
Partial Methane Oxidation in Fuel Cell-Type Reactors for Co-Generation of Energy and Chemicals: A Short Review


Abstract
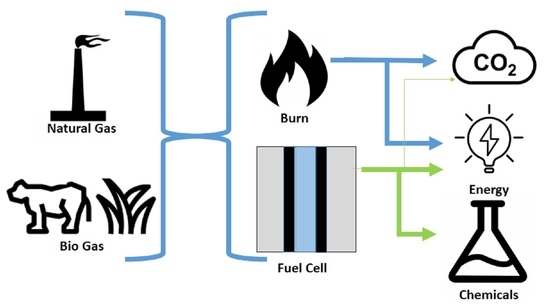
:
1. Introduction
2. Electrochemical Methods for Methane Oxidation
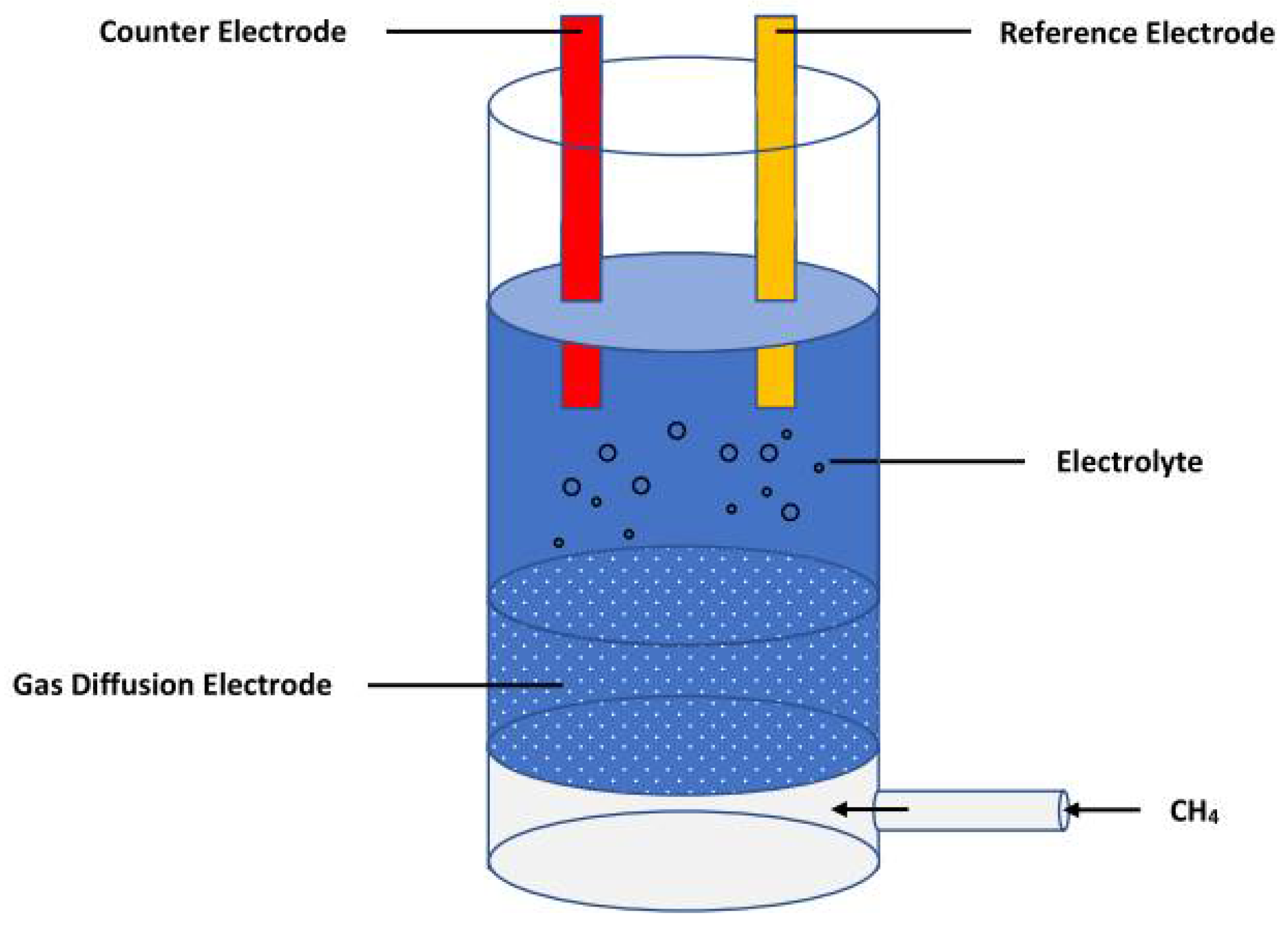
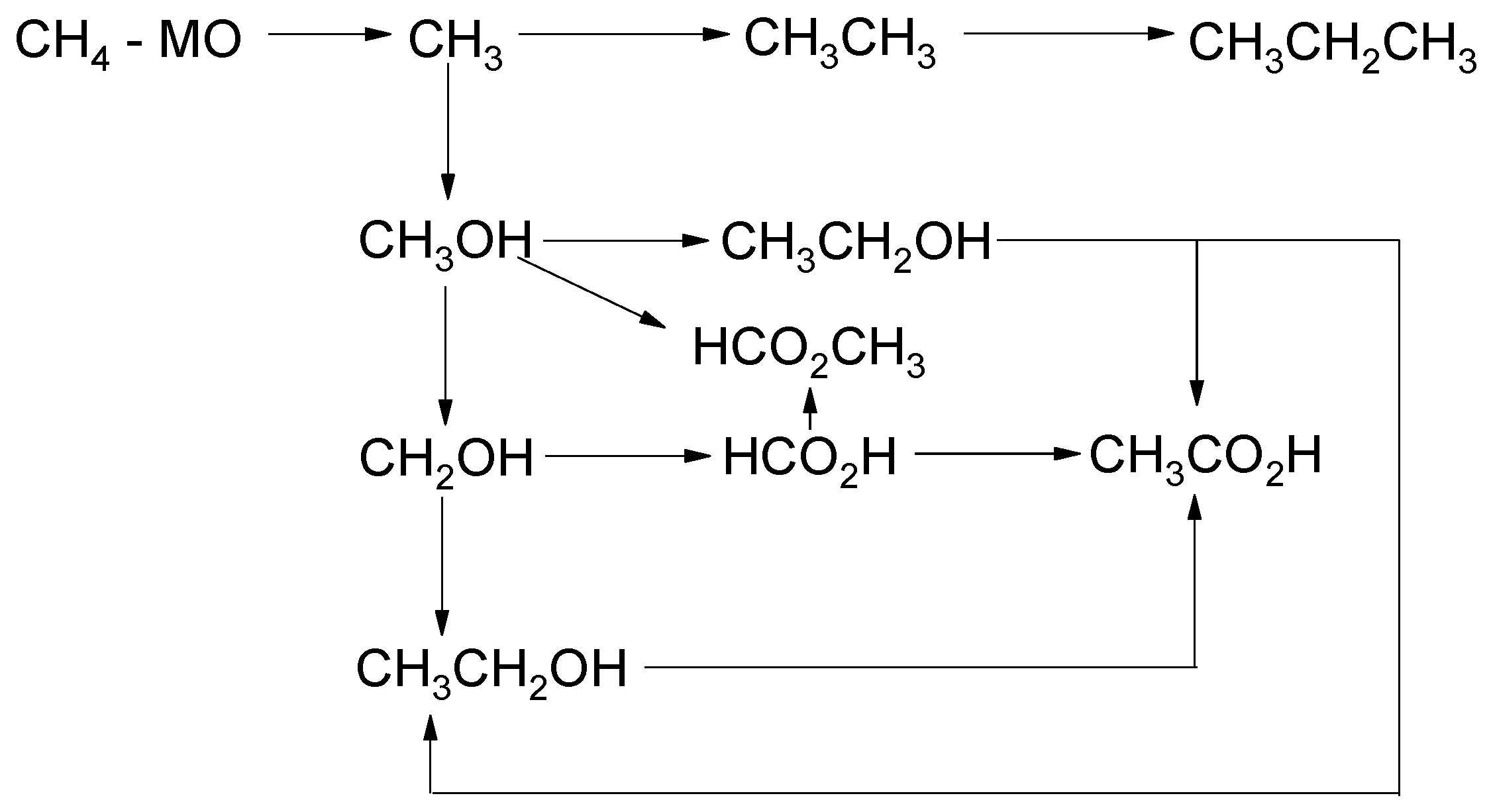
2.1. Faradaic Methane Oxidation
2.2. Non-Faradaic Methane Oxidation
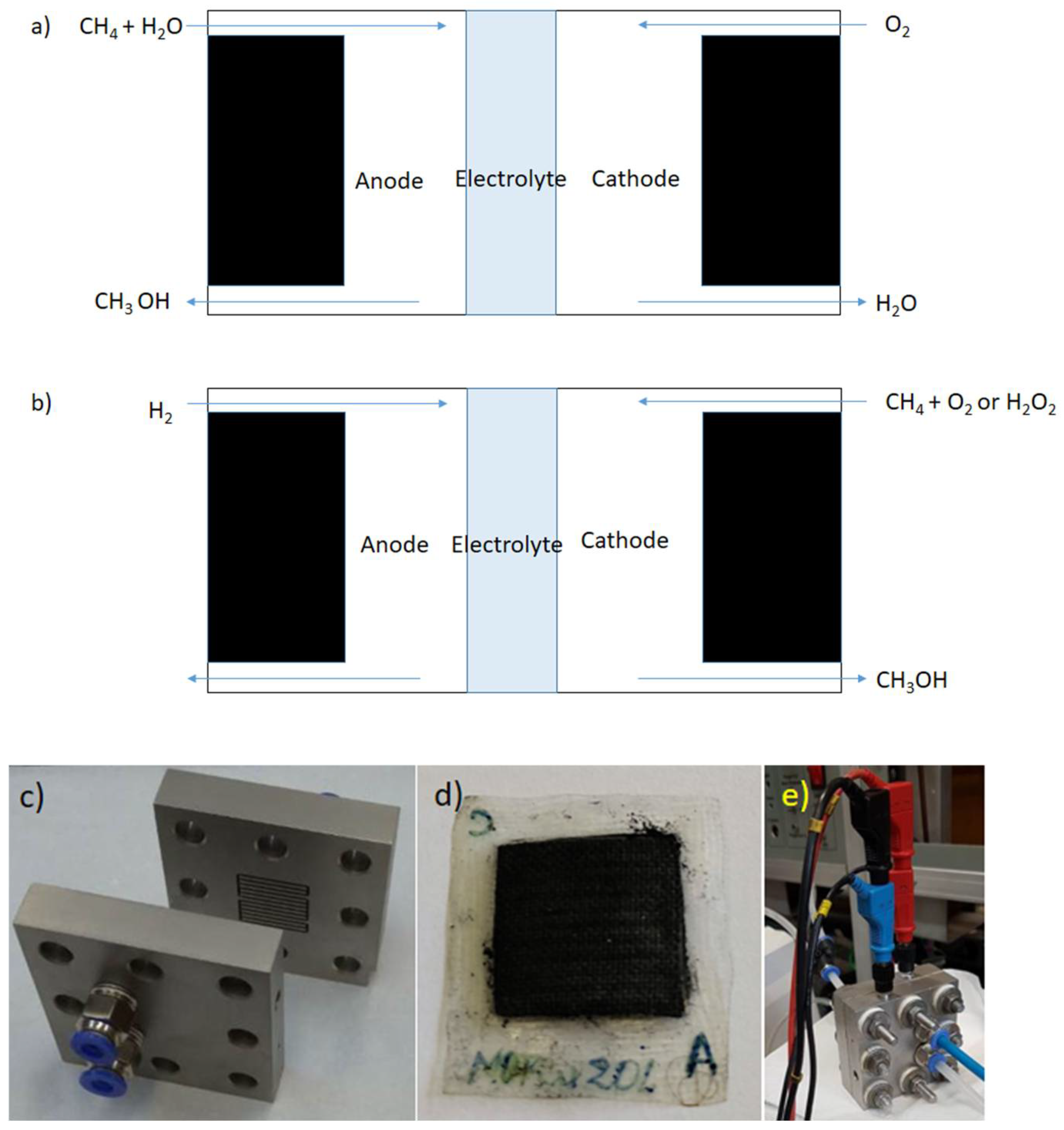
3. Fuel Cells
4. Fuel Cells for Cogeneration of Energy and Chemicals
5. High Temperature Fuel Cells
6. CH4/CO2 Mixtures
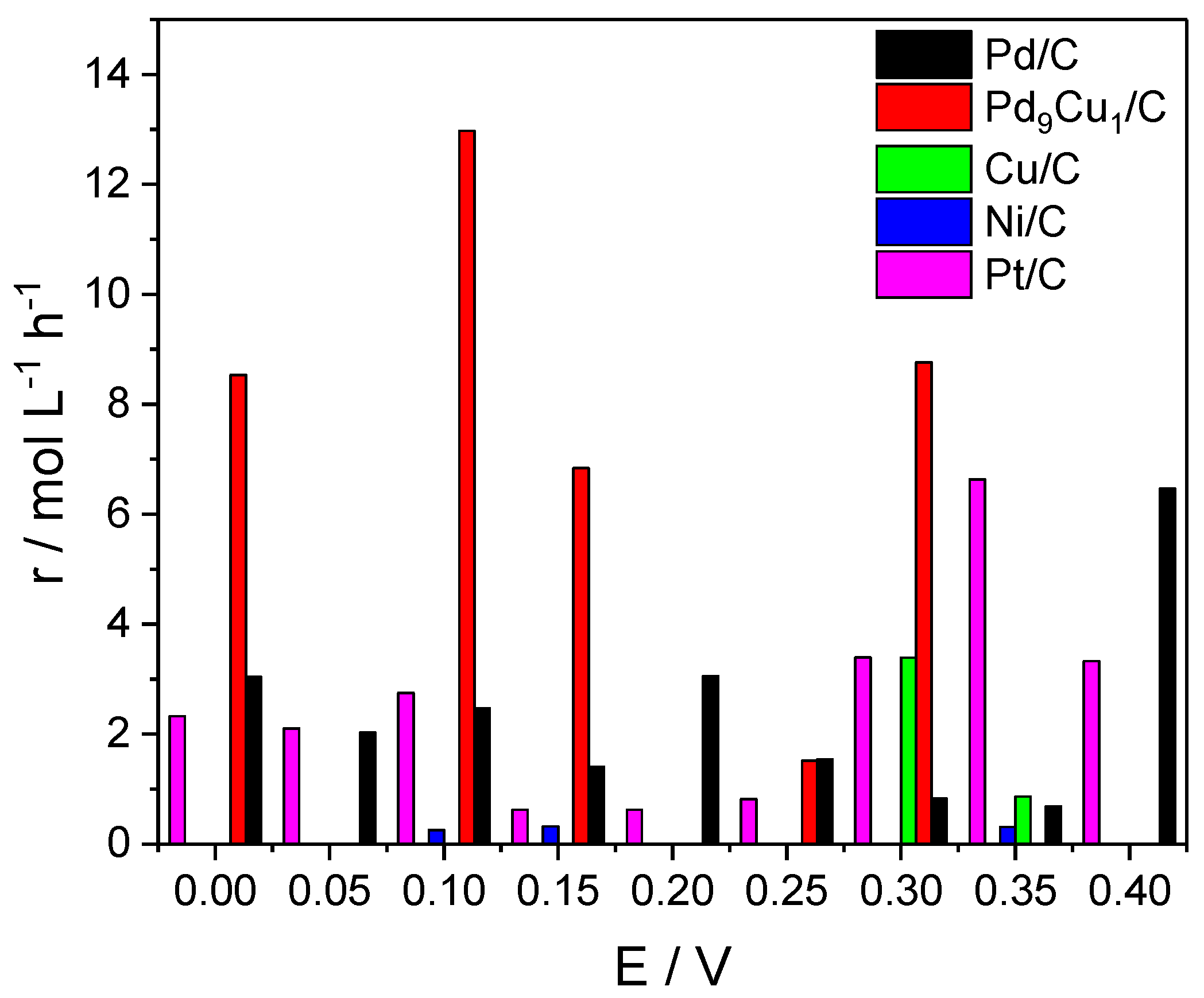
7. Materials for Methane Partial Oxidation Reaction
8. Opportunities and Outlook
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lange, J.-P.; Sushkevich, V.L.; Knorpp, A.J.; van Bokhoven, J.A. Methane-to-Methanol via Chemical Looping: Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Jang, J.; Shen, K.; Morales-Guio, C.G. Electrochemical Direct Partial Oxidation of Methane to Methanol. Joule 2019, 3, 2589–2593. [Google Scholar] [CrossRef]
- Chan, S.I.; Yu, S.S.F.; Liu, C.-C.; Mou, C.-Y. Selective oxidation of light alkanes under mild conditions. Curr. Opin. Green Sustain. Chem. 2020, 22, 39–46. [Google Scholar] [CrossRef]
- Blanco, H.; Nijs, W.; Ruf, J.; Faaij, A. Potential of Power-to-Methane in the EU energy transition to a low carbon system using cost optimization. Appl. Energy 2018, 232, 323–340. [Google Scholar] [CrossRef]
- Li, D.; Xu, R.; Gu, Z.; Zhu, X.; Qing, S.; Li, K. Chemical-Looping Conversion of Methane: A Review. Energy Technol. 2020, 8, 1900925. [Google Scholar] [CrossRef]
- Yang, J.; Hao, J.; Wei, J.; Dai, J.; Li, Y. Visible-light-driven selective oxidation of methane to methanol on amorphous FeOOH coupled m-WO3. Fuel 2020, 266, 117104. [Google Scholar] [CrossRef]
- San-José-Alonso, D.; Juan-Juan, J.; Illán-Gómez, M.J.; Román-Martínez, M.C. Ni, Co and bimetallic Ni–Co catalysts for the dry reforming of methane. Appl. Catal. A Gen. 2009, 371, 54–59. [Google Scholar] [CrossRef]
- Shavi, R.; Hiremath, V.; Seo, J.G. Radical-initiated oxidative conversion of methane to methanol over metallic iron and copper catalysts. Mol. Catal. 2018, 445, 232–239. [Google Scholar] [CrossRef]
- Lee, B.; Hibino, T. Efficient and selective formation of methanol from methane in a fuel cell-type reactor. J. Catal. 2011, 279, 233–240. [Google Scholar] [CrossRef]
- Xie, S.; Lin, S.; Zhang, Q.; Tian, Z.; Wang, Y. Selective electrocatalytic conversion of methane to fuels and chemicals. J. Energy Chem. 2018, 27, 1629–1636. [Google Scholar] [CrossRef] [Green Version]
- Nimkar, S.C.; Mewada, R.K.; Rosen, M.A. Exergy and exergoeconomic analyses of thermally coupled reactors for methanol synthesis. Int. J. Hydrogen Energy 2017, 42, 28113–28127. [Google Scholar] [CrossRef]
- Kim, S.; Kim, M.; Kim, Y.T.; Kwak, G.; Kim, J. Techno-economic evaluation of the integrated polygeneration system of methanol, power and heat production from coke oven gas. Energy Convers. Manag. 2019, 182, 240–250. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. Synthetic methylotrophy: Strategies to assimilate methanol for growth and chemicals production. Curr. Opin. Biotechnol. 2019, 59, 165–174. [Google Scholar] [CrossRef]
- Sharma, R.; Poelman, H.; Marin, G.B.; Galvita, V.V. Approaches for Selective Oxidation of Methane to Methanol. Catalysts 2020, 10, 194. [Google Scholar] [CrossRef] [Green Version]
- Sehested, J. Industrial and scientific directions of methanol catalyst development. J. Catal. 2019, 371, 368–375. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Karakaya, C.; Kee, R.J. Progress in the direct catalytic conversion of methane to fuels and chemicals. Prog. Energy Combust. Sci. 2016, 55, 60–97. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.S.; Santos, M.C.L.; Godoi, C.M.; Oliveira Neto, A.; De Souza, R.F.B. Obtaining C2 and C3 Products from Methane Using Pd/C as Anode in a Solid Fuel Cell-type Electrolyte Reactor. ChemCatChem 2020, 12, 4517–4521. [Google Scholar] [CrossRef]
- Álvarez, M.; Marín, P.; Ordóñez, S. Direct oxidation of methane to methanol over Cu-zeolites at mild conditions. Mol. Catal. 2020, 487, 110886. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef]
- Zhu, J.; Sushkevich, V.L.; Knorpp, A.J.; Newton, M.A.; Mizuno, S.C.M.; Wakihara, T.; Okubo, T.; Liu, Z.; van Bokhoven, J.A. Cu-Erionite Zeolite Achieves High Yield in Direct Oxidation of Methane to Methanol by Isothermal Chemical Looping. Chem. Mater. 2020, 32, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Ikuno, T.; Zheng, J.; Vjunov, A.; Sanchez-Sanchez, M.; Ortuño, M.A.; Pahls, D.R.; Fulton, J.L.; Camaioni, D.M.; Li, Z.; Ray, D.; et al. Methane Oxidation to Methanol Catalyzed by Cu-Oxo Clusters Stabilized in NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2017, 139, 10294–10301. [Google Scholar] [CrossRef] [PubMed]
- Vitillo, J.G.; Bhan, A.; Cramer, C.J.; Lu, C.C.; Gagliardi, L. Quantum Chemical Characterization of Structural Single Fe(II) Sites in MIL-Type Metal–Organic Frameworks for the Oxidation of Methane to Methanol and Ethane to Ethanol. ACS Catal. 2019, 9, 2870–2879. [Google Scholar] [CrossRef]
- Doan, H.A.; Li, Z.; Farha, O.K.; Hupp, J.T.; Snurr, R.Q. Theoretical insights into direct methane to methanol conversion over supported dicopper oxo nanoclusters. Catal. Today 2018, 312, 2–9. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; van Bokhoven, J.A. Kinetic study and effect of water on methane oxidation to methanol over copper-exchanged mordenite. Catal. Sci. Technol. 2020, 10, 382–390. [Google Scholar] [CrossRef]
- Dalton, H. The Leeuwenhoek Lecture 2000 The natural and unnatural history of methane-oxidizing bacteria. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1207–1222. [Google Scholar] [CrossRef]
- Serra-Maia, R.; Michel, F.M.; Kang, Y.; Stach, E.A. Decomposition of Hydrogen Peroxide Catalyzed by AuPd Nanocatalysts during Methane Oxidation to Methanol. ACS Catal. 2020, 10, 5115–5123. [Google Scholar] [CrossRef]
- Lee, H.W.; Dang, H.T.; Kim, H.; Lee, U.; Ha, J.-M.; Jae, J.; Cheong, M.; Lee, H. Pt black catalyzed methane oxidation to methyl bisulfate in H2SO4-SO3. J. Catal. 2019, 374, 230–236. [Google Scholar] [CrossRef]
- Kvande, K.; Pappas, D.K.; Borfecchia, E.; Lomachenko, K.A. Advanced X-ray Absorption Spectroscopy Analysis to Determine Structure-Activity Relationships for Cu-Zeolites in the Direct Conversion of Methane to Methanol. ChemCatChem 2020, 12, 2385–2405. [Google Scholar] [CrossRef]
- Ridruejo, C.; Alcaide, F.; Alvarez, G.; Brillas, E.; Sires, I. On-site H2O2 electrogeneration at a CoS2-based air-diffusion cathode for the electrochemical degradation of organic pollutants. J. Electroanal. Chem. 2018, 808, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Delparish, A.; Kanungo, S.; van der Schaaf, J.; Neira d’Angelo, M.F. Towards coupling direct activation of methane with in situ generation of H2O2. Catal. Sci. Technol. 2019, 9, 5142–5149. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.; Lüke, C.; Cao, Z.; Frenzel, P. Ageing well: Methane oxidation and methane oxidizing bacteria along a chronosequence of 2000 years. Environ. Microbiol. Rep. 2011, 3, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.H. Methane activation by a single copper center in particulate methane monooxygenase: A computational study. Inorg. Chim. Acta 2020, 503, 119441. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, Y.-C.; Shen, G.; Wang, Y.; Liu, X.; Duan, Z.; Pan, L.; Zhang, X.; Zou, J.-J. Photoinduced composite of Pt decorated Ni(OH)2 as strongly synergetic cocatalyst to boost H2O activation for photocatalytic overall water splitting. Appl. Catal. B Environ. 2019, 243, 253–261. [Google Scholar] [CrossRef]
- Kiss, J.; Kukovecz, A.; Konya, Z. Beyond Nanoparticles: The Role of Sub-nanosized Metal Species in Heterogeneous Catalysis. Catal. Lett. 2019, 149, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- López-Martín, Á.; Caballero, A.; Colón, G. Photochemical methane partial oxidation to methanol assisted by H2O2. J. Photochem. Photobiol. A Chem. 2017, 349, 216–223. [Google Scholar] [CrossRef]
- Shi, S.; Sun, Z.; Bao, C.; Gao, T.; Hu, Y.H. The special route toward conversion of methane to methanol on a fluffy metal-free carbon nitride photocatalyst in the presence of H2O2. Int. J. Energy Res. 2020, 44, 2740–2753. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Zhang, G.; Wang, K.; Wu, X. Selective Photocatalytic Oxidation of Low Concentration Methane over Graphitic Carbon Nitride-Decorated Tungsten Bronze Cesium. ACS Sustain. Chem. Eng. 2019, 7, 4382–4389. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Blanco, D.E.; Lee, B.; Modestino, M.A. Optimizing organic electrosynthesis through controlled voltage dosing and artificial intelligence. Proc. Natl. Acad. Sci. USA 2019, 116, 17683–17689. [Google Scholar] [CrossRef] [Green Version]
- Rocha, R.S.; Reis, R.M.; Lanza, M.R.V.; Bertazzoli, R. Electrosynthesis of methanol from methane: The role of V2O5 in the reaction selectivity for methanol of a TiO2/RuO2/V2O5 gas diffusion electrode. Electrochim. Acta 2013, 87, 606–610. [Google Scholar] [CrossRef]
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Dhiman, S.S.; Shrestha, N.; David, A.; Basotra, N.; Johnson, G.R.; Chadha, B.S.; Gadhamshetty, V.; Sani, R.K. Producing methane, methanol and electricity from organic waste of fermentation reaction using novel microbes. Bioresour. Technol. 2018, 258, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Arminio-Ravelo, J.A.; Escudero-Escribano, M. Strategies towards the sustainable electrochemical oxidation of methane to methanol. Curr. Opin. Green Sustain. Chem. 2021, 30, 100489. [Google Scholar] [CrossRef]
- Sustersic, M.; Triaca, W.; Arvía, A. The electrosorption of methane and its potentiodynamic electrooxidation on platinized platinum. J. Electrochem. Soc. 1980, 127, 1242. [Google Scholar] [CrossRef]
- Nandenha, J.; Fontes, E.H.; Piasentin, R.M.; Fonseca, F.C.; Neto, A.O. Direct oxidation of methane at low temperature using Pt/C, Pd/C, Pt/C-ATO and Pd/C-ATO electrocatalysts prepared by sodium borohydride reduction process. J. Fuel Chem. Technol. 2018, 46, 1137–1145. [Google Scholar] [CrossRef]
- Boyd, M.J.; Latimer, A.A.; Dickens, C.F.; Nielander, A.C.; Hahn, C.; Nørskov, J.K.; Higgins, D.C.; Jaramillo, T.F. Electro-Oxidation of Methane on Platinum under Ambient Conditions. ACS Catal. 2019, 9, 7578–7587. [Google Scholar] [CrossRef]
- Hahn, F.; Melendres, C.A. Anodic oxidation of methane at noble metal electrodes: An ‘in situ’ surface enhanced infrared spectroelectrochemical study. Electrochim. Acta 2001, 46, 3525–3534. [Google Scholar] [CrossRef]
- Arnarson, L.; Schmidt, P.S.; Pandey, M.; Bagger, A.; Thygesen, K.S.; Stephens, I.E.L.; Rossmeisl, J. Fundamental limitation of electrocatalytic methane conversion to methanol. Phys. Chem. Chem. Phys. 2018, 20, 11152–11159. [Google Scholar] [CrossRef] [Green Version]
- Rocha, R.S.; Camargo, L.M.; Lanza, M.R.V.; Bertazzoli, R. A Feasibility Study of the Electro-recycling of Greenhouse Gases: Design and Characterization of a (TiO2/RuO2)/PTFE Gas Diffusion Electrode for the Electrosynthesis of Methanol from Methane. Electrocatalysis 2010, 1, 224–229. [Google Scholar] [CrossRef]
- Cook, R.L.; Sammells, A.F. Ambient Temperature Methane Activation to Condensed Species under Cathodic Conditions. J. Electrochem. Soc. 1990, 137, 2007–2008. [Google Scholar] [CrossRef]
- Garcia, L.M.S.; Rajak, S.; Chair, K.; Godoy, C.M.; Silva, A.J.; Gomes, P.V.R.; Sanches, E.A.; Ramos, A.S.; De Souza, R.F.B.; Duong, A.; et al. Conversion of Methane into Methanol Using the [6,6′-(2,2′-Bipyridine-6,6′-Diyl)bis(1,3,5-Triazine-2,4-Diamine)](Nitrato-O)Coppe r(II) Complex in a Solid Electrolyte Reactor Fuel Cell Type. ACS Omega 2020, 5, 16003–16009. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.W. Partial electrochemical oxidation of methane under mild conditions. Langmuir 1991, 7, 13–15. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Kim, R.S.; Oh, S.; Surendranath, Y. Catalytic Methane Monofunctionalization by an Electrogenerated High-Valent Pd Intermediate. ACS Cent. Sci. 2017, 3, 1174–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlquist, M.; Nielsen, R.J.; Periana, R.A.; Goddard, W.A., III. Product Protection, the Key to Developing High Performance Methane Selective Oxidation Catalysts. J. Am. Chem. Soc. 2009, 131, 17110–17115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Huang, R.; Deng, D. Catalytic conversion of C1 molecules under mild conditions. EnergyChem 2021, 3, 100050. [Google Scholar] [CrossRef]
- Santos, M.C.L.; Nunes, L.C.; Silva, L.M.G.; Ramos, A.S.; Fonseca, F.C.; de Souza, R.F.B.; Neto, A.O. Direct Alkaline Anion Exchange Membrane Fuel Cell to Converting Methane into Methanol. ChemistrySelect 2019, 4, 11430–11434. [Google Scholar] [CrossRef]
- Ogura, K.; Migita, C.T.; Ito, Y. Combined Photochemical and Electrochemical Oxidation of Methane. J. Electrochem. Soc. 1990, 137, 500–503. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.-S.; Gong, J. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Jackson, B.A.; Miliordos, E. Weak-field ligands enable inert early transition metal oxides to convert methane to methanol: The case of ZrO. Phys. Chem. Chem. Phys. 2020, 22, 6606–6618. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef] [PubMed]
- Velin, P.; Ek, M.; Skoglundh, M.; Schaefer, A.; Raj, A.; Thompsett, D.; Smedler, G.; Carlsson, P.-A. Water Inhibition in Methane Oxidation over Alumina Supported Palladium Catalysts. J. Phys. Chem. C 2019, 123, 25724–25737. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Roy, K.; van Bokhoven, J.A.; Artiglia, L. Role of Water on the Structure of Palladium for Complete Oxidation of Methane. ACS Catal. 2020, 10, 5783–5792. [Google Scholar] [CrossRef]
- Ma, C.; Tan, X.; Zhang, H.; Shen, Q.; Sun, N.; Wei, W. Direct conversion of methane to methanol over Cu exchanged mordenite: Effect of counter ions. Chin. Chem. Lett. 2020, 31, 235–238. [Google Scholar] [CrossRef]
- Agarwal, N.; Freakley, S.J.; Armstrong, R.D.; Dimitratos, N.; He, Q.; Douthwaite, M.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Low temperature selective oxidation of methane using unsupported gold-palladium colloidal catalysts. Am. Chem. Soc. 2019, 258, COLL-0112. [Google Scholar]
- Tomboc, G.M.; Choi, S.; Kwon, T.; Hwang, Y.J.; Lee, K. Potential Link between Cu Surface and Selective CO2 Electroreduction: Perspective on Future Electrocatalyst Designs. Adv. Mater. 2020, 32, 1908398. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lee, I.; Khramenkova, E.; Wang, M.; Peng, B.; Gutierrez, O.; Fulton, J.L.; Camaioni, D.; Khare, R.; Jentys, A.; et al. Importance of methane chemical potential for its conversion to methanol on Cu-exchanged mordenite. Chem. Eur. J. 2020, 26, 7563–7567. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Kiani, D.; Sourav, S.; Wachs, I.E. Single site vs crystalline: Promoted WO3/SiO2 catalyst design for oxidative coupling of methane (OCM). Am. Chem. Soc. 2019, 258, CATL-0154. [Google Scholar]
- Hassan, M.A.; Miyao, T.; Komiyama, M. Catalytic oxidative coupling of methane in supercritical water: Investigations on a catalytically active species. J. Supercrit. Fluids 2019, 144, 8–13. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- He, Y.; Luan, C.; Fang, Y.; Feng, X.; Peng, X.; Yang, G.; Tsubaki, N. Low-temperature direct conversion of methane to methanol over carbon materials supported Pd-Au nanoparticles. Catal. Today 2020, 339, 48–53. [Google Scholar] [CrossRef]
- Kiratzis, N.; Stoukides, M. The Synthesis of Hydrogen Cyanide in a Solid Electrolyte Fuel Cell. J. Electrochem. Soc. 1987, 134, 1925–1929. [Google Scholar] [CrossRef]
- Tomita, A.; Nakajima, J.; Hibino, T. Direct Oxidation of Methane to Methanol at Low Temperature and Pressure in an Electrochemical Fuel Cell. Angew. Chem. Int. Ed. 2008, 47, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Nandenha, J.; Piasentin, R.M.; Silva, L.M.G.; Fontes, E.H.; Neto, A.O.; de Souza, R.F.B. Partial oxidation of methane and generation of electricity using a PEMFC. Ionics 2019, 25, 5077–5082. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Kyriazis, C.J.C. Cogeneration Electricity + Chemicals. 1. Solid Electrolytes. J. Chemtech. 1991, 21, 422–428. [Google Scholar]
- Yuan, S.; Li, Y.; Peng, J.; Questell-Santiago, Y.M.; Akkiraju, K.; Giordano, L.; Zheng, D.J.; Bagi, S.; Román-Leshkov, Y.; Shao-Horn, Y. Conversion of Methane into Liquid Fuels—Bridging Thermal Catalysis with Electrocatalysis. Adv. Energy Mater. 2020, 10, 2002154. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Fornaciari, J.C.; Primc, D.; Kawashima, K.; Wygant, B.R.; Verma, S.; Spanu, L.; Mullins, C.B.; Bell, A.T.; Weber, A.Z. A Perspective on the Electrochemical Oxidation of Methane to Methanol in Membrane Electrode Assemblies. ACS Energy Lett. 2020, 5, 2954–2963. [Google Scholar] [CrossRef]
- Niedrach, L.W. Galvanostatic and Volumetric Studies of Hydrocarbons Adsorbed on Fuel Cell Anodes. J. Electrochem. Soc. 1964, 111, 1309. [Google Scholar] [CrossRef]
- Niedrach, L.W.; Gilman, S.; Weinstock, I. Studies of Hydrocarbon Fuel Cell Anodes by the Multipulse Potentiodynamic Method. J. Electrochem. Soc. 1965, 112, 1161. [Google Scholar] [CrossRef]
- Niedrach, L.W.; Tochner, M. Studies of Hydrocarbon Fuel Cell Anodes by the Multipulse Potentiodynamic Method. J. Electrochem. Soc. 1967, 114, 17. [Google Scholar] [CrossRef]
- Binder, H.; Köhling, A.; Krupp, H.; Richter, K.; Sandstede, G. Electrochemical Oxidation of Certain Hydrocarbons and Carbon Monoxide in Dilute Sulfuric Acid. J. Electrochem. Soc. 1965, 112, 355. [Google Scholar] [CrossRef]
- Tagawa, T.; Moe, K.K.; Ito, M.; Goto, S. Fuel cell type reactor for Chemicals-energy co-generation. Chem. Eng. Sci. 1999, 54, 1553–1557. [Google Scholar] [CrossRef]
- Tyagi, S.; Ganesh, A.; Aghalayam, P. Direct Methane Proton Exchange Membrane Fuel Cell. ECS Trans. 2019, 6, 371–378. [Google Scholar] [CrossRef]
- Nandenha, J.; Nagahama, I.; Yamashita, J.; Fontes, E.; Ayoub, J.; de Souza, R.; Fonseca, F.; Neto, A.O. Activation of methane on PdZn/C electrocatalysts in an acidic electrolyte at low temperatures. Int. J. Electrochem. Sci. 2019, 14, 10819–10834. [Google Scholar] [CrossRef]
- De Moura Souza, F.; de Souza, R.F.B.; Batista, B.L.; dos Santos, M.C.; Fonseca, F.C.; Neto, A.O.; Nandenha, J. Methane activation at low temperature in an acidic electrolyte using PdAu/C, PdCu/C, and PdTiO2/C electrocatalysts for PEMFC. Res. Chem. Intermed. 2020, 46, 2481–2496. [Google Scholar] [CrossRef]
- Otsuka, K.; Yamanaka, I. Electrochemical cells as reactors for selective oxygenation of hydrocarbons at low temperature. Catal. Today 1998, 41, 311–325. [Google Scholar] [CrossRef]
- Spinner, N.; Mustain, W.E. Electrochemical Methane Activation and Conversion to Oxygenates at Room Temperature. J. Electrochem. Soc. 2013, 160, F1275–F1281. [Google Scholar] [CrossRef]
- Santos, M.C.L.; Godoi, C.M.; Kang, H.S.; de Souza, R.F.B.; Ramos, A.S.; Antolini, E.; Neto, A.O. Effect of Ni content in PdNi/C anode catalysts on power and methanol co-generation in alkaline direct methane fuel cell type. J. Colloid Interface Sci. 2020, 578, 390–401. [Google Scholar] [CrossRef]
- Nidheesh, P.V.; Zhou, M.; Oturan, M.A. An overview on the removal of synthetic dyes from water by electrochemical advanced oxidation processes. Chemosphere 2018, 197, 210–227. [Google Scholar] [CrossRef]
- Nogami, G.; Nishiyama, Y.; Nakamura, H. New Approach to a Rotating Ring Disk Electrode. J. Electrochem. Soc. 1988, 135, 877–884. [Google Scholar] [CrossRef]
- Godoi, C.M.; Santos, M.C.L.; Silva, A.J.; Tagomori, T.L.; Ramos, A.S.; de Souza, R.F.B.; Neto, A.O. Methane conversion to higher value-added product and energy co-generation using anodes OF PdCu/C in a solid electrolyte reactor: Alkaline fuel cell type monitored by differential mass spectroscopy. Res. Chem. Intermed. 2021, 47, 743–757. [Google Scholar] [CrossRef]
- Lee, B.; Sakamoto, Y.; Hirabayashi, D.; Suzuki, K.; Hibino, T. Direct oxidation of methane to methanol over proton conductor/metal mixed catalysts. J. Catal. 2010, 271, 195–200. [Google Scholar] [CrossRef]
- Guo, X.-M.; Hidajat, K.; Ching, C.-B. An experimental study of oxidative coupling of methane in a solid oxide fuel cell with 1 wt%Sr/La2O3-Bi2O3-Ag-YSZ membrane. Korean J. Chem. Eng. 1998, 15, 469–473. [Google Scholar] [CrossRef]
- Kiatkittipong, W.; Goto, S.; Tagawa, T.; Assabumrungrat, S.; Praserthdam, P. Simulation of Oxidative Coupling of Methane in Solid Oxide Fuel Cell Type Reactor for C2 Hydrocarbon and Electricity Co-Generation. J. Chem. Eng. Jpn. 2005, 38, 841–848. [Google Scholar] [CrossRef]
- Wiyaratn, W.; Appamana, W.; Charojrochkul, S.; Kaewkuekool, S.; Assabumrungrat, S. Au/La1−xSrxMnO3 nanocomposite for chemical-energy cogeneration in solid oxide fuel cell reactor. J. Ind. Eng. Chem. 2012, 18, 1819–1823. [Google Scholar] [CrossRef]
- Otsuka, K.; Suga, K.; Yamanaka, I. Oxidative coupling of methane applying a solid oxide fuel cell system. Catal. Today 1990, 6, 587–592. [Google Scholar] [CrossRef]
- Yamada, T.; Hiei, Y.; Akbay, T.; Ishihara, T.; Takita, Y. Simultaneous generation of synthesis gas and electric power by internal reforming fuel cells utilizing LaGaO3 based electrolytes. Solid State Ion. 1998, 113–115, 253–258. [Google Scholar] [CrossRef]
- Ishihara, T.; Yamada, T.; Akbay, T.; Takita, Y. Partial oxidation of methane over fuel cell type reactor for simultaneous generation of synthesis gas and electric power. Chem. Eng. Sci. 1999, 54, 1535–1540. [Google Scholar] [CrossRef]
- Sobyanin, V.A.; Belyaev, V.D. Gas-phase electrocatalysis: Methane oxidation to syngas in a solid oxide fuel cell reactor. Solid State Ion. 2000, 136–137, 747–752. [Google Scholar] [CrossRef]
- Zhang, X.; Ohara, S.; Chen, H.; Fukui, T. Conversion of methane to syngas in a solid oxide fuel cell with Ni–SDC anode and LSGM electrolyte. Fuel 2002, 81, 989–996. [Google Scholar] [CrossRef]
- Zhan, Z.; Lin, Y.; Pillai, M.; Kim, I.; Barnett, S.A. High-rate electrochemical partial oxidation of methane in solid oxide fuel cells. J. Power Sources 2006, 161, 460–465. [Google Scholar] [CrossRef]
- Pillai, M.R.; Bierschenk, D.M.; Barnett, S.A. Electrochemical Partial Oxidation of Methane in Solid Oxide Fuel Cells: Effect of Anode Reforming Activity. Catal. Lett. 2008, 121, 19–23. [Google Scholar] [CrossRef]
- Paloukis, F.; Neophytides, S.G. Numerical simulation of methane fuelled cogenerative SOFCs for the production of synthesis gas and electrical energy. Chem. Eng. Sci. 2007, 62, 3868–3881. [Google Scholar] [CrossRef]
- Brousas, T.; Chiang, P.H.; Eng, D.; Stoukides, M. Technical and economic evaluation of a methane solid oxide fuel cell. Ionics 1995, 1, 328–337. [Google Scholar] [CrossRef]
- Nematollahi, P.; Neyts, E.C. Direct methane conversion to methanol on M and MN4 embedded graphene (M = Ni and Si): A comparative DFT study. Appl. Surf. Sci. 2019, 496, 143618. [Google Scholar] [CrossRef]
- Chen, B.; Xu, H.; Sun, Q.; Zhang, H.; Tan, P.; Cai, W.; He, W.; Ni, M. Syngas/power cogeneration from proton conducting solid oxide fuel cells assisted by dry methane reforming: A thermal-electrochemical modelling study. Energy Convers. Manag. 2018, 167, 37–44. [Google Scholar] [CrossRef]
- Pujare, N.U.; Sammells, A.F. Methane Activation to C2 Hydrocarbon Species in Solid Oxide Fuel Cell. J. Electrochem. Soc. 1988, 135, 2544–2545. [Google Scholar] [CrossRef]
- Hua, B.; Yan, N.; Li, M.; Zhang, Y.-Q.; Sun, Y.-F.; Li, J.; Etsell, T.; Sarkar, P.; Chuang, K.; Luo, J.-L. Novel layered solid oxide fuel cells with multiple-twinned Ni0.8Co0.2 nanoparticles: The key to thermally independent CO2 utilization and power-chemical cogeneration. Energy Environ. Sci. 2016, 9, 207–215. [Google Scholar] [CrossRef]
- Ma, H.-B.; Sheng, T.; Yu, W.-S.; Ye, J.-Y.; Wan, L.-Y.; Tian, N.; Sun, S.-G.; Zhou, Z.-Y. High Catalytic Activity of Pt(100) for CH4 Electrochemical Conversion. ACS Catal. 2019, 9, 10159–10165. [Google Scholar] [CrossRef]
- Zhang, X.; Savara, A.; Getman, R.B. A Method for Obtaining Liquid–Solid Adsorption Rates from Molecular Dynamics Simulations: Applied to Methanol on Pt(111) in H2O. J. Chem. Theory Comput. 2020, 16, 2680–2691. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tao, L.; Cheng, Y.; Yang, F.; Jin, Y.; Zhou, C.; Yu, H.; Yang, Y. Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts 2019, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, S.; Chen, K. Anodic oxidation of methane. J. Electrochem. Soc. 1977, 124, 1171. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liang, J.; Imai, Y.; Ueda, K.; Li, H.; Guo, X.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Highly selective synthesis of methanol from methane over carbon materials supported Pd-Au nanoparticles under mild conditions. Catal. Today 2020, 352, 104–110. [Google Scholar] [CrossRef]
- McVicker, R.; Agarwal, N.; Freakley, S.J.; He, Q.; Althahban, S.; Taylor, S.H.; Kiely, C.J.; Hutchings, G.J. Low temperature selective oxidation of methane using gold-palladium colloids. Catal. Today 2020, 342, 32–38. [Google Scholar] [CrossRef]
- Cortés Ortiz, W.G.; Delgado, D.; Guerrero Fajardo, C.A.; Agouram, S.; Sanchís, R.; Solsona, B.; López Nieto, J.M. Partial oxidation of methane and methanol on FeOx-, MoOx- and FeMoOx -SiO2 catalysts prepared by sol-gel method: A comparative study. Mol. Catal. 2020, 491, 110982. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef] [Green Version]
- Zuo, H.; Klemm, E. Selective oxidation of methane with H2O2 over Fe-silicalite-1: An investigation of the influence of crystal sizes, calcination temperatures and acidities. Appl. Catal. A Gen. 2019, 583, 117121. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, E.; Orozco, I.; Liao, W.; Palomino, R.M.; Rui, N.; Duchoň, T.; Nemšák, S.; Grinter, D.C.; Mahapatra, M.; et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 2020, 368, 513–517. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, R.; Kathalikkattil, A.C.; Roshan, R.; Tharun, J.; Kim, D.-W.; Park, D.-W. Dual-porous metal organic framework for room temperature CO2 fixation via cyclic carbonate synthesis. Green Chem. 2016, 18, 232–242. [Google Scholar] [CrossRef]
- Lustemberg, P.G.; Palomino, R.M.; Gutiérrez, R.A.; Grinter, D.C.; Vorokhta, M.; Liu, Z.; Ramírez, P.J.; Matolín, V.; Ganduglia-Pirovano, M.V.; Senanayake, S.D.; et al. Direct Conversion of Methane to Methanol on Ni-Ceria Surfaces: Metal–Support Interactions and Water-Enabled Catalytic Conversion by Site Blocking. J. Am. Chem. Soc. 2018, 140, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, C.; Chen, H. Nature of Cu active sites in zeolite-based catalysts for selective catalytic oxidation of methane. Res. Chem. Intermed. 2019, 45, 5849–5861. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Selective Oxidation of Methane over Fe-Zeolites by In Situ Generated H2O2. Catalyst 2020, 10, 299. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fuel Cell Type | Anode//Cathode | Methane Input | Eletrolyte | T/°C | Chemicals | MPD [a] /mW cm−2 | Ref. |
|---|---|---|---|---|---|---|---|
| PEMFC [b] | Pd/C//Pt/C Pd10%)Au(10%)/C//Pt/C | Anode | Phosphoric acid doped Nafion membrane | 110 | - | 0.8 2.5 | [84] |
| PEMFC | Pd/C//Pt/C | Anode + H2O(g) | Nafion | 80 | Formate | 0.55 | [46] |
| PEMFC | PdZn/C//Pt/C | Anode+ H2O(g) | Nafion | 80 | Methanol, formate, formic acid | 0.6 | [85] |
| PEMFC | PdAu/C//Pt/C | Anode+ H2O(g) | Nafion | 80 | Methanol, formate, formic acid | 1.0 | [86] |
| PEMFC | Pt/C//PdAu/C | Cathode+ H2O or O2 | Sn0.9In0.1P2O7 | 50–250 | Methanol and CO2 | - | [73] |
| PEMFC | Pt/C//Pd–Au–Cu/C | Cathode + H2O or O2 | Sn0.9In0.1P2O7 | 50–450 | methanol | - | [93] |
| PEMFC | Pt/C//Pt/C | Anode + H2O | Sn0.9In0.1P2O7 | 100–300 | Methanol | - | [9] |
| PEMFC | Pt/C//Pt/C | Cathode + H2O2 | Nafion | 80 | Methanol, formic acid and formaldehyde | 70 | [74] |
| AAEMFC [c] | Pt/C//Pt/C | Anode + KOH | KOH doped Nafion membrane | 25 | Methanol, formate | 0.3 | [57] |
| AAEMFC | PdNi/C//Pt/C | Anode + KOH | KOH doped Nafion membrane | 25 | Methanol, formate | 0.13 | [89] |
| AAEMFC | [6,6′-(2,2′- Bipyridine-6,6′-Diyl)bis(1,3,5-Triazine-2,4-Diamine)](Nitrato- O)Copper(II) Complex/C//Pt/C | Anode + KOH | KOH doped Nafion membrane | 25 | Methanol, formate | - | [52] |
| AAEMFC | Pt/Cu//Pt/C | Anode + KOH | KOH doped Nafion membrane | 25 | Methanol, formate | 0.12 | [92] |
| Anode//Cathode | Methane Input | Eletrolyte | T/°C | Chemicals | MPD [a] /mW cm−2 | Ref. |
|---|---|---|---|---|---|---|
| Pt or Rh//Pt/C | Anode + NH3 | YSZ | 800–1000 | HCN | 10 | [72] |
| Sm2O3-LaSrMnO2//LaSrMnO3 | Anode | YSZ | 760 | C2 hydrocarbons | - | [108] |
| Sr/La2O3-Bi2O3-Ag//Ag | Anode | YSZ | 730 | C2 hydrocarbons | 6 | [94] |
| Au/LaSrMnO3//LaSrMnO3 | Anode | YSZ | 850 | C2 hydrocarbons | 4.4 | [96] |
| Ni//La0.6Sr0.4CoO3 | Anode + N2 | LaGaO3 | 1000 | Syngas | 526 | [99] |
| Ni–SDC//Sm0.6Sr0.4CoO3 | Anode | La0.9Sr0.1Ga0.8Mg0.2O3–δ | 800 | Syngas | 90 | [101] |
| Ni-YSZ//La0.8Sr0.2MnO3 | Anode | YSZ | 750 | Syngas | 700 | [102] |
| Ni//NdBa0.75Ca0.25Co2O5+d | Anode + CO2 | BaZr0.1Ce0.7Y0.1Yb0.1O3-d | 700 | CO-enriched syngas | 910 | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, R.F.B.; Florio, D.Z.; Antolini, E.; Neto, A.O. Partial Methane Oxidation in Fuel Cell-Type Reactors for Co-Generation of Energy and Chemicals: A Short Review. Catalysts 2022, 12, 217. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020217
de Souza RFB, Florio DZ, Antolini E, Neto AO. Partial Methane Oxidation in Fuel Cell-Type Reactors for Co-Generation of Energy and Chemicals: A Short Review. Catalysts. 2022; 12(2):217. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020217
Chicago/Turabian Stylede Souza, Rodrigo F. B., Daniel Z. Florio, Ermete Antolini, and Almir O. Neto. 2022. "Partial Methane Oxidation in Fuel Cell-Type Reactors for Co-Generation of Energy and Chemicals: A Short Review" Catalysts 12, no. 2: 217. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020217