
Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study


1
Department of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia
2
Department of Chemistry, National Taiwan University, Taipei 10617, Taiwan
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(2), 133; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020133
Submission received: 27 December 2021
/
Revised: 15 January 2022
/
Accepted: 18 January 2022
/
Published: 21 January 2022
(This article belongs to the Special Issue Kinetics and Mechanism of Catalytic Reactions—Integrity of Experiment and Theory)
Abstract
:In this work, we used ab initio/DFT method coupled with statistical rate theory to answer the question of whether or not formic acid (HCOOH) and water molecules can catalyze the most important atmospheric and combustion prototype reaction, i.e., ·OH (OH radical) + CH4. The potential energy surface for ·OH + CH4 and ·OH + CH4 (+X) (X = HCOOH, H2O) reactions were calculated using the combination of hybrid-density functional theory and coupled-cluster theory with Pople basis set [(CCSD(T)/ 6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)]. The results of this study show that the catalytic effect of HCOOH (FA) and water molecules on the ·OH + CH4 reaction has a major impact when the concentration of FA and H2O is not included. In this situation the rate constants for the CH4 + HO···HCOOH (3 × 10−9 cm3 molecule−1 s−1) reaction is ~105 times and for CH4 + H2O···HO reaction (3 × 10−14 cm3 molecule−1 s−1 at 300 K) is ~20 times higher than ·OH + CH4 (~6 × 10−15 cm3 molecule−1 s−1). However, the total effective rate constants, which include the concentration of both species in the kinetic calculation has no effect under atmospheric condition. As a result, the total effective reaction rate constants are smaller. The rate constants when taking the account of the FA and water for CH4 + HO···HCOOH (4.1 × 10−22 cm3 molecule−1 s−1) is at least seven orders magnitude and for the CH4 + H2O···HO (7.6 × 10−17 cm3 molecule−1 s−1) is two orders magnitude smaller than ·OH + CH4 reaction. These results are also consistent with previous experimental and theoretical studies on similar reaction systems. This study helps to understand how FA and water molecules change the reaction kinetic under atmospheric conditions for ·OH + CH4 reaction.
1. Introduction
The tropospheric concentration of important greenhouse gases, i.e., methane (CH4), is steadily increasing [1,2,3,4]. In the atmosphere, ·OH (OH radical) plays a crucial role in removing the CH4 [1,2,3,4]. The atmospheric lifetime of methane is due to loss by ·OH is ∼12–17 years [1,2,3,4]. This reaction is also important in high-temperature combustion kinetics [5]. Due to large uncertainty in the rate constants data, many experimental measurements and theoretical studies have been performed [1,2,3,4,5]. To avoid the repetition of previous studies, only a few studies have been discussed here [1,2,3,4,5]. Various experimental measurements were used to estimate the rate constant for ·OH + CH4 reaction into the atmosphere condition and reported its atmospheric lifetime due to loss with the ·OH [1,2,3,4]. Experimental measurement on ·OH + CH4 reaction was done by Vaghjian et al. [2] in the temperature range of 295 to 400 K. Due to the limited number of high temperature studies, Srinivasan et al. measured the rate constants of ·OH + CH4 using the reflected shock tube method [5]. The ·OH + CH4 reaction has been extensively studied by using various theoretical approaches [6,7,8,9]. Most of the theoretical studies have focused on calculating the rate constants using a high-level ab initio methods and statistical rate theory [6,7,8,9]. Ellingson et al. [9] proposed the rate constants for OH + CH4 reaction and its 12C/13C kinetic isotopes effect using harmonic, hindered, and free rotor approaches. Their calculated rate constants were in good agreement with experimentally measurement values. Li and Gua calculated the thermal rate constants for isotopic effects for OH + CH4 using UCCSD(T)-F12a/aug-cc-pVTZ and canonical variational transition state theory (CVT) using the free rotor approach [6,8]. In this study, we have re-calculated the rate constants for ·OH + CH4 using a similar level of theories to validate our theoretical approach and compare the rate constants for the effect of HCOOH/H2O on ·OH + CH4 reaction.
It is believed that volcanic eruptions emitted various gases into the Earth’s atmosphere [10,11,12]. Toxic acids such as hydrochloric (HCl), formic acid (HCOOH), and sulfuric (H2SO4) acid in the atmosphere are produced from volcanic activities [10,11,12]. The source of HCOOH (FA) in atmospheric reactions has been extensively studied in recent years [10,11,12]. FA is mainly produced by oxidation of volatile organic compounds (VOCs), soil, burning of fossil fuels, and terrestrial vegetation [10,11,12]. FA also influences pH-dependent chemical reactions in clouds [11]. FA is one of the most abundant organic acid present in the atmosphere [10]. The atmospheric abundance of FA varies from 0.01 to 10 ppbv depending upon the altitudes [13,14,15,16,17,18,19]. FA can catalyze many important atmospheric reactions such as CH3O + O2 [13], ·OH + HCl [14], H2O + CH2COO [15], HO2 + Cl [16], H2O + CH2O [17], and ·OH + CH2O [19]. These studies suggest that FA can form ring types of complexes and transition states, which lower the barrier heights and increase the reaction rate constant under atmospheric conditions [13,14,15,16,17,18,19]. However, in realistic atmospheric conditions, the effective rate constants decrease and the catalytic effect does not favor the reaction [12,13,14,15,16,17,18,19]. To the best of our knowledge, the acidolysis of ·OH + CH4 by a FA molecule has not been studied. Therefore, we have investigated the effect of the FA molecule on the most important atmospheric and combustion prototype of reaction, i.e., ·OH + CH4. The focus of the present work is to explore the potential energy surface for the effect of FA on ·OH + CH4 and calculate the rate constants under atmospheric conditions.
The oxidation of CH4 in the stratosphere is an important source of water vapor in this region. Over the last few years, many experimental and theoretical studies have been conducted on the catalytic effect of a single water molecule on many important atmospheric reactions, such as ·OH + CH2O, ·OH + CH2CH2, ·OH + CH2NH, ·OH + CH3CHO, ·OH + CH3OH, OH + HCl, O2 + CH3O, and O2 + CH2OH [20,21,22,23,24,25,26,27,28,29,30,31,32]. Experimental measurements on some of the reactions, i.e., OH + CH3OH (+H2O) [20] and OH + CH3CHO (+H2O)] [22], have been performed. Jara et al. [20] and Vöhringer-Martinez [22] have measured reaction rate constants and suggested that the catalytic role of water molecules dominated at lower temperatures. Recently, Wu et al. [21], have proposed reaction kinetic data using a high-level ab initio method and an advanced kinetic model for ·OH + CH3OH. Their calculated rate constants were in good agreement with the experimentally measured values of Jara et al. [20]. Most of the theoretical studies suggest that a single water molecule formed a water-bounded ring type of pre-reactive complexes (PRCs) and transition states (TSs), which reduces the energy barrier but does not increase the rate constant under atmospheric conditions. In 2012, Thomsen et al [23]. proposed the effect of a single water molecule on ·OH + CH4 using ab initio/DFT method [23]. They suggested that the role of water molecules in OH + CH4 reaction is less important under atmospheric conditions. To the best of our knowledge, the detailed kinetics analysis of this reaction is still not known, and the effect of temperature-dependent water concentration on ·OH + CH4 has not been reported. In our previous works, we proposed the catalytic effect of H2O molecules in the atmospheric reactions, i.e., ·OH + CH2O, ·OH + CH2CH2, ·OH + CH2NH, ·OH + CH3OH, CH2NH + H2O, and CH2OH + O2 reactions [25,26,27,28]. We have also suggested that the catalytic role of water is less important under atmospheric conditions.
Because of CH4 long lifetime and a good model for the most important atmospheric and combustion reaction prototype molecule, the effect of FA and water molecules was investigated. The rate constant for the effect of FA and water molecules on ·OH + CH4 were investigated at different atmospheric concentrations of FA and water molecules at different temperatures. The results of this study may be useful for the benchmark performance for other higher chain alkanes. The comparison of reaction energies and rate constants with the literature values provides more confidence in our results.
2. Theoretical Methodology
2.1. Computational Method
All the electronic structure calculations were done with the Gaussian 09 suite of programs (Revision A.02, Gaussian, Inc., Wallingford, CT, USA, 2009) [33]. Optimized geometries for the species involved in ·OH + CH4, ·OH + CH4 (+HCOOH), and ·OH + CH4 (+H2O) reactions were computed using M06-2X/6-311++G(3df,3pd) level [34] (see Table S1). The harmonic vibrational frequency of all the species was calculated to obtain the zero-point corrections (ZPE). The frequency analysis shows that all the reactants, complexes and products have all positive values whereas all the transition states (TSs) have a single imaginary (see Table S2). Intrinsic reaction coordinate (IRC) calculations were performed to confirm the identity of pre-reactive complex and post-reactive complex for every TS. The single point energies were calculated using CCSD(T)/6-311++G(3df,3pd) (CC) level of theory at optimized geometries of M06-2X/6-311++G(3df,3pd)(M06-2X). As discussed in the previous works, the combination of CC//M06-2X typically provides results that are accurate to ~1–2 kcal/mol [23,24,25,26]. Therefore, we believe the CC//M06-2X provides accurate energies//rotational vibrational parameters for ·OH + CH4, ·OH + CH4 (+HCOOH), and ·OH + CH4 (+H2O) reactions. The ZPE, electronic energies, thermal correction, enthalpy correction, and free energy correction for all the species are tabulated in Table S3.
2.2. Chemical Kinetics Calculations
The temperature-dependent rate constants were calculated by Equations (1) and (2):
where is generalized rate constants and are the temperature dependent rate canonical variational transition state theory (CVT) rate constants, VMEP is the barrier height without zero-point correction, is a reaction path degeneracy, h is Planck’s constant, kB is the Boltzmann constant, and and are the total partition functions for the transition state and the reactants, respectively, is the small curvature tunneling (SCT) correction as implemented in Polyrate [35]. The rate constants were calculated using a dual dynamic approach with CVT and the interpolated single point energies (ISPE) correction calculated using dual level direct dynamic approach CVT/SCT with interpolated single point energies (ISPE) as discussed in the reference [35,36]. PolyRate and GaussRate suite of programs were used to calculate the temperature-dependent bimolecular and unimolecular rate constants based on CVT/SCT (Table S4) approach [35,36].
The Multiwell Thermo code [37] was used calculate the equilibrium constant (Keq) for the formation of complexes as given in Equation (3):
The equilibrium constants () for the formation complexes calculated by Equation (3) were tabulated in Tables S5 and S6.
3. Results and Discussion
3.1. Reaction Pathways for OH + CH4
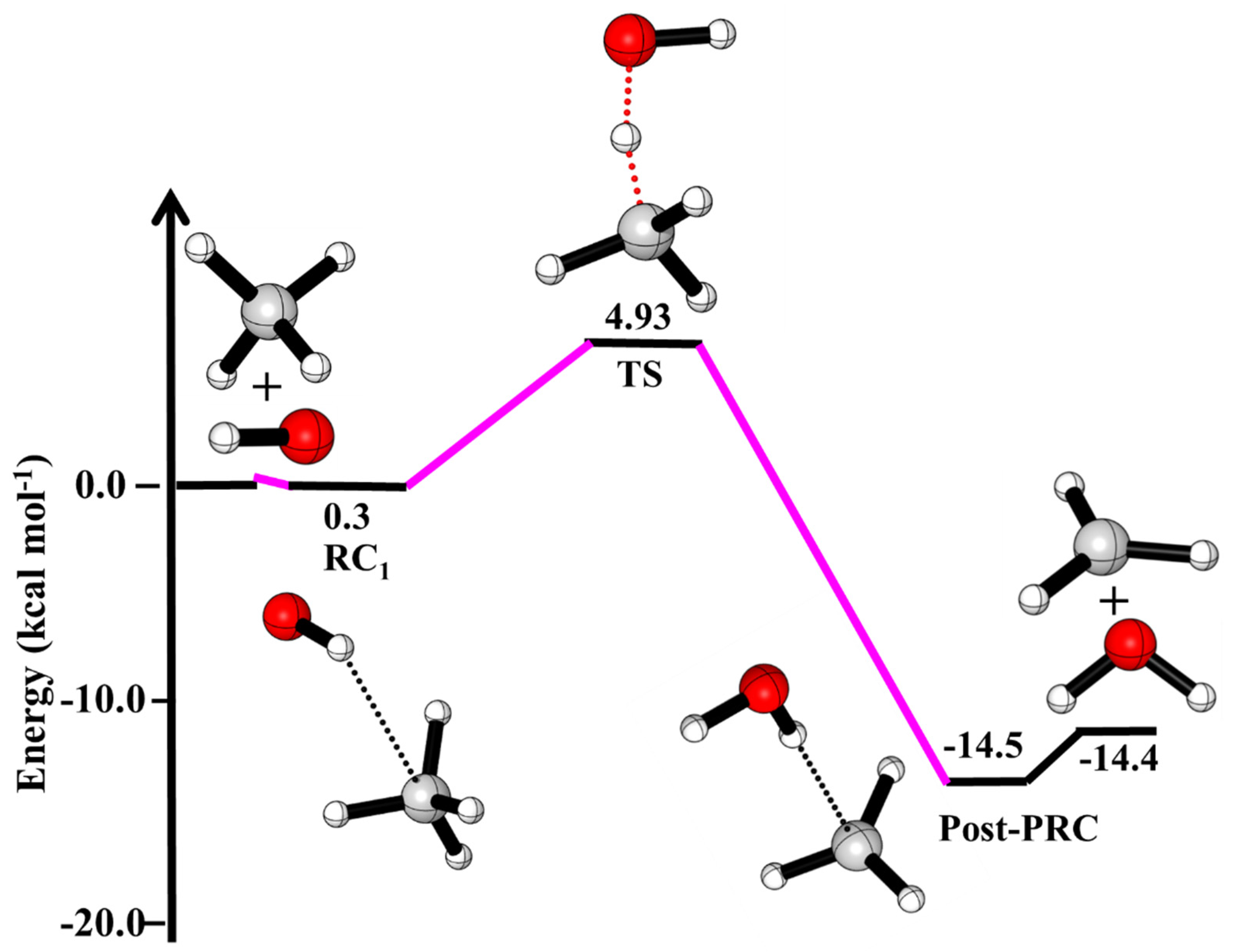
The potential energy surface (PES) for ·OH + CH4 reaction is shown in Figure 1. The rotational vibrational parameters of CH4, ·OH and TS and products are given in Table S2. As shown in Figure 1, the reaction proceeds via the formation of a pre-reactive complex (RC1) whose energy is higher than the reactants. This is due to fact that the orientation of H of ·OH is toward the carbon atom of CH4. The role of complex in ·OH + CH4 reaction is unimportant as suggested in earlier studies [8]. The barrier height for ·OH + CH4 is ~5.0 kcal/mol is in very good agreement with the value reported by earlier theoretical studies (5–6 kcal/mol) [6,7,8,9].
3.1.1. Reaction Pathways for ·OH + CH4 (+HCOOH)
As discussed in the earlier studies, [13,14,25,26,27,28], under true conditions, it is very unlikely that ·OH, CH4, and HCOOH collide simultaneously, therefore the probability of a termolecular reaction is negligible. It is expected that either a CH4···HCOOH or ·OH···HCOOH and CH4···HO· will form first, followed by an attack on this complex by third molecule ·OH or CH4 or HCOOH. In these three cases, the probable reactions are shown in Equations (4)–(6):
CH4···HCOOH + OH →·CH3 + (H2O) + HCOOH
CH4···HO + HCOOH →·CH3 + (H2O) + HCOOH
CH4 + HCOOH···HO →·CH3 + (H2O) + HCOOH
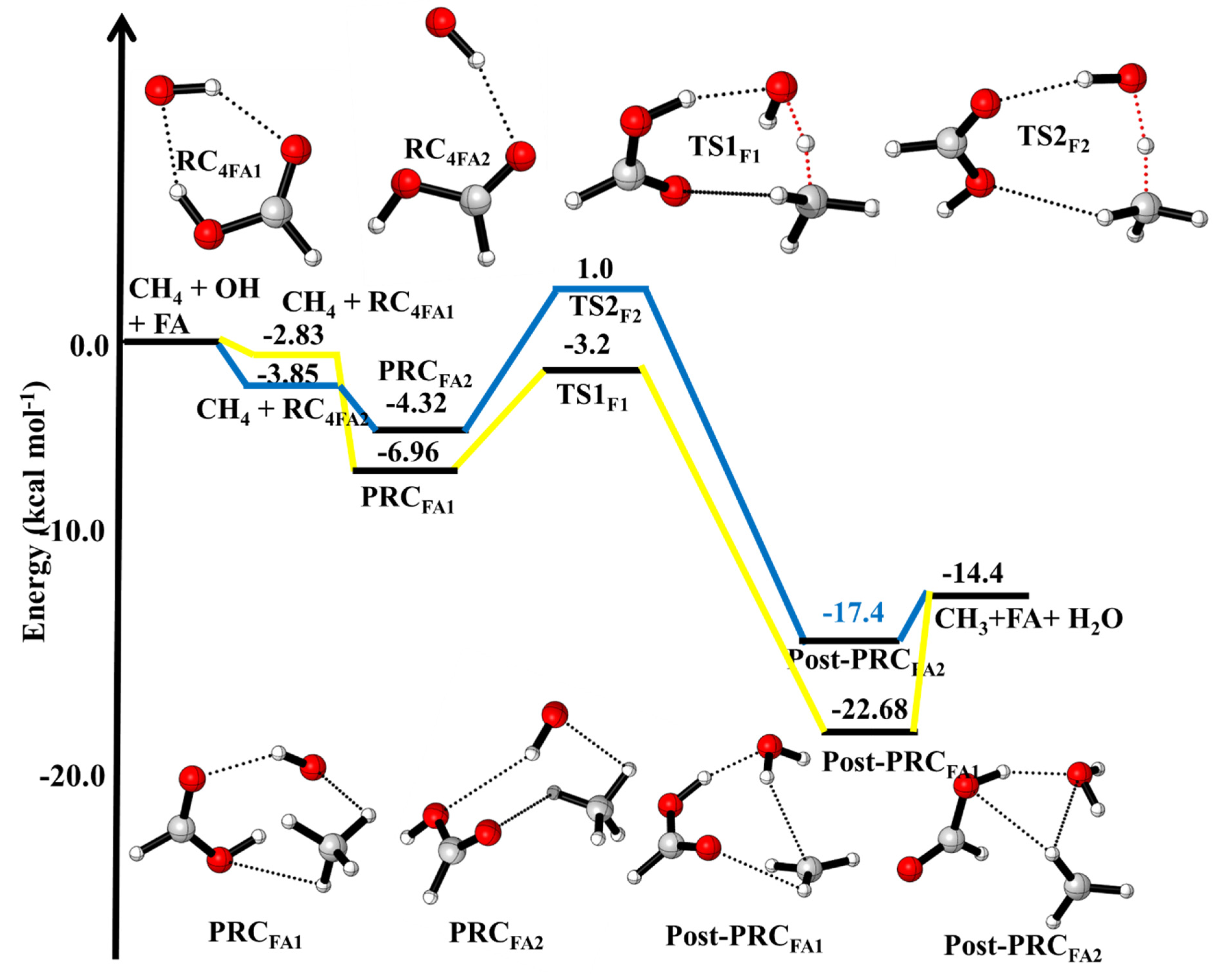
The potential energy surface (PES) for the effect of FA on ·OH + CH4 reaction is shown in Figure 2. The optimized geometries and rotational vibrational parameters of CH4, ·OH, and TS and products are given in Tables S1 and S2. The calculated binding energy (BE) between ·OH and FA1 (−3.85 kcal/mol) is in very good agreement with the value (−3.59 kcal/mol) reported previously [19]. The BE of FA2···OH (−2.83 kcal/mol) is 1 kcal/mol higher than the BE of FA11···OH (−3.85 kcal/mol). This is due to fact that the cis and trans orientation of FA. The trans form of FA with ·OH leading more strong hydrogen bonding than the cis form. The B.E. of CH4···OH (+0.3 kcal/mol) and CH4···HCOOH (+0.3 kcal/mol) is very small compared to B.E. of HCOOH···OH, therefore, we have neglected these complexes. As shown in Figure 2, beginning with the CH4 + HCOOH···OH, two three-body complexes PRCFA-1 and PRCFA-2 were formed due to the trans and cis orientation of FA (Figure 2). The relevant TS structures connected to these PRCs are shown in Figure 2. The other PRCFA was also optimized and found that they are less stable than PRCFA-1 and PRCFA-2, therefore not included in the PES of CH4 + ·OH···HCOOH reaction (see Figure S1).
The BE of PRCFA-1 (−6.96 kcal/mol) and PRCFA-2 (−4.32 kcal/mol) is the result of C···H and O···H interactions. Starting from PRCFA-1 and PRCFA-2, we have identified two reaction pathways, i.e., hydrogen abstraction by ·OH on two different orientations of FA. Transition state (TSF1) corresponds to H-abstraction reaction from methane hydrogen via PRCFA-1. The calculated barrier heights for this pathway (~3.76 kcal/mol), which are lower than the barrier height for ·OH + CH4 reaction (~5 kcal/mol). The transition state (TSF2) corresponds to the H abstraction reaction from methane hydrogen via PRCFA-2. The calculated barrier height TSF2 (~ 5.32 kcal/mol) which leads to form a product FA-2 (cis-Hydrogen). The barrier height of TSF1 (~4 kcal/mol,) is ~1 kcal/mol lower than the value of TSF2 (~5 kcal/mol) expected to play a more important role in the kinetic calculations [13,14,19].
3.1.2. Reaction Pathways for ·OH + CH4 (+H2O)
As suggested in the FA case, the simultaneous collisions of ·OH, CH4, and H2O, are very unlikely, therefore the termolecular reaction probability is negligible under real conditions. It is expected that either a CH4···H2O or ·OH···H2O or CH4···OH will form first, followed by an attack on this complex by the third molecule ·OH or CH4 or H2O to this complex. In these three cases, the most probable reactions are given in Equations (7)–(9):
CH4···H2O + ·OH → ·CH3 + (H2O)2
CH4···HO· + H2O → ·CH3 + (H2O)2
CH4 + H2O···HO· → ·CH3 + (H2O)2
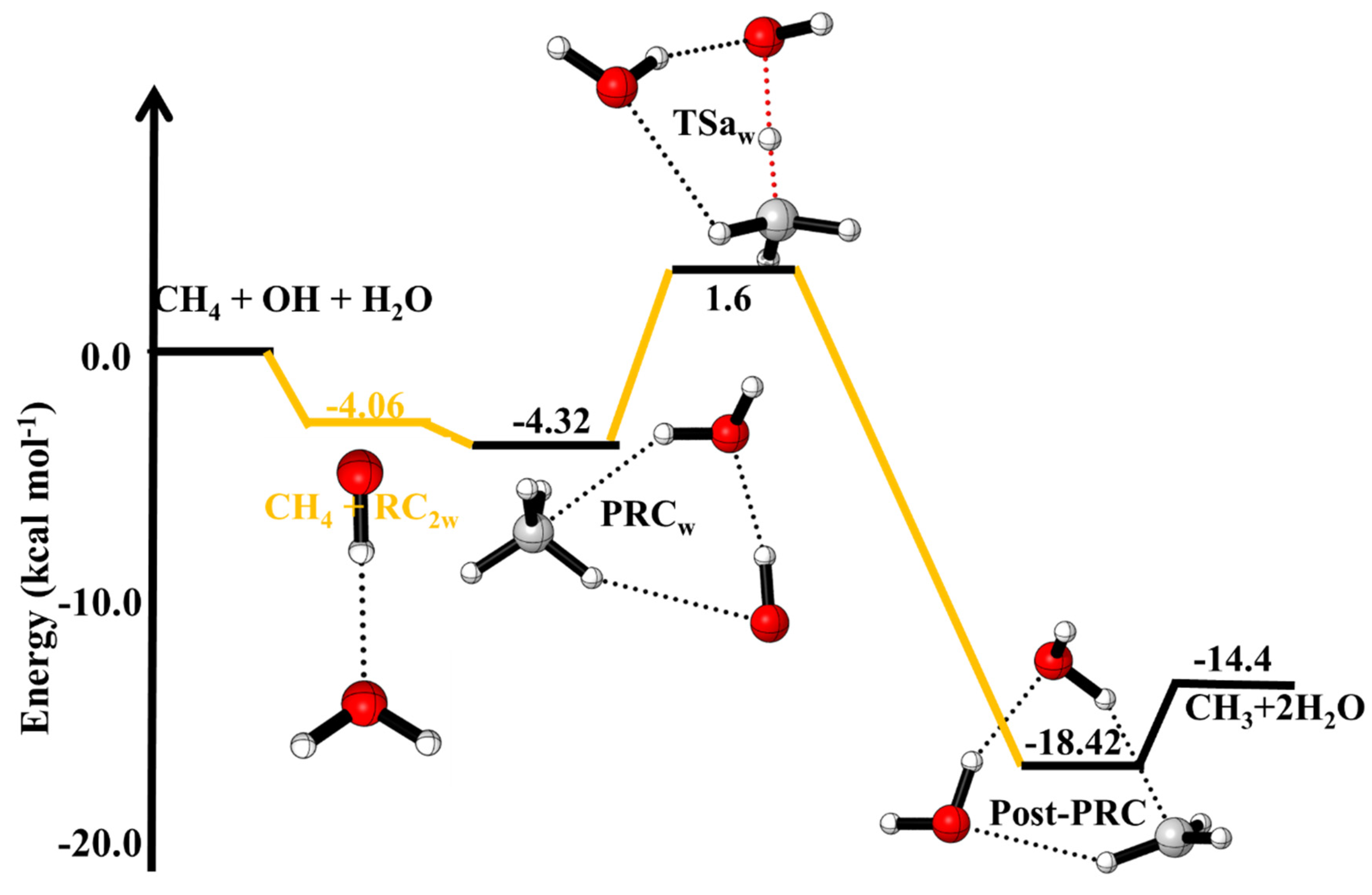
Out of three, only one OH···H2O (−4.1 kcal/mol) as a two-body complex is found to be more stable than CH4···H2O (−0.3 kcal/mol) and CH4···OH (+0.3 kcal/mol), therefore the reactions (7) and (8) are not considered in the potential energy surface plot. The potential energy surface (PES) for the effect of water on ·OH + CH4 reaction is shown in Figure 3. As shown in Figure 3, beginning with the CH4 + ·OH···H2O reaction, the three-body complex i.e., PRCw was formed depending on the approach of the hydrogen atom in the ·OH towards the water oxygen or the hydrogen of ·OH (Figure 3). We tried different way of projecting OH and H2O in the reaction, which led to two different PRCws, i.e., PRCw-1 (−4.3 kcal/mol) and PRCw-2 (−3.1 kcal/mol). The optimized structure is given in the supporting information Figure S1. As suggested in earlier work on a similar reaction system [21], only stable PRCw was considered for the rate constant calculations. Therefore, we restricted our discussion to only stable PRCw-1.
3.2. Enthalpies of Reactions (∆Hrxn (0 K))
The enthalpies (∆H(0 K)) relative to the reactants for ·OH + CH4, ·OH + CH4 (+HCOOH) and ·OH + CH4 (+H2O) reactions are given in Table 1. The calculated ∆H(0 K) of ·OH + CH4→CH3 + H2O (−14.4 kcal/mol) is in excellent agreement with the ATcT thermochemical data (−14.3 kcal/mol) base [38,39,40] and is in very good agreement with previous theoretical calculation (−13.9 kcal/mol) [7,9]. As shown in Figure 2, the formation of the complex in ·OH···CH4···HCOOH (PRCFA1) and ·OH···CH4···HCOOH (PRCFA2), are in trans- and cis orientation of HCOOH. Due to the different orientations of OH and HCOOH molecules, the BE of PRCFA1 (−6.96 kcal/mol) and PRCFA2 (−4.32 kcal/mol) are different (see Figure 2 and Table 1). The orientation of the H-atom of HCOOH makes a strong hydrogen bond between ·OH···CH4···HCOOH leading to more stable PRCFA1. The calculated BE of ·OH···CH4···H2O (PRCw) (−4 kcal/mol) is lower than BE of PRCFA (−6.96 kcal/mol) due to the presence of weak hydrogen bonds. It is clear from Table 1 that barrier height for the effect of FA and water molecules on ·OH + CH4 reaction is smaller than the barrier height of the free ·OH + CH4 reaction, which gives the indication of catalytic behavior of FA and water on ·OH + CH4 reaction.
3.3. Rate Constants
·OH + CH4 Reaction
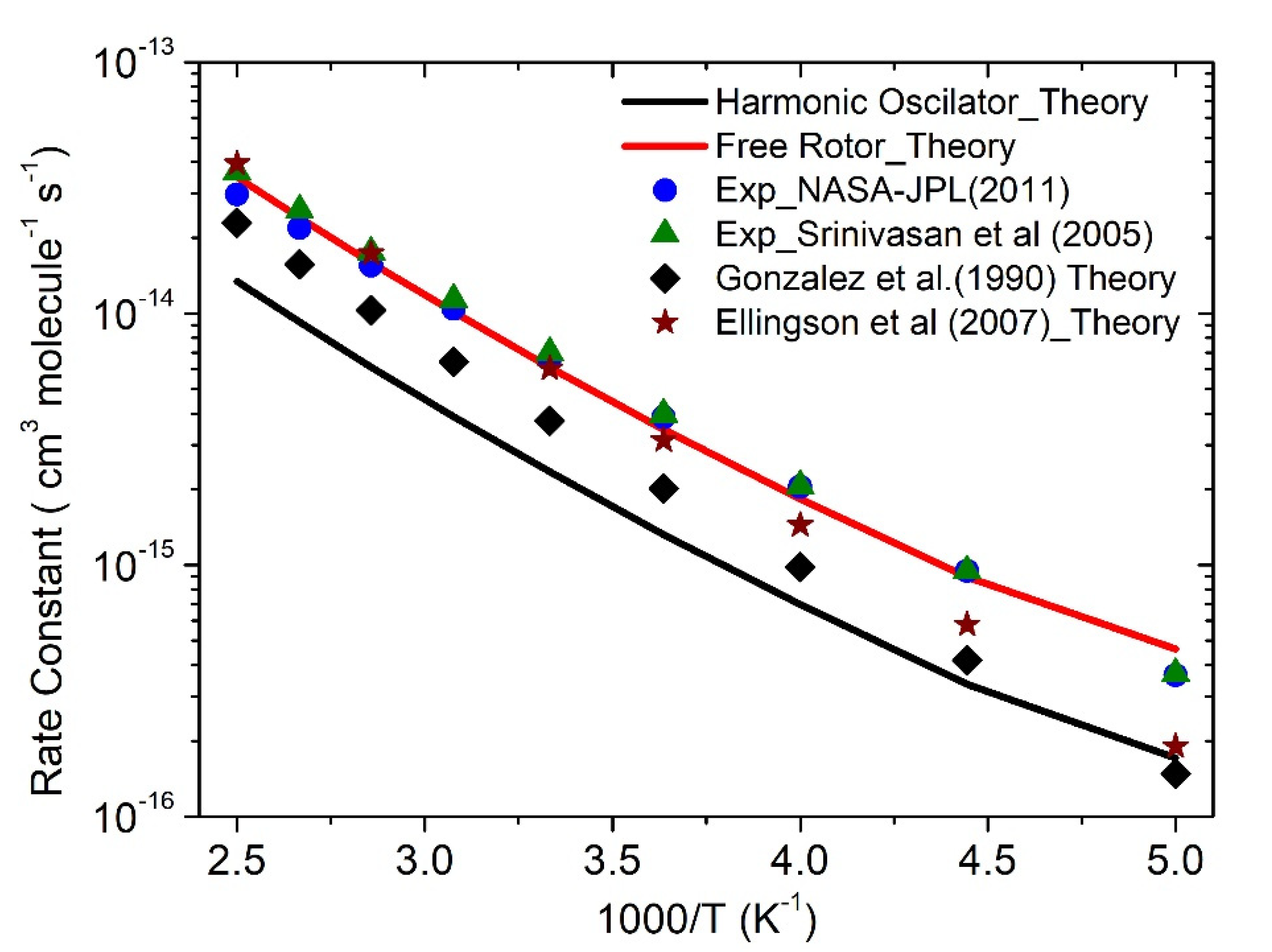
The CVT/SCT rate constant for ·OH + CH4 reaction is calculated and tabulated in Table 2 and shown in Figure 4. The calculated value using harmonic oscillator (HO) approximation is a factor of ~2 lower than experimentally measured values over the entire temperature range (Table 2). The rate constant calculated using HO approximation (2.3 × 10−15 cm3 molecule−1 s−1 at 300 K) is in very good agreement with the HO approximation of Ellingson et al [9] (2.3 × 10−15 cm3 molecule−1 s−1 at 300 K).
As suggested by Ellingson et al. [9], the free-rotor approximation is the correct choice, which is consistent with our study. The calculated value using free-rotor approximation at 300 K (6.1 × 10−15 cm3 molecule−1 s−1) is in excellent agreement with the experimentally measured [2,41] value (6.7 × 10−15 and 6.9 × 10−15 cm3 molecule−1 s−1) and good agreement with theoretically calculated ones (4 × 10−15 cm3 molecule−1 s−1 and 6 × 10−15 cm3 molecule−1 s−1) [7,9]. Our value is in very good agreement over the entire temperature range with the experimentally measured values. The calculated value is also in very good agreement with Srinivasan et al. (Figure 4) [5]. The calculations of rate constant for ·OH + CH4 reaction provide more confidence in our theoretical approach CC//M06-2X with CVT/SCT method. To avoid the repetition of the ·OH + CH4 reaction, we have restricted our comparison to only a few experimental and theoretical values [2,5,7,9,41].
3.4. Formic Acid Assisted ·OH + CH4 Reaction
As suggested in the thermochemistry section, the binding energy of CH4···HCOOH, and CH4···OH smaller than that of HCOOH··· HO· which will result in a shorter lifetime. Therefore, the formation of CH4···HCOOH, CH4···OH is almost negligible compared to HCOOH··· HO· and only one channel, i.e., CH4 + HCOOH··· HO· is considered for the rate constants calculations.
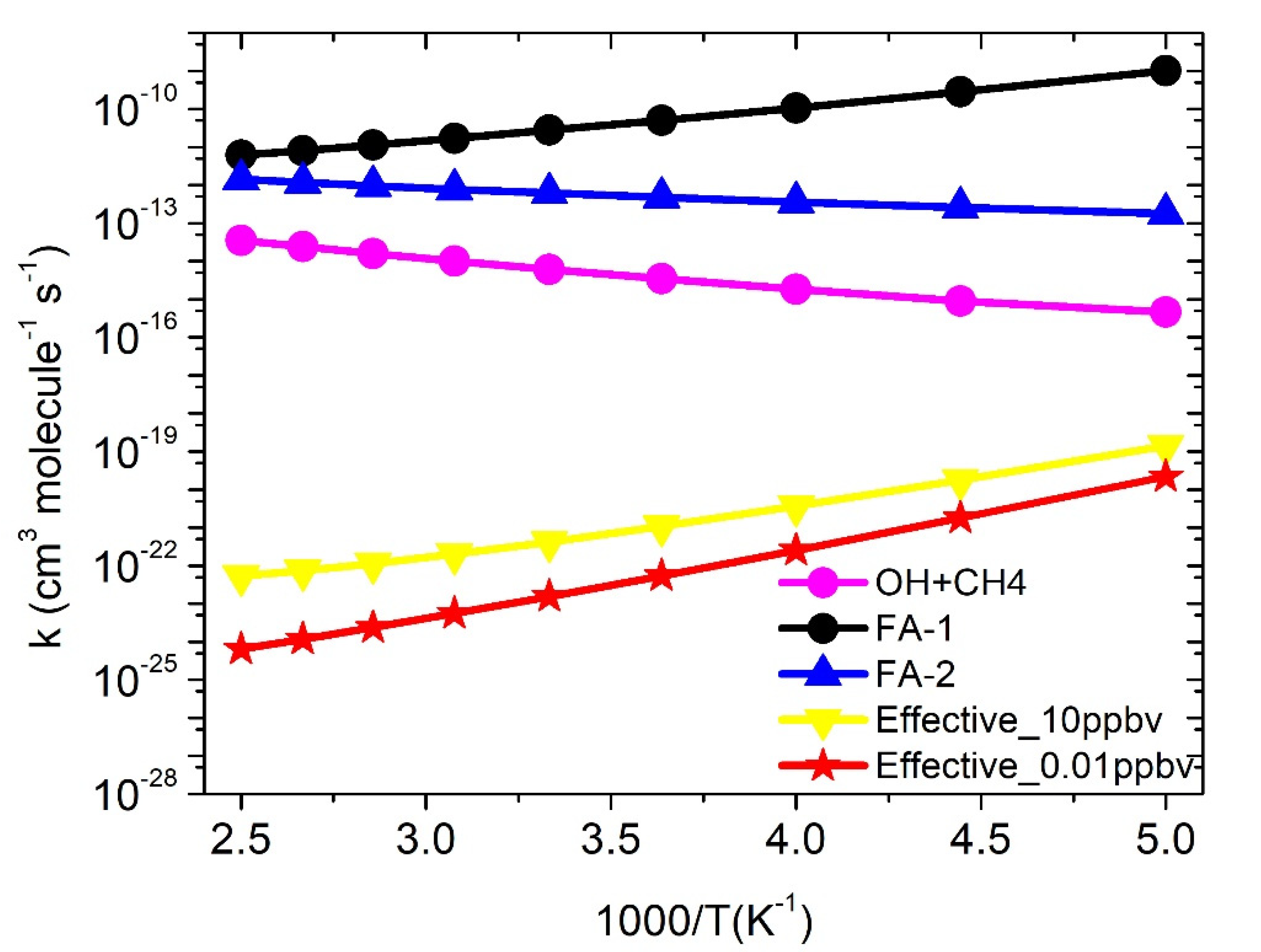
The temperature-dependent rate constants for the reactions of OH + CH4 (+HCOOH) were calculated based on the high-pressure limit condition. Locating the TS of backward reaction, i.e., PRCFA → ·OH···HCOOH + CH4 is complicated, in that case, to account for the presence of forward and backward reactions () equilibrium approach was used. This kinetic model is reasonably correct as discussed in our earlier studies [25,26,27,28]. The rate constants (s−1) (k2f) were computed using the CVT/SCT approach are presented in supporting information, Table S4. The equilibrium constants () are shown in Tables S5 and S6. The rate constants , for ·OH + CH4 (+HCOOH) were calculated in the temperature range of 200 to 400 K and are shown in Figure 5. are bimolecular rate constants of Pathway FA-1 and are bimolecular rate constants for Pathway FA-2, where and are equilibrium constants for CH4 + ·OH···HCOOH →PRCFA1 and CH4 + ·OH···HCOOH →PRCFA2, reactions, respectively (see Table S6). The rate constant of Pathway FA-1 (3 × 10−9 cm3 molecule−1 s−1 at 300 K) is higher than Pathway FA-2 (6 × 10−13 cm3 molecule−1 s−1 at 300 K), which shows that Pathway FA-1 is more kinetically favorable than Pathway FA-2.
The rate constant of FA-assisted reaction is at least four to five orders of magnitude higher (1 × 10−9 to 6 × 10−12 cm3 molecule−1 s−1) than the ·OH + CH4 (5 × 10−16 to 3.5 × 10−16 cm3 molecule−1 s−1) in the temperature range of 200 to 400 K. Our result is consistent with previous theoretical studies on similar reaction systems [13,14]. The calculations suggest that the catalytic effect of FA takes place if the concentration of FA is not included. As suggested earlier [25,26,27,28], the effective rate constant which include the concentration of FA is calculated by Equation (11):
where and are equilibrium constants of ·OH+HCOOH →RC4F1 and ·OH+HCOOH →RC4F2, reactions, respectively (see Table S5) and [HCOOH] is FA concentration used at 10 ppbv and 0.01 ppbv are based on previous studies [13,14]. The total effective rate constant for ·OH + CH4 (+HCOOH) (4 × 10−22 cm3 molecule−1 s−1 at 300 K) is ~7 order magnitude lower than ·OH + CH4 reaction (6 × 10−15 cm3 molecule−1 s−1). This result is similar to FA-assisted reaction ·OH + HCHO (+HCOOH): 7 × 10−20 cm3 molecule−1 s−1 at 300 K) as reported in the earlier study [19]. The rate constants show positive temperature-dependent (Figure 5). The similar kind of behavior was observed in the literature on the effect of FA on atmospheric reactions [13].
3.5. Water-Assisted ·OH + CH4 Reaction
As discussed in the case of FA assisted reaction, only one CH4 + ·OH···H2O channel is considered for the rate constants calculations. The other channels, i.e., the formation of CH4···H2O, CH4···OH, are almost negligible.
The reaction pathways for the effect of a water molecule on ·OH + CH4 starting with ·OH···H2O are presented in Equation (12):
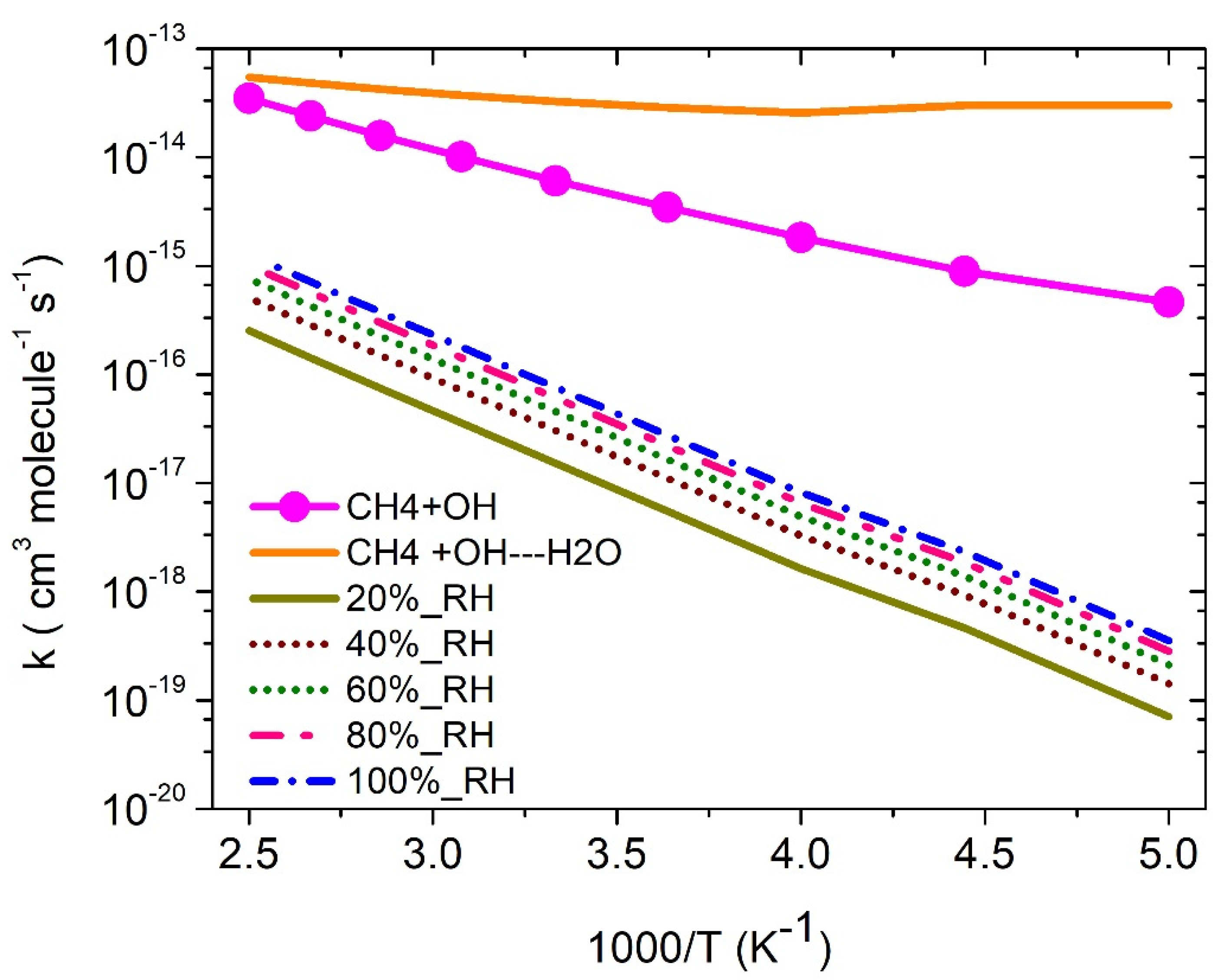
The rate constants (s−1) for ·OH + CH4 (+H2O) (PRCw → TS → P) were tabulated in Table S4. The rate constants (cm3 molecule−1 s−1) were calculated using , where is in equilibriumconstants involved in the ·OH···H2O + CH4 reaction. The rate constants in the temperature range of 200 K to 400 K for with and without water concentration are shown in Figure 6. The rate constants for CH4 + H2O···OH reaction (3 × 10−14 cm3 molecule−1 s−1 at 300 K) is ~20 times higher than ·OH + CH4 reaction (6 × 10−15 cm3 molecule−1 s−1 at 300 K. As shown in Figure 6, the water-catalyzed reaction has higher rate constants than a water-free reaction in the entire temperature range 200–400 K.
As suggested in our earlier studies [25,26,27], the concentration of water at varying humidity levels must be used to estimate the accurate rate constant. Therefore, the correct expression to calculate the total effective rate constants is given by Equation (13)
where are equilibrium constants of H2O +·OH →RC2w reactions, [H2O] is water concentration as discussed in earlier studies [21,27]. The effective rate constant (7 × 10−17 cm3 molecule−1 s−1 at 300 K) is factor of ~100 lower than the water-free OH + CH4 reaction (~6 × 10−15 cm3 molecule−1 s−1 at 300 K) at 100% humidity. The result is also consistent with similar atmospheric reactions, i.e., ·OH + CH2NH, ·OH + CH2CH2, ·OH + CH2O, and OH + CH3OH [25,26,27].
In order to gain correct insight into the effect of FA/water on ·OH + CH4 reaction, the free energy profiles for all the channels were computed and shown in supporting information Figure S2. Due to the high entropy of activation of (ΔS‡) of FA/H2O assisted reaction than free OH + CH4 reaction, rate constants are higher. It is also important to point out that, in the gas phase reaction, the barrier height ΔE‡ and ΔG‡ are calculated as the energies difference between the transition state and those of the two-body complex, i.e., RC complex, while in the water-assisted and FA-assisted reaction, ΔE‡ and ΔG‡ are calculated from energies difference of transition state and termolecular complex i.e., PRCs. Thus, if step 0 is ignored and the rate constant is calculated as keff = Keq × k2 and the reaction rate constant is higher in the presence of FA/H2O and catalytic effect is favorable for ·OH + CH4
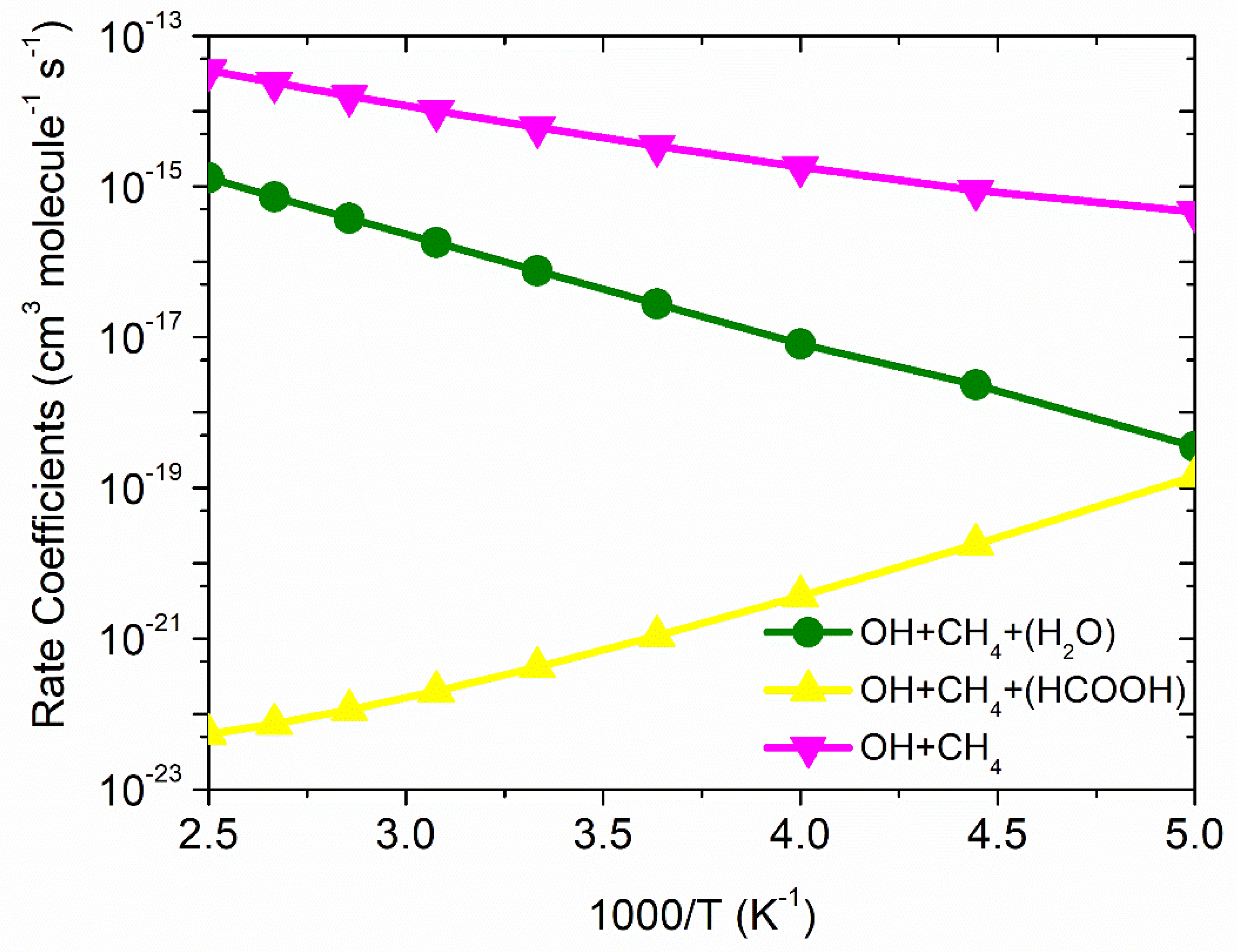
The total rate constants for ·OH + CH4 and effective rate constants for ·OH + CH4 (+HCOOH) and ·OH + CH4 (+H2O) reactions are tabulated in Table 3 and shown in Figure 7.
The result of this study suggests that the catalytic effect of FA and water takes place if the concentration of these molecules were not included, which is unrealistic. Therefore, to calculate the correct reaction rate constants we must include the concentration of HCOOH and H2O in the final rate constant calculations. In that situation, the results explain that the total effective rate constants for systems ·OH + CH4 (+HCOOH) (~7 order) and ·OH + CH4 (+H2O) (2 order) are magnitudes smaller than the free situation. The total effective rate constants ·OH + CH4 (+HCOOH) (~4 × 10−22 cm3 molecule−1 s−1 at 300 K) and ·OH + CH4 (+H2O) (~7 × 10−17 cm3 molecule−1 s−1 at 300 K) are smaller than ·OH + CH4 (~6 × 10−15 cm3 molecule−1 s−1 at 300 K). Similar results were reported in an earlier study [13,25,26,27,28]. It is clear from geometries of PRCs and TSs, which are different in OH + CH4 (+HCOOH) than reaction ·OH + CH4(+H2O) resulted in different computed enthalpies and rate constants. Because of that, the kinetics of ·OH + CH4 (+HCOOH) are quite different from those ·OH + CH4 (+H2O) reaction systems. In the case of the FA, the rate constants show positive temperature dependence, and in the case of water, the rate constants show negative temperature dependence. This is due to water concentration varying greatly with temperature, whereas HCOOH concentration is nearly temperature independent.
In the case of FA-assisted reaction, the PRCFA has the larger stability than that of PRCW, which results in a larger equilibrium constant of CH4 + HCOOH···HO (4 × 10−21 at 300 K) than CH4 + H2O···HO (1 × 10−21 at 300 K). The barrier height for FA-assisted reaction is lower than water-assisted reaction results larger rate constants for FA-assisted reaction (3 × 10−11 cm3 molecule−1 s−1) than water-assisted reaction (3.3 × 10−14 cm3 molecule−1 s−1). However, the concentrations of the FA are much lower than H2O in the atmosphere, which then makes the rate constants smaller for FA case (4.2 × 10−22 cm3 molecule−1 s−1 300 K) than water (7.6 × 10−17 cm3 molecule−1 s−1 at 300 K).
In general, the effective rate constants of the FA and water-assisted reaction is smaller than the ·OH + CH4 reaction system. As a result, the catalytic effect of FA/H2O on ·OH + CH4 reaction is of minor importance in gas-phase atmospheric reaction chemistry. This result is consistent with previously reported results on similar reaction system [13,25,26,27,28]. To understand the upper troposphere consequences of CH4, we have calculated the lifetime of CH4 at 250 K at an altitude of ∼11 km. The averaged concentration of [·OH] ~ 1 × 106 molecule cm−3 and the rate constant (2 × 10−15 cm3 molecule−1 s−1 at 250 K) were used to calculate the lifetime of CH4 as τ = 1/{[OH] × k}. The calculated lifetime of CH4 (16 years) is in good agreement with the value reported by Kulongoski et al. (~12–17 years) [42].
As discussed in this study, the rate constants for the effect of FA and water are almost negligible as compared to the naked reaction. It is important to mention that the comparison of effective rate constant with naked reaction does not provide the complete picture for the degradation mechanism of CH4. Experimental measurement is required to validate the current study. Based on this study, we believe that the effect of FA and water on the formation of formaldehyde will even be slower. We believe the current finding provides better insights into the gas-phase catalytic activity of FA and water molecules on ·OH + CH4.
4. Conclusions
The effect of FA and water molecules on ·OH + CH4 was explored. The potential energy surfaces for OH + CH4, CH4 + HO··· HCOOH, and CH4 + HO···H2O have been explored using CC//M06-2X. The rate constants for these reactions were computed using CVT/SCT approach.
In the presence of FA, the two different channels of the hydrogen abstraction reaction were identified. In the case of the water reaction, only one reaction pathway was identified. Under the atmospheric condition, the kinetics of ·OH + CH4 (+HCOOH) is quite different from those of ·OH + CH4 (+H2O). This difference is possibly due to the FA concentrations being much lower than H2O. Our results demonstrate that a FA and water molecule has the potential to catalyze the gas-phase reaction if the atmospheric concentration of these species is not included in the kinetic calculations. However, as suggested, the total effective rate constant must include the concentration of FA and water. In that situation, the total rate is constants for the effect of FA and water than the ·OH + CH4 reaction is lower. The present study provides a comprehensive model of how the acidic nature of these catalysts affects the gas-phase reaction kinetics. The atmospheric degradation mechanism suggests that CH3 can further react with O2 molecules to form the formaldehyde under atmospheric and combustion conditions. The effect of FA and water molecules could be even slower in the formation of formaldehyde. These kinds of results are interesting and can be used to identify the effect of FA and water on higher chain alkane compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12020133/s1, Figure S1: Three body complexes for the effect of FA/H2O on OH + CH4 reaction; Figure S2: Free energy profiles for the CH4 + OH (___), CH4 + OH + H2O (___) and CH4 + OH + HCOOH (___) reactions; Table S1: Optimized geometries of reactants, complexes, products and transition states obtained using M06-2X/6-311++G(3df,3pd); Table S2: Rotational -vibrational parameters obtained from vibrational analysis; Table S3: Electronic Energies ( Eo), Zero-point correction (ZPE), Thermal Correction (ET), Thermal Correction to Enthalpy (HT),Free energy correction (G), Entropy (cal/mol-K) of all the species involved in the reactions; Table S4: Calculated CVT unimolecular rate constant (k2 in s−1); Table S5: Calculated equilibrium constants (Keq in cm3 molecule−1) for the formation of two-body complex; Table S6: Calculated equilibrium constants (Keq in cm3 molecule−1) for the formation of three-body complex.
Author Contributions
M.A.A. prepared figures and tables and draft of the paper. B.M. done all the Gaussian and chemical kinetic calculations as discussed it in the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deanship of Scientific Research at King Faisal University; Nasher Track #206164 and the Article Processing Charge (APC) was also funded by Nasher (206164) Deanship of Scientific Research at King Faisal University Saudi Arabia.
Data Availability Statement
All data generated through this study are given in Supporting Information file.
Acknowledgments
Mohamad Akbar Ali (MAA) gratefully acknowledges the Deanship of Scientific Research at King Faisal University, Saudi Arabia for financial support under Nasher Track (206164.). MAA thanks the computational support by Department of Chemistry at King Faisal University, Saudi Arabia, and supercomputer facility at department of Chemistry National Taiwan University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crabtree, R.H. Aspects of Methane Chemistry. Chem. Rev. 1995, 95, 987–1007. [Google Scholar] [CrossRef]
- Vaghjiani, G.L.; Ravishankara, A.R. New measurement of the rate coefficient for the reaction of OH with methane. Nature 1991, 350, 406–409. [Google Scholar] [CrossRef]
- Jackson, R.B.; Saunois, M.; Bousquet, P.; Canadell, J.G.; Poulter, B.; Stavert, A.R.; Bergamaschi, P.; Niwa, Y.; Segers, A.; Tsuruta, A. Increasing anthropogenic methane emissions arise equally from agricultural and fossil fuel sources. Environ. Res. Lett. 2020, 15, 071002. [Google Scholar] [CrossRef]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Srinivasan, N.K.; Su, M.C.; Sutherland, J.W.; Michael, J.V. Reflected Shock Tube Studies of High-Temperature Rate Constants for OH + CH4 → CH3 + H2O and CH3 + NO2 → CH3O + NO. J. Phys. Chem. A 2005, 109, 1857–1863. [Google Scholar] [CrossRef]
- Li, J.; Guo, H. Thermal Rate Coefficients and Kinetic Isotope Effects for the Reaction OH + CH4 → H2O + CH3 on an ab Initio-Based Potential Energy Surface. J. Phys. Chem. A 2018, 122, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; McDouall, J.J.W.; Schlegel, H.B. Ab initio study of the reactions between methane and hydroxyl, hydrogen atom, and triplet oxygen atom. J. Phys. Chem. 1990, 94, 7467–7471. [Google Scholar] [CrossRef]
- Li, J.; Guo, H. Communication: An accurate full 15 dimensional permutationally invariant potential energy surface for the OH + CH4 → H2O + CH3 reaction. J. Chem. Phys. 2015, 143, 221103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellingson, B.A.; Pu, J.; Lin, H.; Zhao, Y.; Truhlar, D.G. Multicoefficient Gaussian-3 Calculation of the Rate Constant for the OH + CH4 Reaction and Its 12C/13C Kinetic Isotope Effect with Emphasis on the Effects of Coordinate System and Torsional Treatment. J. Phys. Chem. A 2007, 111, 11706–11717. [Google Scholar] [CrossRef] [PubMed]
- Franco, B.; Blumenstock, T.; Cho, C.; Clarisse, L.; Clerbaux, C.; Coheur, P.F.; De Mazière, M.; De Smedt, I.; Dorn, H.P.; Emmerichs, T.; et al. Ubiquitous atmospheric production of organic acids mediated by cloud droplets. Nature 2021, 593, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Millet, D.B.; Baasandorj, M.; Farmer, D.K.; Thornton, J.A.; Baumann, K.; Brophy, P.; Chaliyakunnel, S.; de Gouw, J.A.; Graus, M.; Hu, L.; et al. A large and ubiquitous source of atmospheric formic acid. Atmos. Chem. Phys. 2015, 15, 6283–6304. [Google Scholar] [CrossRef] [Green Version]
- Keene, W.C.; Galloway, J.N. The biogeochemical cycling of formic and acetic acids through the troposphere: An overview of current understanding. Tellus 1988, 40B, 322–334. [Google Scholar] [CrossRef]
- Kumar, A.; Mallick, S.; Mishra, B.K.; Kumar, P. Effect of ammonia and formic acid on the CH3O˙ + O2 reaction: A quantum chemical investigation. Phys. Chem. Chem. Phys. 2020, 22, 2405–2413. [Google Scholar] [CrossRef]
- Mallick, S.; Sarkar, S.; Bandyopadhyay, B.; Kumar, P. Effect of Ammonia and Formic Acid on the OH• + HCl Reaction in the Troposphere: Competition between Single and Double Hydrogen Atom Transfer Pathways. J. Phys. Chem. A 2017, 122, 350–363. [Google Scholar] [CrossRef]
- Louie, M.K.; Francisco, J.S.; Verdicchio, M.; Klippenstein, S.J.; Sinha, A. Hydrolysis of Ketene Catalyzed by Formic Acid: Modification of Reaction Mechanism, Energetics, and Kinetics with Organic Acid Catalysis. J. Phys. Chem. A 2015, 119, 4347–4357. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Wen, M.; Tang, Z.; Long, B.; Yu, X.; Zhao, C.; Wang, W. Effects of water, ammonia and formic acid on HO2 + Cl reactions under atmospheric conditions: Competition between a stepwise route and one elementary step. RSC Adv. 2019, 9, 21544–21556. [Google Scholar] [CrossRef] [Green Version]
- Hazra, M.K.; Francisco, J.S.; Sinha, A. Gas Phase Hydrolysis of Formaldehyde To Form Methanediol: Impact of Formic Acid Catalysis. J. Phys. Chem. A 2013, 117, 11704–11710. [Google Scholar] [CrossRef]
- Monge-Palacios, M.; Rissanen, M.P.; Wang, Z.; Sarathy, S.M. Theoretical kinetic study of the formic acid catalyzed Criegee intermediate isomerization: Multistructural anharmonicity and atmospheric implications. Phys. Chem. Chem. Phys. 2018, 20, 10806–10814. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Du, B.; Qin, Z. Catalytic Effect of Water, Formic Acid, or Sulfuric Acid on the Reaction of Formaldehyde with OH Radicals. J. Phys. Chem. A 2014, 118, 4797–4807. [Google Scholar] [CrossRef]
- Jara-Toro, R.A.; Hernández, F.J.; Taccone, R.A.; Lane, S.I.; Pino, G.A. Water catalysis of the reaction between methanol and OH at 294 K and the atmospheric implications. Angew. Chem. Int. Ed. 2017, 56, 2166–2170. [Google Scholar] [CrossRef]
- Wu, J.; Gao, L.G.; Varga, Z.; Xu, X.; Ren, W.; Truhlar, D.G. Water catalysis of the Reaction of Methanol with OH Radical in the Atmosphere is Negligible. Angew. Chem. Int. Ed. 2020, 59, 10826–10830. [Google Scholar] [CrossRef]
- Vöhringer-Martinez, E.; Hansmann, B.; Hernandez, H.; Francisco, J.S.; Troe, J.; Abel, B. Water Catalysis of a Radical-Molecule Gas-Phase Reaction. Science 2007, 315, 497–501. [Google Scholar] [CrossRef]
- Thomsen, D.L.; Kurtén, T.; Jørgensen, S.; Wallington, T.J.; Baggesen, S.B.; Aalling, C.; Kjaergaard, H.G. On the possible catalysis by single water molecules of gas-phase hydrogen abstraction reactions by OH radicals. Phys. Chem. Chem. Phys. 2012, 14, 12992–12999. [Google Scholar] [CrossRef]
- Inaba, S. Catalytic Role of H2O Molecules in Oxidation of CH3OH in Water. Catalysts 2018, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Balaganesh, M.; Lin, K.C. Catalytic effect of a single water molecule on the OH + CH2NH reaction. Phys. Chem. Chem. Phys. 2018, 20, 4297–4307. [Google Scholar] [CrossRef]
- Ali, M.A.; Balaganesh, M.; Jang, S. Can a single water molecule catalyze the OH+CH2CH2 and OH+CH2O reactions? Atmos. Environ. 2019, 207, 82–92. [Google Scholar] [CrossRef]
- Ali, M.A.; Balaganesh, M.; Al-Odail, F.A.; Lin, K.C. Effect of ammonia and water molecule on OH + CH3OH reaction under tropospheric condition. Sci. Rep. 2021, 11, 12185. [Google Scholar] [CrossRef]
- Ali, M.A. Computational studies on the gas phase reaction of methylenimine (CH2NH) with water molecules. Sci. Rep. 2020, 10, 10995. [Google Scholar] [CrossRef]
- Buszek, R.J.; Torrent-Sucarrat, M.; Anglada, J.M.; Francisco, J.S. Effects of a Single Water Molecule on the OH + H2O2 Reaction. J. Phys. Chem. A 2012, 116, 5821–5829. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Vivier-Bunge, A. On the possible catalytic role of a single water molecule in the acetone + OH gas phase reaction: A theoretical pseudo-second-order kinetics study. Theor. Chem. Accounts 2011, 129, 209–217. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Reyes, L.; Vivier-Bunge, A. Can a Single Water Molecule Really Catalyze the Acetaldehyde + OH Reaction in Tropospheric Conditions? J. Phys. Chem. Lett. 2010, 1, 3112–3115. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, W.; Li, C.; Du, Y.; Lü, J. Catalytic effect of a single water molecule on the atmospheric reaction of HO2 + OH: Fact or fiction? A mechanistic and kinetic study. RSC Adv. 2013, 3, 7381–7391. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhang, S.; Lynch, B.J.; Corchado, J.C.; Chuang, Y.-Y.; Fast, P.L.; Hu, W.P.; Liu, Y.P.; Lynch, G.C.; Nguyen, K.A.; et al. POLYRATE, Version 2008; University of Minnesota: Minneapolis, MN, USA, 2009. [Google Scholar]
- Zheng, J.; Zhang, S.; Corchado, J.C.; Chuang, Y.Y.; Coitino, E.L.; Ellingson, B.A.; Truhlar, D.G. GAUSSRATE, Version 2009-A; University of Minnesota: Minneapolis, MN, USA, 2010. [Google Scholar]
- Barker, J.R.; Ortiz, N.F.; Preses, J.M.; Lohr, L.L.; Maranzana, A.; Stimac, P.J.; Lam, N.T.; Dhilip Kumar, T.J. MultiWell-2016 Software; University of Michigan: Ann Arbor, MI, USA, 2016. [Google Scholar]
- Ruscic, B.; Pinzon, R.E.; Morton, M.L.; von Laszevski, G.; Bittner, S.J.; Nijsure, S.G.; Amin, K.A.; Minkoff, M.; Wagner, A.F. Introduction to Active Thermochemical Tables: Several “Key” Enthalpies of Formation Revisited. J. Phys. Chem. A 2004, 108, 9979–9997. [Google Scholar] [CrossRef]
- Ruscic, B.; Pinzon, R.E.; Von Laszewski, G.; Kodeboyina, D.; Burcat, A.; Leahy, D.; Montoy, D.; Wagner, A.F. Active Thermochemical Tables: Thermochemistry for the 21st century. J. Phys. Conf. Ser. 2005, 16, 561–570. [Google Scholar] [CrossRef]
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT) Enthalpies of Formation Values Based on ver. 1.112d of the Thermochemical Network. 2018. Available online: https://atct.anl.gov/ (accessed on 26 December 2021).
- Sander, S.P.; Abbatt, J.P.D.; Barker, J.R.; Burkholder, J.B.; Friedl, R.R.; Golden, D.M.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; Moortgat, G.K.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 17; JPL Publication 10-6; Jet Propulsion Laboratory: Pasadena, CA, USA, 2011. [Google Scholar]
- Kulongoski, J.T.; McMahon, P.B. Methane emissions from groundwater pumping in the USA. npj Clim. Atmos. Sci. 2019, 2, 11. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Potential energy surface for the ·OH + CH4 reaction leading to form methyl radical and water. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.
Figure 1.
Potential energy surface for the ·OH + CH4 reaction leading to form methyl radical and water. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.

Figure 2.
Potential-energy surface for the effect of FA on ·OH + CH4 reaction leading to form methyl radical, water, and formic acid. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.
Figure 2.
Potential-energy surface for the effect of FA on ·OH + CH4 reaction leading to form methyl radical, water, and formic acid. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.

Figure 3.
Potential energy surface profile for the effect of water molecules on ·OH + CH4 reaction leading to form methyl radical and water molecules. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.
Figure 3.
Potential energy surface profile for the effect of water molecules on ·OH + CH4 reaction leading to form methyl radical and water molecules. The stationary point was computed at the CC//M06-2X level and ZPE correction obtained from vibrational analysis using M06-2X.

Figure 4.
Rate constants for ·OH + CH4 reaction. The rate constants were calculated using harmonic oscillator and free-rotor approximation.
Figure 4.
Rate constants for ·OH + CH4 reaction. The rate constants were calculated using harmonic oscillator and free-rotor approximation.

Figure 5.
Rate constants for ·OH + CH4 and ·OH + CH4 (+HCOOH) reactions. The presented rate constants are for assumed the concentration of HCOOH at 0.01 ppbv (2 × 108 cm−3 molecule) and 10 ppbv (2 × 1011 cm−3 molecule). FA-1 and FA-2 are rate constants without FA concentration.
Figure 5.
Rate constants for ·OH + CH4 and ·OH + CH4 (+HCOOH) reactions. The presented rate constants are for assumed the concentration of HCOOH at 0.01 ppbv (2 × 108 cm−3 molecule) and 10 ppbv (2 × 1011 cm−3 molecule). FA-1 and FA-2 are rate constants without FA concentration.

Figure 6.
Rate constants for ·OH + CH4 + (H2O) with different relative humidity. CH4 + H2O···OH is the rate constants without water concentration. The water concentration at this humidity is taken from our earlier studies [21,27]. The presented rates are the relative humidity of water from 20% to 100%.
Figure 6.
Rate constants for ·OH + CH4 + (H2O) with different relative humidity. CH4 + H2O···OH is the rate constants without water concentration. The water concentration at this humidity is taken from our earlier studies [21,27]. The presented rates are the relative humidity of water from 20% to 100%.

Figure 7.
Comparison between rate constants for ·OH + CH4, ·OH + CH4 + (HCOOH) and ·OH + CH4 + (H2O).
Figure 7.
Comparison between rate constants for ·OH + CH4, ·OH + CH4 + (HCOOH) and ·OH + CH4 + (H2O).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enthalpies of reaction (∆Hrxn (0 K)) in kcal mol−1 of ·OH + CH4 reaction.
| ·OH + CH4→ | This Work | Literature a,b,c |
|---|---|---|
| ·OH···CH4 (RC1) | 0.30 | |
| HO···HCH3 (TS) | 4.93 | |
| ·CH3 + H2O | −14.4 | −14.3 a, −13.9 b, −13.4 c |
| ·OH + CH4 + HCOOH → | This Work | |
| ·OH···CH4···HCOOH (PRCFA1) | −6.96 | |
| ·OH···CH4···HCOOH(PRCFA) | −4.32 | |
| ·OH···HCH3···HCOOH (TSFA1) | −3.2 | |
| ·OH···HCOOH···HCH3 (TSFA2) | +1.0 | |
| H2O···HCOOH···CH3 (Post-PRCFA1) H2O···HCOOH···CH3 (Post-PRCFA2) CH3 + H2O + FA | −17.4 −22.7 −14.4 | |
| ·OH + CH4 +H2O→ | This Work | |
| ·OH···CH4···H2O (PRCw) | −4.32 | |
| ·OH···HCH3···H2O (TSw) | +1.6 | |
| H2O···H2O···CH3(Post-PRCw) | −18.42 | |
| ·CH3 + 2H2O | −14.4 |
Table 2.
Comparison of calculated rate constants (cm3 molecule−1 s−1) and literature rate constants for the ·OH + CH4→CH3 + H2O.
Table 2.
Comparison of calculated rate constants (cm3 molecule−1 s−1) and literature rate constants for the ·OH + CH4→CH3 + H2O.
| Temp (K) | k-HO | k-FR | Exp [2,39] | Exp [5] | Theory [9] |
|---|---|---|---|---|---|
| 200 | 1.7 × 10−16 | 4.6 × 10−16 | 3.6 × 10−16 | 3.7 × 10−16 | 1.5 × 10−16 |
| 225 | 3.4 × 10−16 | 8.9 × 10−16 | 9.5 × 10−16 | 9.5 × 10−16 | 4.2 × 10−16 |
| 250 | 7.0 × 10−16 | 1.8 × 10−15 | 2.0 × 10−15 | 2.1 × 10−15 | 9.8 × 10−16 |
| 275 | 1.3 × 10−15 | 3.5 × 10−15 | 3.9 × 10−15 | 4.0 × 10−15 | 2.0 × 10−15 |
| 300 | 2.3 × 10−15 | 6.1 × 10−15 | 6.6 × 10−15 | 6.9 × 10−15 | 3.8 × 10−15 |
| 325 | 3.9 × 10−15 | 1.0 × 10−14 | 1.0 × 10−14 | 1.1 × 10−14 | 6.4 × 10−15 |
| 350 | 6.1 × 10−15 | 1.6 × 10−14 | 1.5 × 10−14 | 1.8 × 10−14 | 1.0 × 10−14 |
| 375 | 9.2 × 10−15 | 2.4 × 10−14 | 2.2 × 10−14 | 2.6 × 10−14 | 1.6 × 10−14 |
| 400 | 1.3 × 10−14 | 3.5 × 10−14 | 2.9 × 10−14 | 3.6 × 10−14 | 2.3 × 10−14 |
| k = ATn exp(−B/T) | A = 1.2 × 10−28 N = 5.9 B = 134 | A = 3 × 10−29 N = 5.9 B = 134 | A = 3 × 10−14 N = 0.67 B = 1575 | A = 1.6 × 10−18 N = 2.1 B = 1231 | A = 3.7 × 10−13 N = 2.3 B = 1377 |
Table 3.
Calculated rate constants ( cm3 molecule−1 s−1) for the ·OH + CH4, ·OH + CH4 (+HCOOH) and ·OH + CH4 (+H2O).
Table 3.
Calculated rate constants ( cm3 molecule−1 s−1) for the ·OH + CH4, ·OH + CH4 (+HCOOH) and ·OH + CH4 (+H2O).
| Temp (K) | kCH4+OH | keff FA | keffW |
|---|---|---|---|
| 200 | 4.6 × 10−16 | 1.4 × 10−19 | 3.5 × 10−19 |
| 225 | 8.9 × 10−16 | 1.8 × 10−20 | 4.9 × 10−18 |
| 250 | 1.8 × 10−15 | 3.7 × 10−21 | 8.2 × 10−18 |
| 275 | 3.5 × 10−15 | 1.1 × 10−21 | 2.8 × 10−17 |
| 300 | 6.1 × 10−15 | 4.2 × 10−22 | 7.6 × 10−17 |
| 325 | 1.0 × 10−14 | 2.0 × 10−22 | 1.8 × 10−16 |
| 350 | 1.6 × 10−14 | 1.1 × 10−22 | 3.8 × 10−16 |
| 375 | 2.4 × 10−14 | 7.4 × 10−23 | 7.1 × 10−16 |
| 400 | 3.5 × 10−14 | 5.4 × 10−23 | 1.3 × 10−15 |
| k = ATn exp(−B/T) | A = 3.0 × 10−29 N = 5.9 B = 134 | A = 1.0 × 10−54 N = 9.7 B = −5872 | A = 7.4 × 10−18 N = 2.0 B = 2717 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ali, M.A.; Muthiah, B. Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study. Catalysts 2022, 12, 133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020133
AMA Style
Ali MA, Muthiah B. Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study. Catalysts. 2022; 12(2):133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020133
Chicago/Turabian StyleAli, Mohamad Akbar, and Balaganesh Muthiah. 2022. "Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study" Catalysts 12, no. 2: 133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020133
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.