catal. 2017, 7, 2248–2256. [Google Scholar] [CrossRef]
- Dabral, S.; Bayarmagnai, B.; Hermsen, M.; Schießl, J.; Mormul, V.; Hashmi, A.S.K.; Schaub, T. Silver-Catalyzed Carboxylative Cyclization of Primary Propargyl Alcohols with CO2. Org. Lett. 2019, 21, 1422–1425. [Google Scholar] [CrossRef]
- Jung, M.E.; Piizzi, G. gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev. 2005, 105, 1735–1766. [Google Scholar] [CrossRef]
- Islam, S.S.; Salam, N.; Molla, R.A.; Riyajuddin, S.; Yasmin, N.; Das, D.; Ghosh, K.; Islam, S.M. Zinc (II) incorporated porous organic polymeric material (POPs): A mild and efficient catalyst for synthesis of dicoumarols and carboxylative cyclization of propargyl alcohols and CO2 in ambient conditions. Mol. Catal. 2019, 477, 110541. [Google Scholar] [CrossRef]
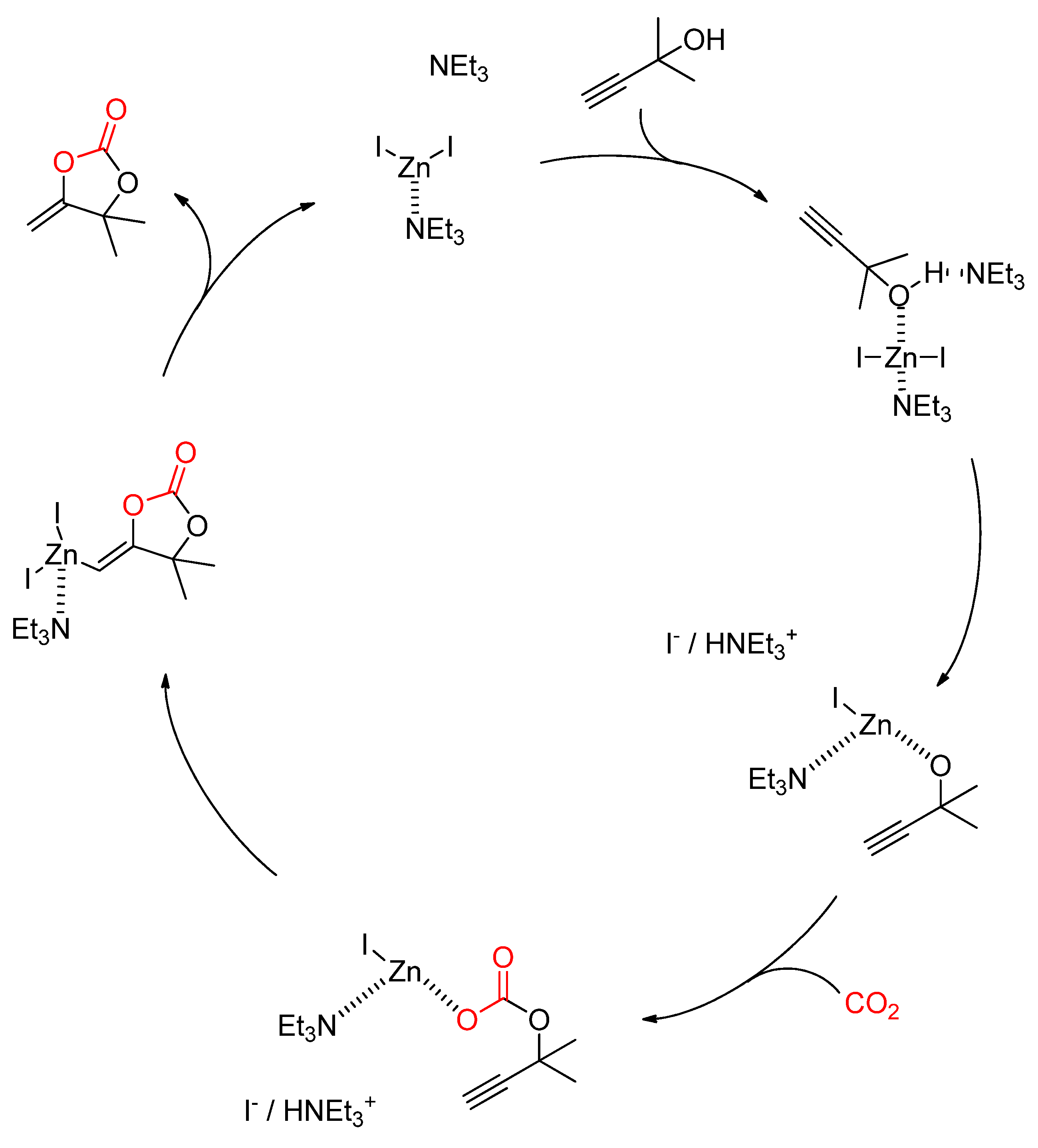
- Hu, J.; Ma, J.; Zhu, Q.; Qian, Q.; Han, H.; Mei, Q.; Han, B. Zinc(ii)-catalyzed reactions of carbon dioxide and propargylic alcohols to carbonates at room temperature. Green Chem. 2016, 18, 382–385. [Google Scholar] [CrossRef]
- Ma, J.; Lu, L.; Mei, Q.; Zhu, Q.; Hu, J.; Han, B. ZnI2/NEt3-Catalyzed Cycloaddition of CO2 with Propargylic Alcohols: Computational Study on Mechanism. ChemCatChem 2017, 9, 4090–4097. [Google Scholar] [CrossRef]
- Tamura, M.; Ito, K.; Honda, M.; Nakagawa, Y.; Sugimoto, H.; Tomishige, K. Direct Copolymerization of CO2 and Diols. Sci. Rep. 2016, 6, 24038. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Matsuda, K.; Nakayama, A.; Tamura, M.; Nakagawa, Y.; Tomishige, K. Direct Synthesis of Alternating Polycarbonates from CO2 and Diols by Using a Catalyst System of CeO2 and 2-Furonitrile. ACS Sustain. Chem. Eng. 2019, 7, 6304–6315. [Google Scholar] [CrossRef]
- Gong, Z.-J.; Li, Y.-R.; Wu, H.-L.; Lin, S.D.; Yu, W.-Y. Direct copolymerization of carbon dioxide and 1,4-butanediol enhanced by ceria nanorod catalyst. Appl. Catal. B Environ. 2020, 265, 118524. [Google Scholar] [CrossRef]
- Kadokawa, J.-I.; Habu, H.; Fukamachi, S.; Karasu, M.; Tagaya, H.; Chiba, K. Direct polycondensation of carbon dioxide with xylylene glycols: A new method for the synthesis of polycarbonates. Macromol. Rapid Commun. 1998, 19, 657–660. [Google Scholar] [CrossRef]
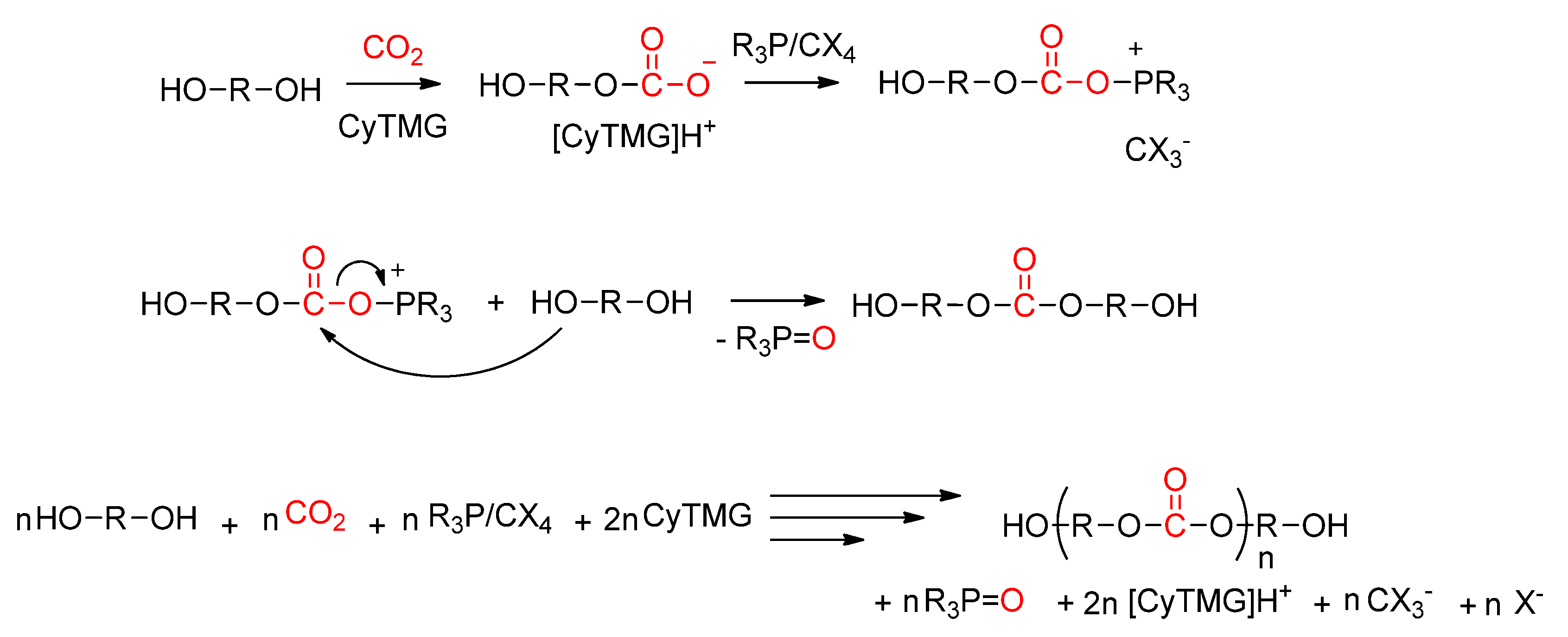
- Kadokawa, J.-I.; Fukamachi, S.; Tagaya, H.; Chiba, K. Direct Polycondensation of Carbon Dioxide with Various Diols Using the Triphenylphosphine/Bromotrichloromethane/N-Cyclohexyl-N′,N′,N″,N″-tetramethylguanidine System as Condensing Agent. Polym. J. 2000, 32, 703. [Google Scholar] [CrossRef] [Green Version]
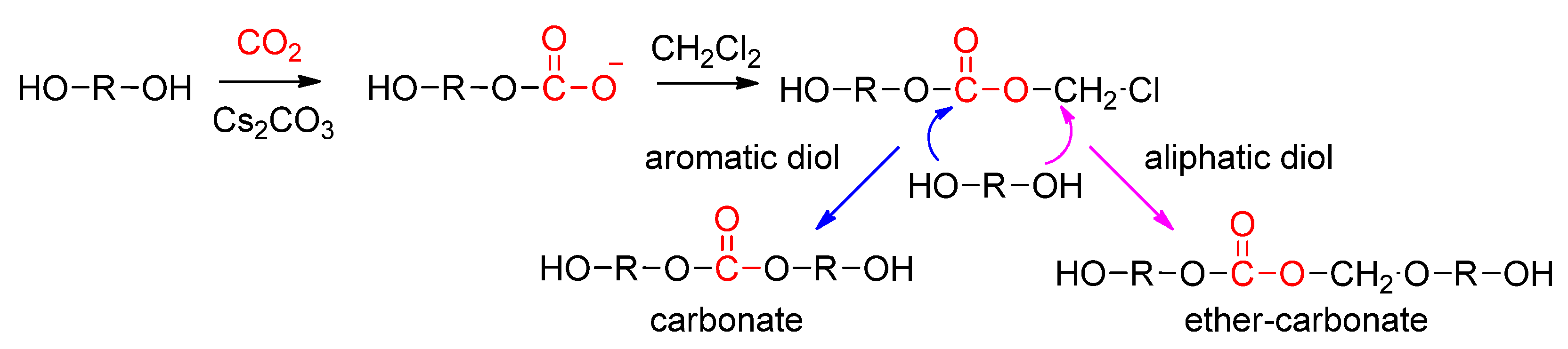
- Bian, S.; Pagan, C.; Andrianova Artemyeva, A.A.; Du, G. Synthesis of Polycarbonates and Poly(ether carbonate)s Directly from Carbon Dioxide and Diols Promoted by a Cs2CO3/CH2Cl2 System. ACS Omega 2016, 1, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Chen, Z.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. Synthesis of polyglycocarbonates through polycondensation of glucopyranosides with CO2. Polym. Chem. 2017, 8, 2640–2646. [Google Scholar] [CrossRef]
- Soga, K.; Toshida, Y.; Hosoda, S.; Ikeda, S. A convenient synthesis of a polycarbonate. Makromol. Chem. 1977, 178, 2747–2751. [Google Scholar] [CrossRef]
- Soga, K.; Toshida, Y.; Hosoda, S.; Ikeda, S. Synthesis of a polycarbonate directly from carbon dioxide, alkali metal diolates and α,ω-dihalo compounds. Makromol. Chem. 1978, 179, 2379–2386. [Google Scholar] [CrossRef]
- Chen, Z.; Hadjichristidis, N.; Feng, X.; Gnanou, Y. Cs2CO3-promoted polycondensation of CO2 with diols and dihalides for the synthesis of miscellaneous polycarbonates. Polym. Chem. 2016, 7, 4944–4952. [Google Scholar] [CrossRef]
- Bian, S.; Andrianova, A.A.; Kubatova, A.; Du, G. Effect of dihalides on the polymer linkages in the Cs2CO3-promoted polycondensation of 1 atm carbon dioxide and diols. Mater. Today Commun. 2019, 18, 100–109. [Google Scholar] [CrossRef]
- Höcker, H.; Keul, H.; Kühling, S.; Hovestadt, W.; Müller, A.; Wurm, B. Ring-opening polymerization and copolymerization of cyclic carbonates with a variety of initiating systems. Makromol. Chem. Macromol. Symp. 1993, 73, 1–5. [Google Scholar] [CrossRef]
- Rokicki, G. Aliphatic cyclic carbonates and spiroorthocarbonates as monomers. Prog. Polym. Sci. 2000, 25, 259–342. [Google Scholar] [CrossRef]
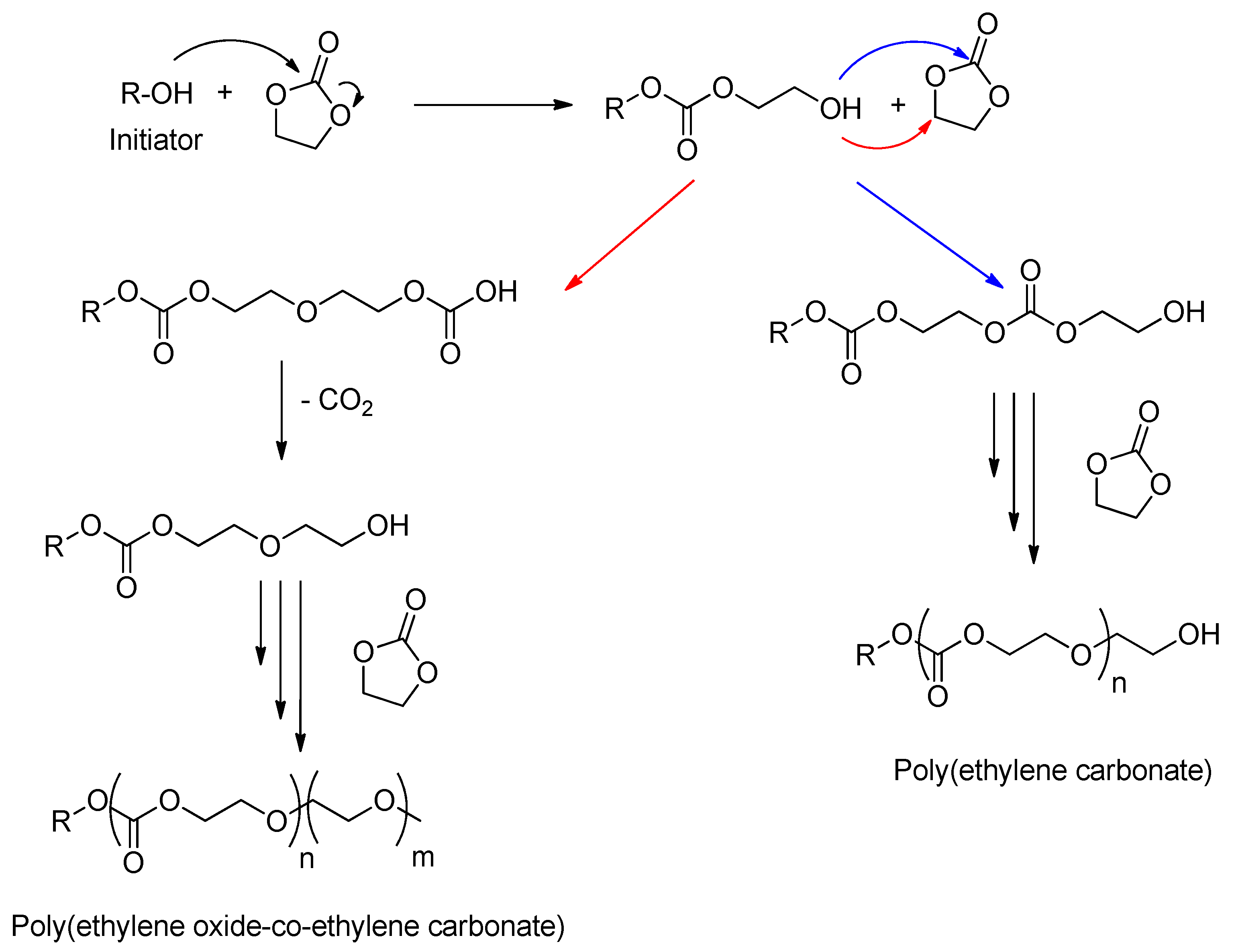
- Vogdanis, L.; Heitz, W. Carbon dioxide as a monomer, 3. The polymerization of ethylene carbonate. Die Makromol. Chem. Rapid Commun. 1986, 7, 543–547. [Google Scholar] [CrossRef]
- Vogdanis, L.; Martens, B.; Uchtmann, H.; Hensel, F.; Heitz, W. Synthetic and thermodynamic investigations in the polymerization of ethylene carbonate. Die Makromol. Chem. 1990, 191, 465–472. [Google Scholar] [CrossRef]
- Storey, R.F.; Hoffman, D.C. Formation of poly(ethylene ether carbonate) diols: Proposed mechanism and kinetic analysis. Macromolecules 1992, 25, 5369–5382. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Jonté, J.M.; Berl, M. Polylactones 3. Copolymerization of glycolide with L, L-lactide and other lactones. Die Makromol. Chem. 1985, 12, 25–38. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Berl, M.; Scharnagl, N. Poly(lactones). 9. Polymerization mechanism of metal alkoxide initiated polymerizations of lactide and various lactones. Macromolecules 1988, 21, 286–293. [Google Scholar] [CrossRef]
- Clements, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Lee, J.-C.; Litt, M.H. Ring-Opening Polymerization of Ethylene Carbonate and Depolymerization of Poly(ethylene oxide-co-ethylene carbonate). Macromolecules 2000, 33, 1618–1627. [Google Scholar] [CrossRef]
- Yang, H.; Yan, M.; Pispas, S.; Zhang, G. Synthesis of Poly[(ethylene carbonate)-co-(ethylene oxide)] Copolymer by Phosphazene-Catalyzed ROP. Macromol. Chem. Phys. 2011, 212, 2589–2593. [Google Scholar] [CrossRef]
- Soga, K.; Tazuke, Y.; Hosoda, S.; Ikeda, S. Polymerization of propylene carbonate. J. Polym. Sci. Polym. Chem. Ed. 1977, 15, 219–229. [Google Scholar] [CrossRef]
- Tezuka, K.; Komatsu, K.; Haba, O. The anionic ring-opening polymerization of five-membered cyclic carbonates fused to the cyclohexane ring. Polym. J. 2013, 45, 1183–1187. [Google Scholar] [CrossRef] [Green Version]
- Haba, O.; Tomizuka, H.; Endo, T. Anionic Ring-Opening Polymerization of Methyl 4,6-O-Benzylidene-2,3-O- carbonyl-α-d-glucopyranoside: A First Example of Anionic Ring-Opening Polymerization of Five-Membered Cyclic Carbonate without Elimination of CO2. Macromolecules 2005, 38, 3562–3563. [Google Scholar] [CrossRef]
- Guerin, W.; Diallo, A.K.; Kirilov, E.; Helou, M.; Slawinski, M.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Enantiopure Isotactic PCHC Synthesized by Ring-Opening Polymerization of Cyclohexene Carbonate. Macromolecules 2014, 47, 4230–4235. [Google Scholar] [CrossRef]
- Azechi, M.; Matsumoto, K.; Endo, T. Anionic ring-opening polymerization of a five-membered cyclic carbonate having a glucopyranoside structure. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1651–1655. [Google Scholar] [CrossRef]
- Rokicki, G.; Parzuchowski, P.G. Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 247–308. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Jenssen, J. Polylactones. 16. Cationic Polymerization of Trimethylene Carbonate and Other Cyclic Carbonates. J. Macromol. Sci. Part A—Chem. 1989, 26, 631–644. [Google Scholar] [CrossRef]
- Ariga, T.; Takata, T.; Endo, T. Cationic Ring-Opening Polymerization of Cyclic Carbonates with Alkyl Halides To Yield Polycarbonate without the Ether Unit by Suppression of Elimination of Carbon Dioxide. Macromolecules 1997, 30, 737–744. [Google Scholar] [CrossRef]
- Delcroix, D.; Martín-Vaca, B.; Bourissou, D.; Navarro, C. Ring-Opening Polymerization of Trimethylene Carbonate Catalyzed by Methanesulfonic Acid: Activated Monomer versus Active Chain End Mechanisms. Macromolecules 2010, 43, 8828–8835. [Google Scholar] [CrossRef]
- Liu, J.; Cui, S.; Li, Z.; Xu, S.; Xu, J.; Pan, X.; Liu, Y.; Dong, H.; Sun, H.; Guo, K. Polymerization of trimethylene carbonates using organic phosphoric acids. Polym. Chem. 2016, 7, 5526–5535. [Google Scholar] [CrossRef]
- Keul, H.; Bächer, R.; Höcker, H. Anionic ring-opening polymerization of 2,2-dimethyltrimethylene carbonate. Die Makromol. Chem. 1986, 187, 2579–2589. [Google Scholar] [CrossRef]
- Sanda, F.; Kamatani, J.; Endo, T. Synthesis and Anionic Ring-Opening Polymerization Behavior of Amino Acid-Derived Cyclic Carbonates. Macromolecules 2001, 34, 1564–1569. [Google Scholar] [CrossRef]
- Kühling, S.; Keul, H.; Höcker, H.; Buysch, H.-J.; Schön, N. Synthesis of poly(2-ethyl-2-hydroxymethyltrimethylene carbonate). Die Makromol. Chem. 1991, 192, 1193–1205. [Google Scholar] [CrossRef]
- Murayama, M.; Sanda, F.; Endo, T. Anionic Ring-Opening Polymerization of a Cyclic Carbonate Having a Norbornene Structure with Amine Initiators. Macromolecules 1998, 31, 919–923. [Google Scholar] [CrossRef]
- Matsuo, J.; Sanda, F.; Endo, T. A novel observation in anionic ring-opening polymerization behavior of cyclic carbonates having aromatic substituents. Macromol. Chem. Phys. 1998, 199, 2489–2494. [Google Scholar] [CrossRef]
- Matsuo, J.; Aoki, K.; Sanda, F.; Endo, T. Substituent Effect on the Anionic Equilibrium Polymerization of Six-Membered Cyclic Carbonates. Macromolecules 1998, 31, 4432–4438. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, X.; Gross, R.A. Polycarbonates from Sugars: Ring-Opening Polymerization of 1,2-O-Isopropylidene-d-Xylofuranose-3,5-Cyclic Carbonate (IPXTC). Macromolecules 1999, 32, 2799–2802. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Ganguly, P.; Billodeaux, D. Ring-Opening Polymerization of Trimethylene Carbonate Using Aluminum(III) and Tin(IV) Salen Chloride Catalysts. Macromolecules 2005, 38, 5406–5410. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Choi, W.; Ganguly, P.; Richers, C.P. Biometal Derivatives as Catalysts for the Ring-Opening Polymerization of Trimethylene Carbonate. Optimization of the Ca(II) Salen Catalyst System. Macromolecules 2006, 39, 4374–4379. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Choi, W.; Richers, C.P. Ring-Opening Polymerization of Cyclic Monomers by Biocompatible Metal Complexes. Production of Poly(lactide), Polycarbonates, and Their Copolymers. Macromolecules 2007, 40, 3521–3523. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Choi, W.; Karroonnirun, O.; Bhuvanesh, N. Ring-Opening Polymerization of Cyclic Monomers by Complexes Derived from Biocompatible Metals. Production of Poly(lactide), Poly(trimethylene carbonate), and Their Copolymers. Macromolecules 2008, 41, 3493–3502. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Mahler, A. Polymers of carbonic acid 18: Polymerizations of cyclobis(hexamethylene carbonate) by means of BuSnCl3 or Sn(II)2-ethylhexanoate. Polymer 1996, 37, 4383–4388. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Stricker, A. Polymers of carbonic acid, 28. SnOct2-initiated polymerizations of trimethylene carbonate (TMC, 1,3-dioxanone-2). Macromol. Chem. Phys. 2000, 201, 2557–2565. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Weegen-Schulz, B. Polymers of carbonic acid. XIV. High molecular weight poly(trimethylene carbonate) by ring-opening polymerization with butyltin chlorides as initiators. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 2193–2201. [Google Scholar] [CrossRef]
- Pêgo, A.P.; Grijpma, D.W.; Feijen, J. Enhanced mechanical properties of 1,3-trimethylene carbonate polymers and networks. Polymer 2003, 44, 6495–6504. [Google Scholar] [CrossRef]
- Ling, J.; Shen, Z.; Huang, Q. Novel Single Rare Earth Aryloxide Initiators for Ring-Opening Polymerization of 2,2-Dimethyltrimethylene Carbonate. Macromolecules 2001, 34, 7613–7616. [Google Scholar] [CrossRef]
- Nederberg, F.; Lohmeijer, B.G.G.; Leibfarth, F.; Pratt, R.C.; Choi, J.; Dove, A.P.; Waymouth, R.M.; Hedrick, J.L. Organocatalytic Ring Opening Polymerization of Trimethylene Carbonate. Biomacromolecules 2007, 8, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.; Thomas, A.W.; Dove, A.P. Highly Polarized Alkenes as Organocatalysts for the Polymerization of Lactones and Trimethylene Carbonate. ACS Macro Lett. 2016, 5, 134–138. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.L.; Jenisch, L.M.; Charles, B.; Kociok-Köhn, G.; Buchard, A. Polymers from Sugars and CO2: Synthesis and Polymerization of a d-Mannose-Based Cyclic Carbonate. Macromolecules 2016, 49, 7165–7169. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.L.; Kociok-Köhn, G.; Buchard, A. Polymers from sugars and CO2: Ring-opening polymerisation and copolymerisation of cyclic carbonates derived from 2-deoxy-d-ribose. Polym. Chem. 2017, 8, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.L.; Hierons, E.M.; Kociok-Köhn, G.; Sharma, R.I.; Buchard, A. CO2-Driven stereochemical inversion of sugars to create thymidine-based polycarbonates by ring-opening polymerisation. Polym. Chem. 2017, 8, 1714–1721. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.L.; Lopez-Vidal, E.M.; Buchard, A. Polymers from sugars: Cyclic monomer synthesis, ring-opening polymerisation, material properties and applications. Chem. Commun. 2017, 53, 2198–2217. [Google Scholar] [CrossRef] [Green Version]
- Pati, D.; Feng, X.; Hadjichristidis, N.; Gnanou, Y. CO2 as versatile carbonation agent of glycosides: Synthesis of 5- and 6-membered cyclic glycocarbonates and investigation of their ring-opening. J. CO2 Util. 2018, 24, 564–571. [Google Scholar] [CrossRef]
- Harris, R.F. Molecular weight advancement of poly(ethylene ether carbonate) polyols. J. Appl. Polym. Sci. 1989, 38, 463–476. [Google Scholar] [CrossRef]
- Pawłowski, P.; Rokicki, G. Synthesis of oligocarbonate diols from ethylene carbonate and aliphatic diols catalyzed by alkali metal salts. Polymer 2004, 45, 3125–3137. [Google Scholar] [CrossRef]
- Rokicki, G.; Kowalczyk, T. Synthesis of oligocarbonate diols and their characterization by MALDI-TOF spectrometry. Polymer 2000, 41, 9013–9031. [Google Scholar] [CrossRef]
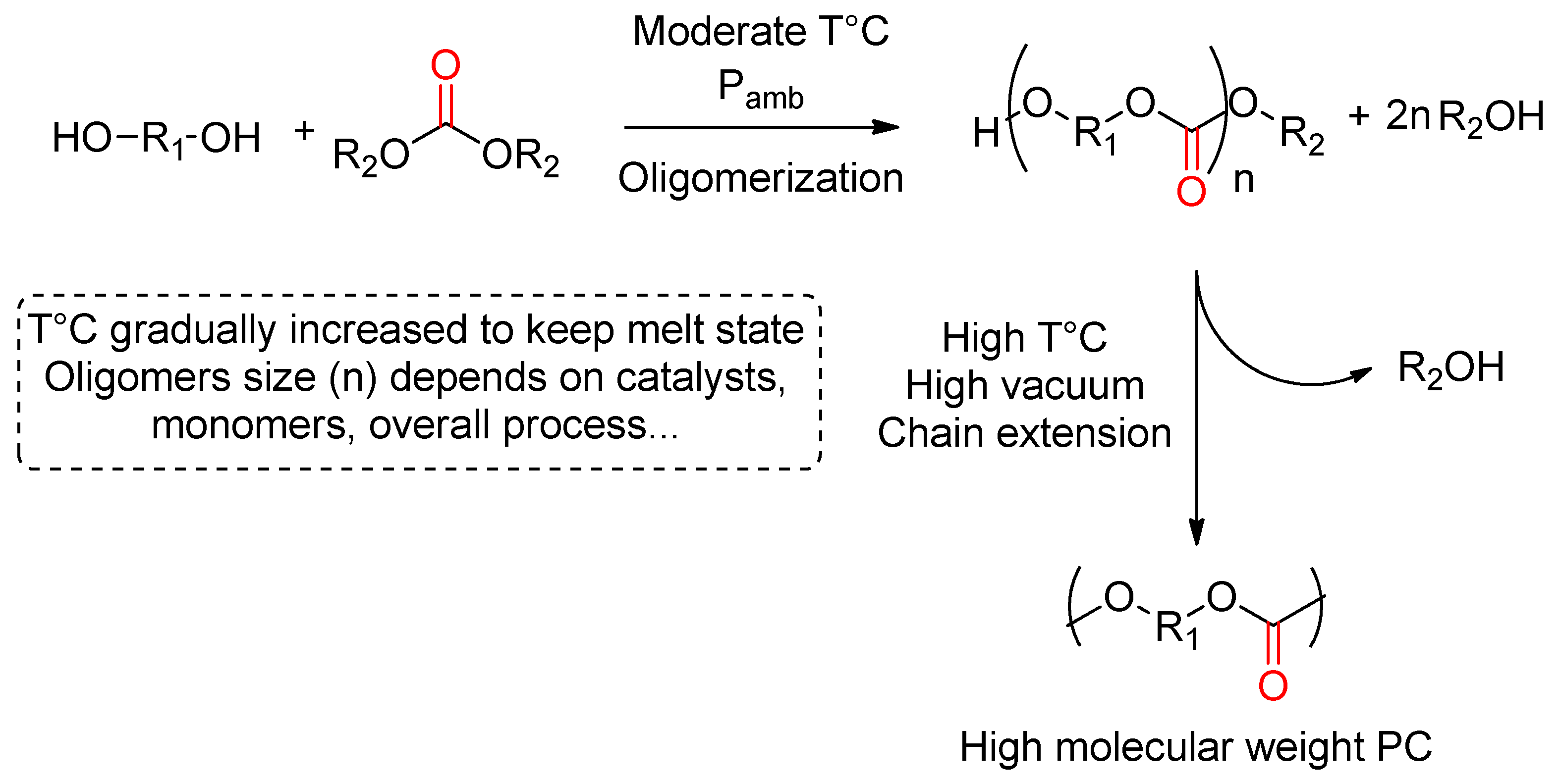
- Xu, J.; Feng, E.; Song, J. Renaissance of aliphatic polycarbonates: New techniques and biomedical applications. J. Appl. Polym. Sci. 2014, 131, 39822. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Zhang, Q.; Liao, S.; Zhao, C.; Xie, X. Understanding cyclic by-products and ether linkage formation pathways in the transesterification synthesis of aliphatic polycarbonates. Eur. Polym. J. 2017, 97, 253–262. [Google Scholar] [CrossRef]
- Sun, J.; Kuckling, D. Synthesis of high-molecular-weight aliphatic polycarbonates by organo-catalysis. Polym. Chem. 2016, 7, 1642–1649. [Google Scholar] [CrossRef] [Green Version]
- Meabe, L.; Lago, N.; Rubatat, L.; Li, C.; Müller, A.J.; Sardon, H.; Armand, M.; Mecerreyes, D. Polycondensation as a Versatile Synthetic Route to Aliphatic Polycarbonates for Solid Polymer Electrolytes. Electrochim. Acta 2017, 237, 259–266. [Google Scholar] [CrossRef]
- Naik, P.U.; Refes, K.; Sadaka, F.; Brachais, C.-H.; Boni, G.; Couvercelle, J.-P.; Picquet, M.; Plasseraud, L. Organo-catalyzed synthesis of aliphatic polycarbonates in solvent-free conditions. Polym. Chem. 2012, 3, 1475–1480. [Google Scholar] [CrossRef]
- Mutlu, H.; Ruiz, J.; Solleder, S.C.; Meier, M.a.R. TBD catalysis with dimethyl carbonate: A fruitful and sustainable alliance. Green Chem. 2012, 14, 1728–1735. [Google Scholar] [CrossRef]
- Park, J.H.; Jeon, J.Y.; Lee, J.J.; Jang, Y.; Varghese, J.K.; Lee, B.Y. Preparation of High-Molecular-Weight Aliphatic Polycarbonates by Condensation Polymerization of Diols and Dimethyl Carbonate. Macromolecules 2013, 46, 3301–3308. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, W.; Li, C.; Zhang, D.; Xiao, Y.; Guan, G.; Zheng, L. Effect of the biobased linear long-chain monomer on crystallization and biodegradation behaviors of poly(butylene carbonate)-based copolycarbonates. RSC Adv. 2015, 5, 2213–2222. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, X.; Li, C.; Xiao, Y.; Zhang, D.; Guan, G. High-molecular-weight aliphatic polycarbonates by melt polycondensation of dimethyl carbonate and aliphatic diols: Synthesis and characterization. Polym. Int. 2011, 60, 1060–1067. [Google Scholar] [CrossRef]
- Zhu, W.; Zhou, W.; Li, C.; Xiao, Y.; Zhang, D.; Guan, G.; Wang, D. Synthesis, Characterization and Degradation of Novel Biodegradable Poly(butylene-co-hexamethylene carbonate) Copolycarbonates. J. Macromol. Sci. Part A 2011, 48, 583–594. [Google Scholar] [CrossRef]
- Sun, J.; Aly, K.I.; Kuckling, D. A novel one-pot process for the preparation of linear and hyperbranched polycarbonates of various diols and triols using dimethyl carbonate. RSC Adv. 2017, 7, 12550–12560. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, X.; Liu, S.; Hu, J.; Zhang, H.; Wang, G. One-pot synthesis of high-molecular-weight aliphatic polycarbonates via melt transesterification of diphenyl carbonate and diols using Zn(OAc)2 as a catalyst. RSC Adv. 2015, 5, 87311–87319. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, X.; Li, J.; Liu, S.; Wang, G. Synthesis of high-molecular-weight aliphatic polycarbonates from diphenyl carbonate and aliphatic diols by solid base. J. Mol. Catal. A Chem. 2016, 424, 77–84. [Google Scholar] [CrossRef]
- Vanderhenst, R.; Miller, S.A. Polycarbonates from biorenewable diols via carbonate metathesis polymerization. Green Mater. 2013, 1, 64–78. [Google Scholar] [CrossRef]
- Haban, O.; Itakura, I.; Ueda, M.; Kuze, S. Synthesis of Polycarbonate from Dimethyl Carbonate and bisphenol-A. J. Polym. Sci. 1998, 37, 2087–2093. [Google Scholar]
- Ignatov, V.N.; Tartari, V.; Carraro, C.; Pippa, R.; Nadali, G.; Berti, C.; Fiorini, M. New Catalysts for Bisphenol A Polycarbonate Melt Polymerisation, 2. Polymer Synthesis and Characterisation. Macromol. Chem. Phys. 2001, 202, 1946–1949. [Google Scholar] [CrossRef]
- Kim, Y.; Choi, K.Y.; Chamberlin, T.A. Kinetics of melt transesterification of diphenyl carbonate and bisphenol A to polycarbonate with lithium hydroxide monohydrate catalyst. Ind. Eng. Chem. Res. 1992, 31, 2118–2127. [Google Scholar] [CrossRef]
- Fukuoka, S.; Kawamura, M.; Komiya, K.; Tojo, M.; Hachiya, H.; Hasegawa, K.; Aminaka, M.; Okamoto, H.; Fukawa, I.; Konno, S. A novel non-phosgene polycarbonate production process using by-product CO2 as starting material. Green Chem. 2003, 5, 497–507. [Google Scholar] [CrossRef]
- Zhang, M.; Lai, W.; Su, L.; Lin, Y.; Wu, G. A synthetic strategy toward isosorbide polycarbonate with a high molecular weight: The effect of intermolecular hydrogen bonding between isosorbide and metal chlorides. Polym. Chem. 2019, 10, 3380–3389. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, L.; An, H.; Li, C.; Zhang, Z.; Fang, W.; Xu, F.; Zhang, S. Cost-Effective Synthesis of High Molecular Weight Biobased Polycarbonate via Melt Polymerization of Isosorbide and Dimethyl Carbonate. ACS Sustain. Chem. Eng. 2020, 8, 9968–9979. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, W.; Li, C.; Guan, G.; Zhang, D.; Xiao, Y.; Zheng, L. A non-phosgene process to homopolycarbonate and copolycarbonates of isosorbide using dimethyl carbonate: Synthesis, characterization, and properties. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1387–1397. [Google Scholar] [CrossRef]
- Eo, Y.S.; Rhee, H.-W.; Shin, S. Catalyst screening for the melt polymerization of isosorbide-based polycarbonate. J. Ind. Eng. Chem. 2016, 37, 42–46. [Google Scholar] [CrossRef]
- Yang, Z.; Li, X.; Xu, F.; Wang, W.; Shi, Y.; Zhang, Z.; Fang, W.; Liu, L.; Zhang, S. Synthesis of bio-based polycarbonate via one-step melt polycondensation of isosorbide and dimethyl carbonate by dual site-functionalized ionic liquid catalysts. Green Chem. 2021, 23, 447–456. [Google Scholar] [CrossRef]
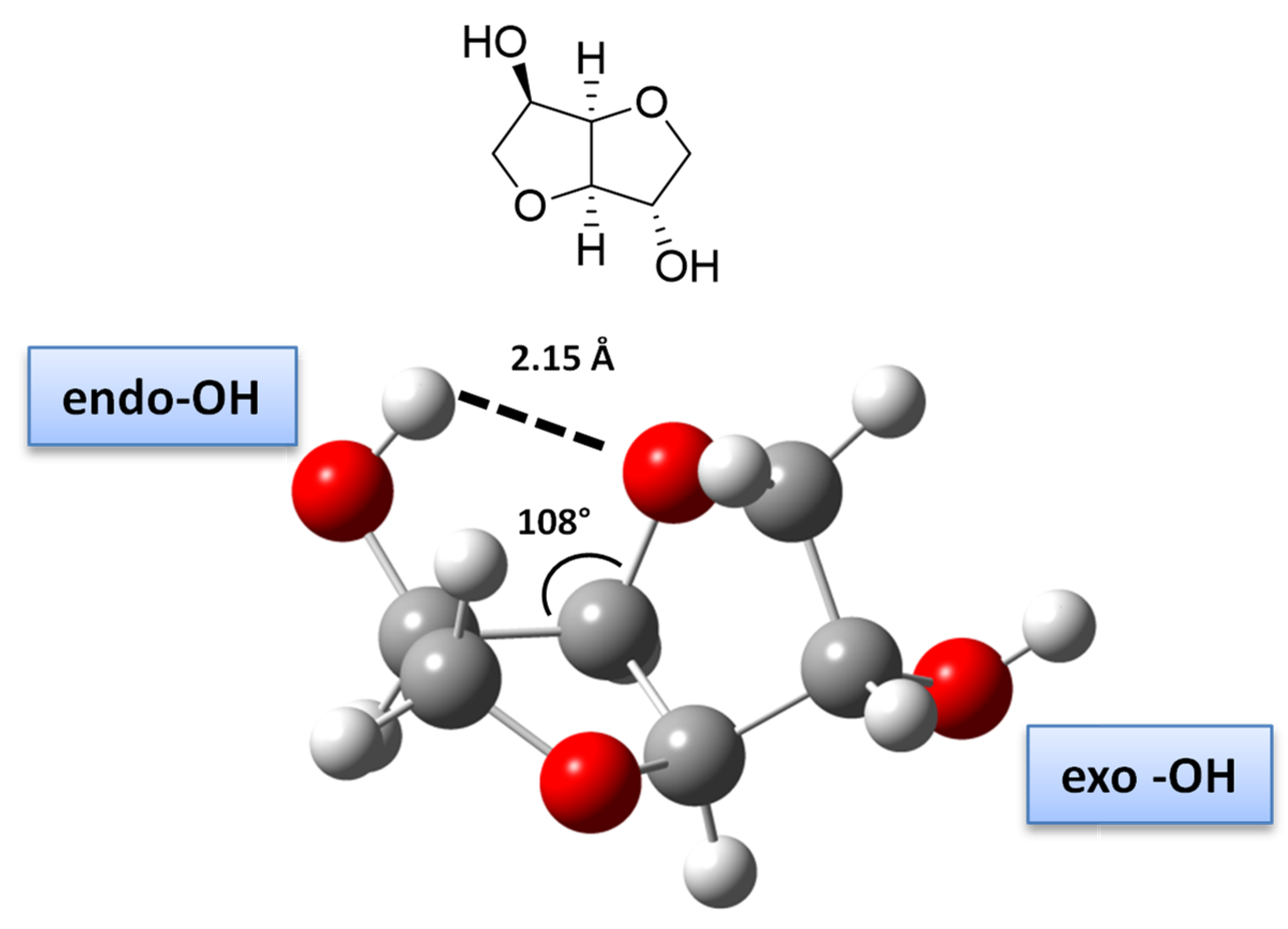
- Qian, W.; Ma, X.; Liu, L.; Deng, L.; Su, Q.; Bai, R.; Zhang, Z.; Gou, H.; Dong, L.; Cheng, W.; et al. Efficient synthesis of bio-derived polycarbonates from dimethyl carbonate and isosorbide: Regulating exo-OH and endo-OH reactivity by ionic liquids. Green Chem. 2020, 22, 5357–5368. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Z.; Yang, Z.; Fang, W.; An, H.; Li, T.; Xu, F. Synthesis of bio-based poly(oligoethylene glycols-co-isosorbide carbonate)s with high molecular weight and enhanced mechanical properties via ionic liquid catalyst. React. Funct. Polym. 2020, 155, 104689. [Google Scholar] [CrossRef]
- Qian, W.; Tan, X.; Su, Q.; Cheng, W.; Xu, F.; Dong, L.; Zhang, S. Transesterification of Isosorbide with Dimethyl Carbonate Catalyzed by Task-Specific Ionic Liquids. ChemSusChem 2019, 12, 1169–1178. [Google Scholar] [CrossRef]
- Qian, W.; Liu, L.; Zhang, Z.; Su, Q.; Zhao, W.; Cheng, W.; Li, D.; Yang, Z.; Bai, R.; Xu, F.; et al. Synthesis of Bioderived Polycarbonates with Molecular Weight Adjustability Catalyzed by Phenolic-derived Ionic Liquids. Green Chem. 2020, 22, 2488–2497. [Google Scholar] [CrossRef]
- Sun, W.; Xu, F.; Cheng, W.; Sun, J.; Ning, G.; Zhang, S. Synthesis of isosorbide-based polycarbonates via melt polycondensation catalyzed by quaternary ammonium ionic liquids. Chin. J. Catal. 2017, 38, 908–917. [Google Scholar] [CrossRef]
- Ma, C.; Xu, F.; Cheng, W.; Tan, X.; Su, Q.; Zhang, S. Tailoring Molecular Weight of Bioderived Polycarbonates via Bifunctional Ionic Liquids Catalysts under Metal-Free Conditions. ACS Sustain. Chem. Eng. 2018, 6, 2684–2693. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, F.; He, H.; Ding, W.; Fang, W.; Sun, W.; Li, Z.; Zhang, S. Synthesis of high-molecular weight isosorbide-based polycarbonates through efficient activation of endo-hydroxyl groups by an ionic liquid. Green Chem. 2019, 21, 3891–3901. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, Z.; Yang, Z.; Zhang, Y.; Xu, F.; Li, C.; An, H.; Song, T.; Luo, Y.; Zhang, S. One-pot synthesis of bio-based polycarbonates from dimethyl carbonate and isosorbide under metal-free condition. Green Chem. 2020, 22, 4550–4560. [Google Scholar] [CrossRef]
- Shen, X.; Liu, S.; Wang, Q.; Zhang, H.; Wang, G. Synthesis of Poly(isosorbide carbonate) via Melt Polycondensation Catalyzed by a KF/MgO Catalyst. Chem. Res. Chin. Univ. 2019, 35, 721–728. [Google Scholar] [CrossRef]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- Shen, X.-L.; Wang, Z.-Q.; Wang, Q.-Y.; Liu, S.-Y.; Wang, G.-Y. Synthesis of Poly(isosorbide carbonate) via Melt Polycondensation Catalyzed by Ca/SBA-15 Solid Base. Chin. J. Polym. Sci. 2018, 36, 1027–1035. [Google Scholar] [CrossRef]
- Zhang, M.; Lai, W.; Su, L.; Wu, G. Effect of Catalyst on the Molecular Structure and Thermal Properties of Isosorbide Polycarbonates. Ind. Eng. Chem. Res. 2018, 57, 4824–4831. [Google Scholar] [CrossRef]
- Ouhib, F.; Meabe, L.; Mahmoud, A.; Eshraghi, N.; Grignard, B.; Thomassin, J.-M.; Aqil, A.; Boschini, F.; Jérôme, C.; Mecerreyes, D.; et al. CO2-sourced polycarbonates as solid electrolytes for room temperature operating lithium batteries. J. Mater. Chem. A 2019, 7, 9844–9853. [Google Scholar] [CrossRef]
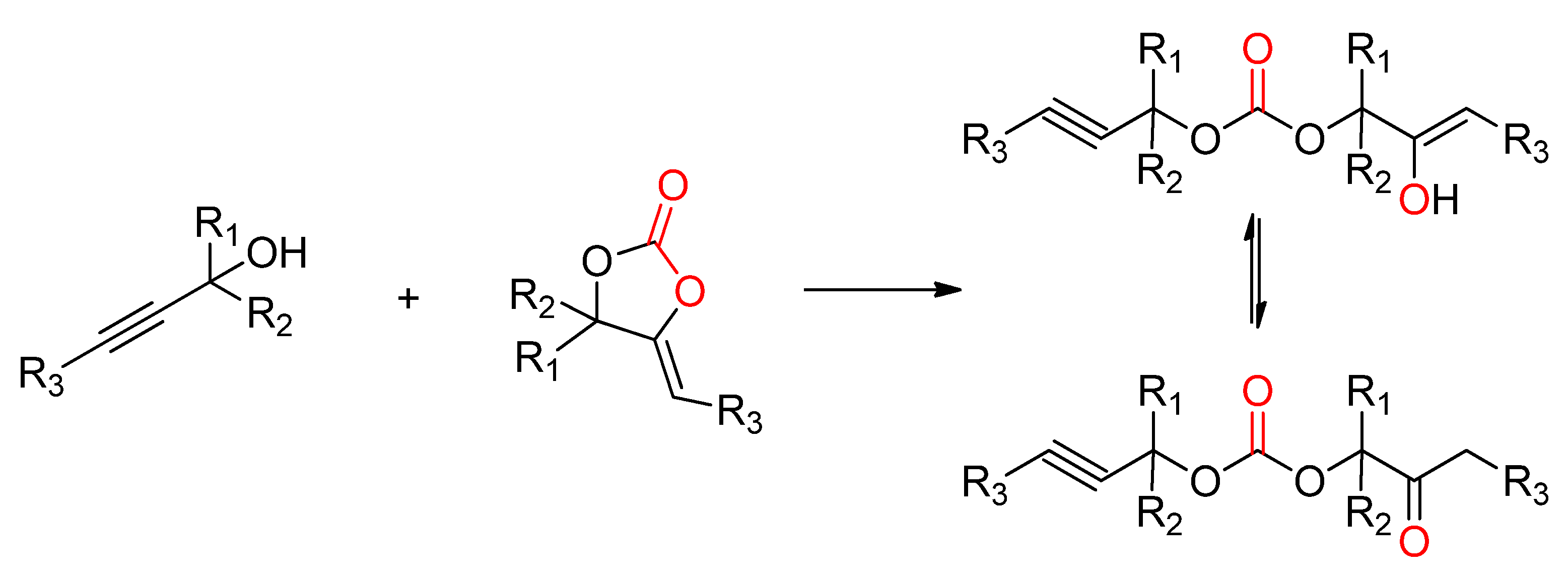
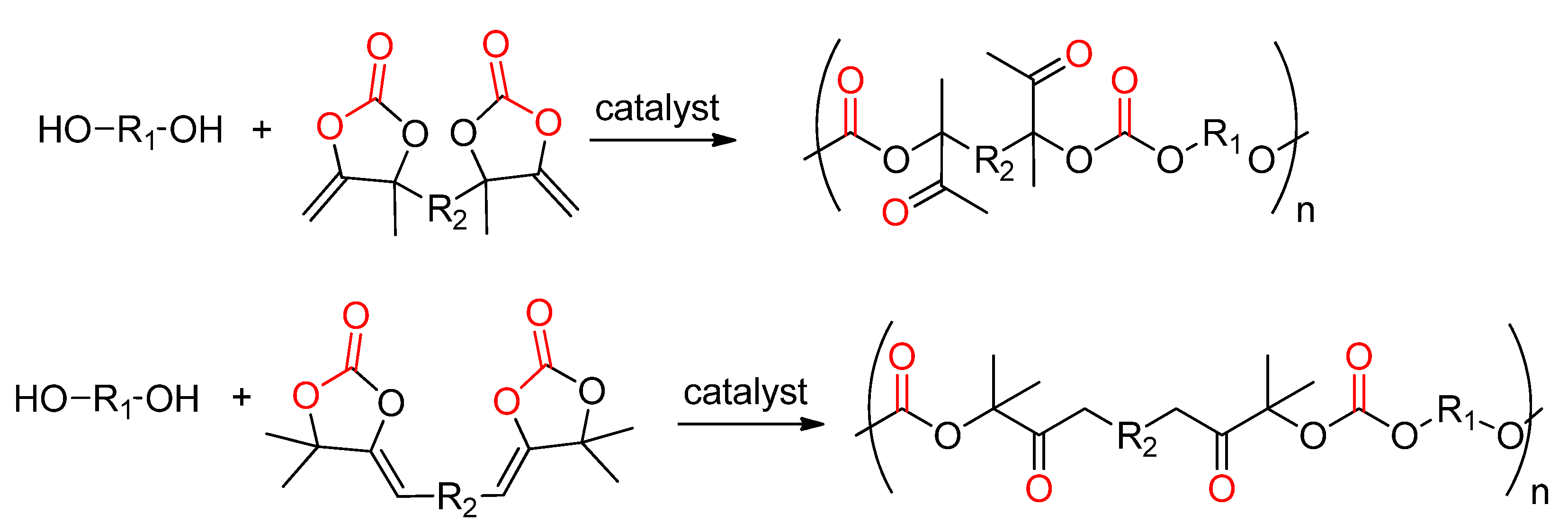
- Ngassam Tounzoua, C.; Grignard, B.; Brege, A.; Jerome, C.; Tassaing, T.; Mereau, R.; Detrembleur, C. A Catalytic Domino Approach toward Oxo-Alkyl Carbonates and Polycarbonates from CO2, Propargylic Alcohols, and (Mono- and Di-)Alcohols. ACS Sustain. Chem. Eng. 2020, 8, 9698–9710. [Google Scholar] [CrossRef]
- Siragusa, F.; Van Den Broeck, E.; Ocando, C.; Müller, A.J.; De Smet, G.; Maes, B.U.W.; De Winter, J.; Van Speybroeck, V.; Grignard, B.; Detrembleur, C. Access to Biorenewable and CO2-Based Polycarbonates from Exovinylene Cyclic Carbonates. ACS Sustain. Chem. Eng. 2021, 9, 1714–1728. [Google Scholar] [CrossRef]

































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brege, A.; Grignard, B.; Méreau, R.; Detrembleur, C.; Jerome, C.; Tassaing, T. En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches. Catalysts 2022, 12, 124. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020124
Brege A, Grignard B, Méreau R, Detrembleur C, Jerome C, Tassaing T. En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches. Catalysts. 2022; 12(2):124. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020124
Chicago/Turabian StyleBrege, Antoine, Bruno Grignard, Raphaël Méreau, Christophe Detrembleur, Christine Jerome, and Thierry Tassaing. 2022. "En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches" Catalysts 12, no. 2: 124. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020124