
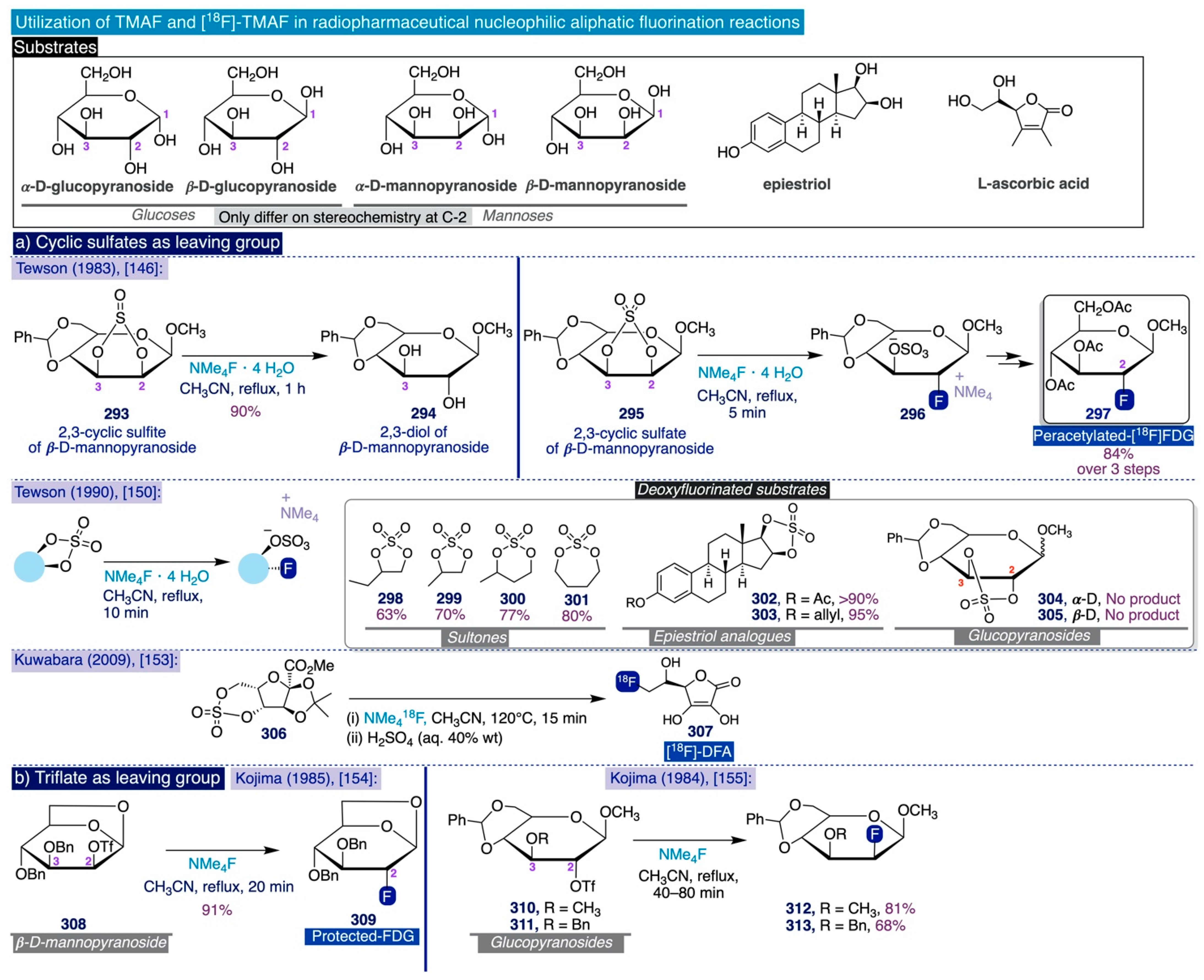
Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base


1
Department of Chemistry, Faculty of Science, University of Helsinki, P.O. Box 55, 00014 Helsinki, Finland
2
VTT Technical Research Centre of Finland Ltd., P.O. Box 1000, FI-02044 Espoo, Finland
*
Authors to whom correspondence should be addressed.
Catalysts 2022, 12(2), 233; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020233
Submission received: 14 January 2022
/
Revised: 14 February 2022
/
Accepted: 15 February 2022
/
Published: 18 February 2022
(This article belongs to the Special Issue Organohalogen Chemistry and Catalysis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Nucleophilic ionic sources of fluoride are essential reagents in the synthetic toolbox to access high added-value fluorinated building blocks unattainable by other means. In this review, we provide a concise description and rationale of the outstanding features of one of these reagents, tetramethylammonium fluoride (TMAF), as well as disclosing the different methods for its preparation, and how its physicochemical properties and solvation effects in different solvents are intimately associated with its reactivity. Furthermore, herein we also comprehensively describe its historic and recent utilization, up to December 2021, in C-F bond-forming reactions with special emphasis on nucleophilic aromatic substitution fluorinations with a potential sustainable application in industrial settings, as well as its use as a base capable of rendering unprecedented transformations.
1. Introduction
Fluorine-containing molecules are increasingly prevalent in last-generation medicines and non-invasive medical imaging agents for the healthcare of the ageing population [1,2,3,4,5], improved agrochemicals enabling optimal and sustainable food production for a global population dramatically escalating [6,7], and materials science (Scheme 1a) [8,9,10]. This trend is significantly pronounced in modern medicine, an area in which around 20% of the administered drugs in 2010 contained at least one fluorine atom [11,12,13], as well as 18 out of the 42 new molecular entities that were approved as marketed drugs by the FDA in 2018 [14]. Indeed, fluorine and fluorine-containing functional groups are considered privileged moieties in fragment-based drug discovery [15], due to their unique ability to impart a synergistic combination of physicochemical properties to drug candidates such as a significant biostability towards oxidative and hydrolytic metabolic pathways, enhanced lipophilicity and membrane permeability, increased drug potency, and/or target specificity [16,17,18,19]. Additionally, the presence of fluorine-based groups can determine the preferred conformation of a drug molecule and promote favorable protein–ligand interactions that facilitate access to challenging areas for drug delivery such as the brain (Scheme 1b) [20,21,22].
Fluorine in the form of fluoride minerals—fluorite, fluorapatite, and cryolite—is the 13th most abundant element on the earth’s crust, the first amongst halogens. Although the presence of several fluoroalkanes has been detected in volcanic and geothermal emissions, the solubility of fluoride anions from these inorganic sources is really limited in the aqueous media where most secondary metabolites initially originated (e.g. the average concentration of fluorides in seawater is 1.3 ppm, 15,000 times inferior to chlorides).
Scheme 1.
(a) Ubiquitous applicability of fluorine-containing molecules, (b) Fluorine-induced conformational control, and (c) Biosynthesis of fluoroacetate: Isolated example of a biosynthetic C-F bond formation.
Scheme 1.
(a) Ubiquitous applicability of fluorine-containing molecules, (b) Fluorine-induced conformational control, and (c) Biosynthesis of fluoroacetate: Isolated example of a biosynthetic C-F bond formation.

Perhaps for that reason, naturally occurring organofluorine molecules only represent 50 out of the 5000 organohalogen compounds documented to date [23,24]. In nature, organohalogen compounds are mainly produced by haloperoxidases, a family of enzymes in which the existence of fluoroperoxidase members remains hitherto unknown [25]. In fact, the biosynthetic origin of the vast majority of naturally occurring organofluorinated molecules remains a mystery, and there is only one single example of the bioenzymatic formation of C-F bonds in nature [26]. It involves the fluorination of S-adenosyl-L-methionine (SAM) in Streptomyces cattleya by a natural fluorinase enzyme to 5′-fluoro-5′-deoxyadenosine (FDA) [27], which is further enzymatically converted to fluoroacetate (Scheme 1c)—a toxin for mammals.
Synthetic organofluorine chemistry has witnessed major progress in recent decades [28,29,30,31,32,33]. As such, it constitutes the ultimate scientific driving force enabling us to overcome the rare occurrence of natural fluorine-containing molecules, and thus satisfy the increasing demands in those areas of major societal importance. Alongside efficiency and sustainability, the selectivity and substrate scope of the developed fluorination synthetic methods are critical to access a wide range of structural diversity. This is particularly important in medicinal chemistry, where fluorine-based functional groups must be judiciously installed at certain positions of the drug scaffold to avoid the possibility of enzymatic release of HF or other toxic metabolites [34,35].
Based on the fluorinating source and reaction mechanism, C-F bond-forming reactions can be categorized into three groups: (i) Nucleophilic [36,37], (ii) electrophilic [38,39,40], and (iii) thermally or photo-induced radical fluorinations [41,42,43,44]. Nucleophilic fluorination reactions are the only class exclusively involving the action of fluoride anions alone or as part of ionic pairs in solution. In this regard, tetramethylammonium fluoride (TMAF) constitutes one of the most representative and versatile nucleophilic fluorinating reagents. Furthermore, a singular combination of properties allows it to successfully participate in other synthetic and catalytic processes. In this review, we will first describe the fundamental physicochemical properties and preparation of this useful reagent, subsequently providing a comprehensive survey of its main synthetic applications (Scheme 2).
2. TMAF: General Physicochemical Properties and Preparation
2.1. “Nakedness” of the Fluoride Anion
The combination of a 1s2, 2s2, 2p5 electronic configuration in fluorine, along with its small size (van der Waals radius: 1.47 Å; ionic radius 1.33 Å) [45,46], makes it the most electronegative of all elements (Pauling value, χ = 4) [47]. As a result, fluorine forms polarized bonds with marked electrostatic character that are amongst the strongest covalent bonds known, such as C-F (e.g. BDE in CF4 is 130 kcal/mol) or Si-F (e.g. BDE up to 166 kcal/mol for SiF4) (Scheme 3a) [48,49]. Another far-reaching consequence of the high electronegativity of fluorine concerns the fact that it only can exist as a free, highly reactive “naked” fluoride ion in an ideal gas phase [50]. In practice, fluoride anions possess such a high charge density that they are inevitably stabilized and deactivated in any chemical environment by strong coulombic electrostatic forces with its countercation or by hydrogen or covalent bonds with any other molecules present nearby [51]—including solvents. Since electrostatic forces are directly proportional to the magnitude of the charges involved and inversely proportional to the square of the distance between them, the “nakedness” and basicity of fluoride anions decrease in ionic compounds with small countercations. This entails the best approximation to obtain highly reactive “naked” fluoride involving the use of salts of large elemental cations—namely, the Cesium effect [52]. However, these salts are largely insoluble due to their high lattice energies [53].
In this context, considerable attention has been paid to study potential sources of highly reactive “naked” fluoride anions with significant solubility in organic solvents [54], as well as to the measurement of their reactivity and structural elucidation [50,53,54,55,56,57,58,59,60]. To that end, the fluoride salts of several large organic cations have been prepared as hydrates and then subsequently dried to their anhydrous form (Scheme 3b). The key factor enabling them to behave as sources of “naked” fluoride is the presence of a shell of methyl hydrogen atoms in the countercation that shelters its positive charge, thus weakening the electrostatic attraction to the fluoride anion. Conversely, these salts lack antiperiplanar β-hydrogen atoms susceptible to undergoing Hofmann (E2) elimination with the fluoride anion (Scheme 3c), such as TMAF (1) [55], N,N-trimethyl-1-adamantylammonium fluoride (2) [61], 1,1,3,3,5,5-hexamethylpiperidinium fluoride (3) [62], N-1-methylhexamethylenetetramine, fluoride (MHAF) (4) [63], tetramethylphosphonium fluoride (TMPF) (5) [64], hexamethylphosphazenium fluoride (6) [65], acyl azolium fluoride (7) [66], and more recently, azabicyclo [2,2,2]octane (8) (Scheme 3b) [67].
Scheme 3.
(a) The fluorine atom, (b) Free naked fluoride, (c) General structural requirement for ionic organic sources of fluoride, and (d) Exception.
Scheme 3.
(a) The fluorine atom, (b) Free naked fluoride, (c) General structural requirement for ionic organic sources of fluoride, and (d) Exception.

Additionally, DiMagno and co-workers demonstrated that the inherent instability of anhydrous tetrabutylammonium fluoride (TBAF) (11)—due to the presence of β-hydrogen atoms—can be circumvented by carefully treating hexafluorobenzene (12) in situ with tetrabutylammonium cyanide in polar aprotic solvents at sub-zero temperatures (Scheme 3d) [68]. Furthermore, organometallic species [69,70] and ionic liquids [71] have also been noted as possible sources of “naked” fluoride anions. All these ionic sources should not be confused with other nucleophilic fluorinating reagents (Scheme 3c)—the discussion of which exceeds the scope of this review—such as organofluorosilicates (e.g. triphenyldifluorosilicate, TBAT (9) [72]) or organofluorinated sulfur compounds (e.g. diethylaminosulfur trifluoride, DAST (10) [73]) where the fluoride anion is only released after the breakage of a weak covalent bond Si-F(F) or N-S-F(F).
2.2. Physicochemical Properties and Behaviour of TMAF in Different Solvents
2.2.1. Physicochemical Properties and Solid Structure
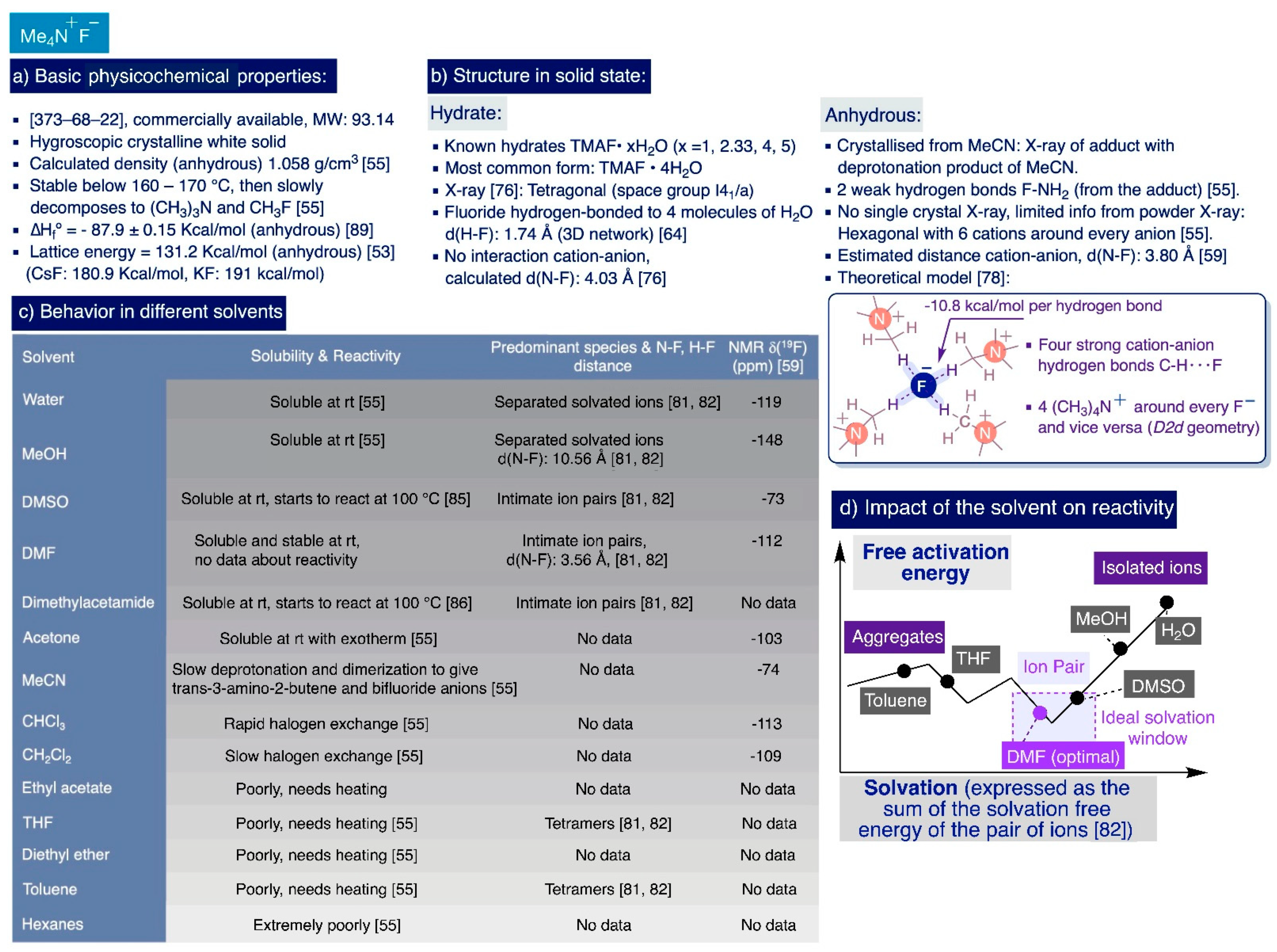
TMAF features a privileged combination of properties amongst sources of “naked” fluoride anions. It is a white hygroscopic crystalline solid that is relatively easy to handle and readily available from commercial sources at a relatively low cost—amenable for multi-gram scale synthesis—in hydrated or anhydrous forms [74]. Furthermore, the chemical inertness of the small and highly symmetric tetramethylammonium cation (ionic radius, 3.22 Å [75]) endows TMAF with a very high thermal stability, whose anhydrous form only starts to slowly decompose into trimethylamine and fluoromethane at temperatures around 160–170 °C [55] (Scheme 4a).
In the solid state, TMAF occurs in the already-mentioned anhydrous form, and four hydrated crystalline forms (Me4N·xH2O with x = 1, 2.33, 4, and 5) whose structures have been determined by X-ray diffractometry [76,77]. Careful examination of the available X-ray data shows that the high charge density of fluoride prevents it from really existing as a totally free “naked” anion in any of the solid-state forms of TMAF, thus attenuating its reactivity in solution. In fact, an X-ray of the most common hydrated form (Me4N·4H2O), reported in 1967 [76], reveals a tetragonal crystalline structure (space group I41/a) where the fluoride anions are hydrogen-bonded to four molecules of H2O (distance (F---H-OH) = 2.63 Å, contact distance (H—F) = 1.74 Å [64]). These units form a three-dimensional network in which the tetramethylammonium cations are embedded without making any direct contact with the fluoride anions (distance (N---F) = 4.03 Å) [76] (Scheme 4b).
In the absence of hydration, the fluoride anion establishes strong hydrogen bonds with any other potential donor in the media. This fact explains why the single-crystal X-ray structural determination of pure anhydrous TMAF remains elusive. However, Christe and co-workers managed to obtain structural information of anhydrous TMAF from its powder X-ray and also elucidate the crystal structure of the adduct of TMAF with trans-3-amino-2-butenenitrile obtained by slow diffusion of hexane into a saturated MeCN solution of anhydrous TMAF.
The crystallographic powder data of anhydrous TMAF suggested a hexagonal unit cell with a calculated density of 1.058 g/cm3 where the N(CH3)4+ cations would be surrounded by six fluoride anions located at the corners of a trigonal prism, and the fluoride anion would be surrounded by six N(CH3)4+ cations and two other fluorides. No further information was obtained from this powder X-ray data regarding the hydrogen bond distances with the fluoride anions. However, subsequent molecular orbital ab initio calculations combined with IR studies suggested the presence of strong hydrogen bonds in anhydrous TMAF where the fluoride anions act as acceptors—10.8 kcal/mol per bond—of four methyl hydrogen atoms, one from each cation [78] (Scheme 4b).
Regarding X-rays of the crystalline adduct of TMAF with trans-3-amino-2-butenenitrile, two weak hydrogen bonds were observed between the fluoride anions and two hydrogen atoms from two NH2 groups of different trans-3-amino-2-butenenitrile molecules at 1.808 and 1.853 Å. The rest of the distances were considerably longer than the sum of the van der Waals radii, indicating the absence of other hydrogen bonds.
2.2.2. Solubility of TMAF in Different Solvents
Studies by Christe et al.—to quantitatively measure the so-called “nakedness” of fluoride anions in different ionic sources [53]—provided the calculated free energy changes in the Born–Haber cycles associated with the transfer of fluoride from one of these sources to a given acceptor. Then, the lattice energy of anhydrous TMAF was determined to be 131.2 kcal/mol, which is remarkably lower than the lattice energy for KF (191 kcal/mol) and CsF (180.9 kcal/mol) (Scheme 4a) [79]. In fact, these data partially explain the higher solubility of TMAF in different solvents compared to KF and CsF since the lattice energy of an ionic solid is closely related to its solubility [80].
In practice, the polarity of the solvent largely determines the degree of solvation and aggregation of TMAF ionic species in solution, which are key for reactivity (Section 2.2.3) [81,82]. In this sense, anhydrous TMAF is highly soluble in polar protic solvents, such as water (ε = 78.5) and alcohols (e.g. MeOH, ε = 32.6) [55], amenable to engaging in strong hydrogen bonds with the fluoride anion that compensate its lattice energy and replacing the hydrogen bonds existing in a solid state between the N(CH3)4+ cations and fluoride anions. In such solvents, the fluoride anions are part of single-solvated ion pairs (e.g. the anion–cation distance in MeOH was determined to be 10.56 Å, [82]). Anhydrous TMAF also dissolves in nitromethane (ε = 35.9), exothermically in acetone (sic) (ε = 21.0), and in high amounts (4.4 wt%) at −80 °C in a relatively chemically inert media such as CHF3 (bp -84.4 °C, mp -160 °C) [55]. Conversely, TMAF exhibits negligible solubility in apolar solvents such as hexane (ε = 1.9), benzene (ε = 2.3), or toluene (ε = 2.4), as well as in slightly polar aprotic solvents such as ethyl acetate (ε = 6), diethyl ether (ε = 4.3), THF (ε = 7.5), or dimethoxyethane (ε = 7.2) (Scheme 4c). Unfortunately, to the best of our knowledge, there are no more-precise values reported in the literature of the solubility of TMAF in different solvents at different temperatures.
However, TMAF is not always chemically inert in solvents, due to the extraordinary basicity and nucleophilicity of the fluoride anion [83]. For instance, TMAF undergoes chlorine–fluorine exchange at room temperature in chlorinated hydrocarbons, such as dichloromethane and chloroform, to afford CH2ClF (slowly from dichloromethane) and a mixture of CHCl2F, CHClF2, and CHF3 products (rapidly from chloroform). Additionally, TMAF can slowly deprotonate MeCN at room temperature to generate the anion CH2CN− that instantaneously rearranges in situ to trans-3-amino-2-butene [55]. NMR studies enabled monitoring the progress of this reaction (5–10% conversion after several hours, up to 30% after several days at room temperature), which is accompanied by the accumulation of HF2− in the solution and a yellow color (Scheme 4c) [83].
The solubility of anhydrous TMAF is quite particular in DMF (ε = 38.2) and DMSO (ε = 47), where the ions are not entirely dissociated at room temperature but rather forming intimate ion pairs in close contact (e.g. anion–cation distance in DMF was calculated to be 3.56 Å, [82]). However, since the basicity of TMAF is in the range of strong Brønsted bases such as NaH or PhLi [74], DMSO is the more stable of the two solvents in alkaline conditions and likely the more convenient to conduct synthetic processes requiring elevated temperatures [84]. In practice, DMSO only starts to react with TMAF at temperatures as high as 100 °C [84,85], producing the bifluoride anion (ca 5% after 1h at 90 °C and 10% after 2.2 h, [84]). Additionally, TMAF in hydrated and anhydrous form is also able to deprotonate N,N-dimethylacetamide (DMA), another aprotic polar solvent that is commonly used as an alternative to DMSO and DMF in nucleophilic aromatic fluorination reactions [86].
CAUTION:The highest temperatures reported in synthetic reactions using TMAF either in DMSO or DMF range between 80 and 100 °C. These solvents can violently decompose at elevated temperatures in the presence of a superbasic reagent such as NaH (which possesses comparable basicity to TMAF) [87,88]. It is advisable to conduct reactions requiring the most active anhydrous form of TMAF, especially in gram-scale, either at lower temperatures or in a different solvent.
Scheme 4.
TMAF: (a) Basic physicochemical properties [89], (b) Structure in solid state, (c) Behavior in solution, and (d) Impact of the solvent on reactivity.
Scheme 4.
TMAF: (a) Basic physicochemical properties [89], (b) Structure in solid state, (c) Behavior in solution, and (d) Impact of the solvent on reactivity.

2.2.3. Impact of the Solvent on the Chemical Reactivity of TMAF
The degree of “nakedness” of the fluoride anion is critical for the reactivity of TMAF, which can be controlled by “tuning” its hydrogen bonding and aggregation state in solution. These factors greatly depend (but not exclusively) on the polarity of the solvent and the degree of hydration of the TMAF used. Theoretical studies involving electronic structure and molecular dynamics calculations on the nucleophilic aromatic substitution (SNAr) of 2-bromobenzonitrile with TMAF on different solvents provide qualitative insight into the influence of the solvent’s polarity on TMAF reactivity [82]. In this study, five solvents covering a wide range of polarity were chosen, namely, MeOH, DMF, pyridine, THF, and benzene. In general, the more polar is the solvent, the higher is the degree of solvation of the ionic species prevalent in solution, and the lower is their aggregation state. Indeed, this theoretical model suggests that TMAF exists predominantly as a tetrameric species in THF and benzene, as intimate ionic pairs in DMF and pyridine, and as separated solvated ionic species in MeOH.
However, the polarity of the solvent and degree of solvation of the species in solution are not the only factors determining the reactivity of TMAF in solution. Indeed, these studies indicated that the transition states with lower activation free energies—by which this SNAr reaction is kinetically easier—correspond to dimeric species in benzene, intimate ionic pairs in THF, DMF, and pyridine, and single-solvated fluoride ions in MeOH [82]. The authors concluded that DMF and pyridine are the optimum solvents for the SNAr reaction of 2-bromobenzonitrile with TMAF by considering (i) the solubility of TMAF, (ii) the relative concentration in which the more-reactive ionic species are present on each solvent, and (iii) the relative profiles of the activation free energies of those species. In other words, DMF and pyridine fall as solvents in the ideal solvation window where there is a larger presence of intimate ion pairs in close contact, which are less solvated and more reactive than the rest of feasible species in solution (Scheme 4d).
A previous report by the same authors deemed that DMSO supersedes DMF as a solvent for SNAr reactions with anhydrous TBAF instead of TMAF [81]. No comparison is yet reported between DMSO and DMF as solvents to conduct ionic reactions with TMAF.
The majority of reactions using TMAF proceed through transition states with a marked ionic character, such as those two SNAr processes previously considered [81,82]. Hence, the considerations concerning the impact of the solvent polarity in the overall reactivity may be applied to other reactions with TMAF. Indeed, as we will discuss in the following sections, most often, the optimal solvents for these reactions are polar aprotic solvents such as DMF and DMSO. Additionally, the reactivity of TMAF may be attenuated if needed for a particular application by increasing its degree of hydration or increasing the ratio of polar protic solvents, such as MeOH, that lead to larger concentrations in a solution of less-reactive isolated hydrated ions. However, in reactions where TMAF acts as a base (Section 4), the relationship between the solvent and reactivity may vary.
2.3. Preparation of TMAF
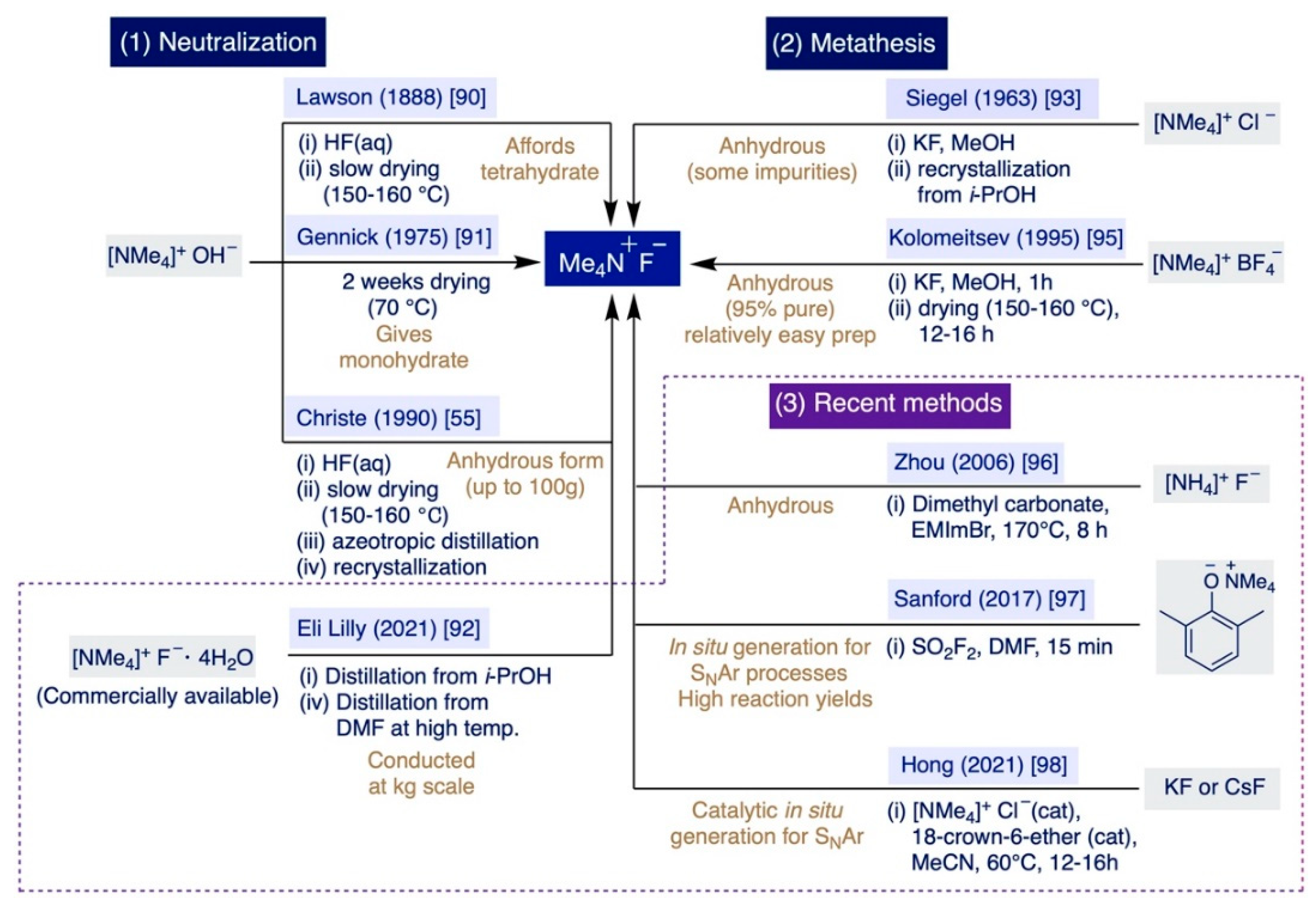
Historically, there has been two main approaches to prepare TMAF: (i) Via neutralization of tetramethylammonium hydroxide (TMAOH) with HF, and (ii) through the metathesis reaction of different ammonium salts with inorganic sources of fluoride such as KF or CsF (Scheme 5). The first documented preparation of TMAF dates back to 1888 by Lawson and Collie, who neutralized TMAOH with an aqueous solution of HF to isolate TMAF as a hydrated crystalline solid [90]. The authors observed that a large portion of H2O could be removed at elevated temperatures under vacuum (150–160 °C), although they also noted that removing the last amounts of H2O was a very slow process with a sound problem due to the already-mentioned fact that TMAF starts to decompose slowly around 160 °C [55].
Lawson–Collie’s method followed by crystallization from an aqueous solution at 0-5 °C preferentially delivers the tetrahydrate salt [76]. Subsequent improvements of Lawson–Collie’s protocol by Harmon and Gennick allowed the isolation of TMAF monohydrate using an equimolar amount of aqueous HF for neutralization, followed by drying the remaining solid for a week at 70 °C under vacuum, and further drying for another week at 60 °C over P2O5 [91]. Starting with this “dried” monohydrate, Harmon and Gennick also selectively prepared the trihydrate and tetrahydrate salts by reaction with 2 equiv and 3 equiv of H2O, respectively, followed by crystallization overnight. Moreover, their simple protocol also enabled the first preparation of TMAF monohydrate-d2 by subjecting anhydrous TMAF to an H-D exchange with an excess of D2O [91].
Subsequent improvements to the Harmon–Gennick method by Christe et al. led to what the authors claimed as the first synthesis of anhydrous TMAF [55]. Remarkably, Christe’s procedure was conducted up to a 100 g scale—facilitating the use of TMAF in bulk applications—and significantly reduced the amounts of impurities of the bifluoride anion or the monohydrate salt that accumulate as a result of the incomplete removal of water or the TMAF decomposition at elevated temperatures. In practice, this protocol involves careful titration of TMAOH in a CO2-free degassed solution—to avoid the formation of bicarbonate salts as impurities—with a precise amount of aqueous HF in Teflon equipment to avoid etching, followed by meticulously keeping the temperature at 150 °C during water removal. The remaining solid was nearly anhydrous, yet next it was azeotropically distilled from isopropanol (i-PrOH) and then also recrystallized from dry i-PrOH.
Scheme 5.
Preparation of TMAF: (1) Neutralization, (2) Metathesis, and (3) Recent methods.

Finally, a very recent report by chemists at Eli Lilly describes the preparation of anhydrous TMAF at a bulk scale (with just <0.2 wt% H2O and 60 ppm of i-PrOH) directly from the readily available TMAF tetrahydrate salt [92]. This process involves azeotropic distillation with i-PrOH, and then DMF at an elevated temperature. Subsequently, the authors prepared 36.8 kg of 4-fluorothiazole via SNAr fluorination of 4-chlorothiazole using 45.1 kg of the anhydrous TMAF produced in that manner.
Regarding the metathesis approach, the first precedent was reported in 1963 and entails the reaction of equimolar quantities of tetramethylammonium chloride (TMACl) and KF in MeOH. The resulting solid was recrystallized from i-PrOH [93]. The operational simplicity of this method compensates for the fact that it furnishes TMAF containing variable amounts of the initial chloride salt as impurities [94]. Remarkably, such a problem can be circumvented by switching from TMACl to pre-dried tetramethylammonium tetrafluoroborate as the starting material for the anion exchange reaction with pre-dried KF (230–250 °C) in MeOH [95]. This method is rather fast (ca. 1h) and straightforward. It only involves filtration of the precipitated KBF4, partial evaporation of the MeOH used as the solvent, and the final addition of diethyl ether to the remaining solution. In these conditions, TMAF precipitates in a nearly anhydrous form, which can be further dried under vacuo at 130–140 °C for just 12–16h rather than the weeks required in previous methods. The authors declared that the final TMAF is up to 95% pure (with only 0.06 wt.% H2O).
In the last two decades, several groups have described alternative approaches that do not imply neutralization or metathesis reactions to prepare TMAF. For instance, full methylation of ammonium fluoride can be accomplished by using dimethyl carbonate as a methylating agent, and an ionic liquid—1-ethyl-3-methylimidazolium bromide—as the catalyst at 170 °C for 8 h [96]. Then, TMAF is isolated by direct filtration washing with acetone. However, little information was provided regarding the level of impurities of the TMAF prepared in this manner.
More recently, Sanford and co-workers have explored the direct generation of TMAF in situ for SNAr fluorination reactions that could proceed under milder conditions, more rapidly, and with the generation of less waste for potential implementation at a large industrial scale (Section 3.2.3) [97]. These elegant studies concluded in the development of an operationally straightforward method—enabling one to avoid the lengthy drying steps inherent to some of the previous methodologies—that rapidly delivers TMAF to the reaction media from relatively inexpensive reagents. In practice, the authors demonstrated that the treatment of tetramethylammonium 2,6-dimethylphenoxide with an equimolar amount of sulfuryl fluoride (SO2F2) in DMF leads to the nearly quantitative formation of anhydrous TMAF in just 15 min at rt.
This work constitutes a significant step forward for the introduction of TMAF in pharmaceutical and agrochemical industrial processes, albeit it also offers some room for improvement due to the toxicity profile of SO2F2 and the need for drying tetramethylammonium 2,6-dimethylphenoxide for three days prior to use.
Furthermore, in 2021, Hong et al. described the first in situ catalytic generation of TMAF in MeCN at 60 °C from KF or CsF by using catalytic amounts of tetramethylammonium chloride and 18-crown-6 ether, which sequestrates the K+ or Cs+ countercations, thus rendering the fluoride anion more reactive in solution (Section 3.2.3) [98].
Finally, during the completion of this review, the Sanford group reported the first in situ generation of [18F]-TMAF for the nucleophilic radiofluorination of heteroaromatic molecules [99]. This method delivered a relatively wide range of [18F]-(hetero)aryl fluoride products with good to excellent yields in the presence of ambient air and moisture, and involves the swift metathesis of an easy-to-dry quaternary methylammonium salt, such as Me4NHCO3, with the well-known source of [18F]fluoride, [18F]KF·K2.2.2, which in practice is much less hygroscopic than the [18F]R4NF salts traditionally used for this purpose.
3. TMAF as Ionic Source of Fluoride for C-F Bond Formation
3.1. Comparison of TMAF with Other Nucleophilic Sources of Fluoride
In terms of chemical bonding, there are two distinctive classes of fluorinating nucleophilic reagents: (i) Ionic sources (Scheme 3b) and (ii) reagents that release fluoride anions after breakage in solution of a labile covalent bond (e.g. S-F or Si-F bonds) (Scheme 6). Interestingly, recent reports suggest that some N-F electrophilic fluorinating reagents can be used in nucleophilic fluorination processes such as the allylic fluorination of alkenes [100,101,102].
TMAF encompasses a unique series of features in the realm of nucleophilic fluorination. Firstly, it holds larger potential applicability in multi-gram-scale industrial processes than most other covalent and ionic fluorinating nucleophilic reagents due to its simplicity, relatively low cost, and the recent development of optimized methodologies to prepare it anhydrously—in large amounts [55,92] or in situ [97,98]—with minimal impurities (Section 2.3). Secondly, TMAF outcompetes almost all the remaining fluorinating nucleophilic reagents in terms of the atom economy and the potential environmental impact inherent to its use. In this sense, the pronounced reactivity of TMAF precludes the need for utilizing large excess of the reagent that would generate huge amounts of waste per fluorinated molecule (Section 3.2.3 and Section 3.3.2).
Additionally, the waste produced in TMAF-mediated processes consists mainly of quaternary ammonium, bifluoride, or methylamine salts [55], which bear substantially less environmental costs than other fluorinating reagents producing toxic waste such as HF, e.g. DMPU-HF, TREAT-HF, or Olah’s reagent (Scheme 6). Furthermore, TMAF is a solid that is relatively easy to handle and thermally stable in a broad range of temperatures (Section 2.1) [55,74]. In fact, only inorganic ionic sources such as KF and CsF are as cost-effective, atom-economical, environmentally sustainable, and operationally simple as TMAF. However, TMAF possesses significantly lower lattice energy than KF and CsF [53], and as such, is substantially more soluble in the organic solvents commonly used for fluorinating reactions (Section 2.2). This fact provides another advantage of using TMAF in nucleophilic fluorination processes. Indeed, the possibility of using several protic and aprotic polar solvents as reaction media allows tuning TMAF reactivity by controlling the degree of hydrogen bonding and the association of the reactive ionic species. In practice, increasing the ratio of polar protic solvents, such as MeOH, leads to larger concentrations in solution of less-reactive, isolated, hydrated ions (Section 2.2).
Scheme 6.
Representative reagents for nucleophilic fluorination and features of TMAF.

Finally, TMAF is one of the nucleophilic fluorinating ionic reagents approaching the optimum in terms of providing free “naked” fluoride anions in solution (Section 2.1). This attribute translates to an intriguing combination of strong basicity and fluorinating power that often leads to the discovery of new chemical processes unattainable by other means (Section 4). Additionally, this rare combination entails a pronounced boost of reactivity that may significantly accelerate many known chemical processes proceeding through transition states with ionic nature.
This is a critical advantage for the syntheses of [18F]-containing radiopharmaceuticals, which demand fast methods allowing the rapid installation of [18F] at a later stage of the synthetic route [3,4] (Section 3.4). TBAF is perhaps the only nucleophilic fluorinating reagent that shares most of the characteristics of TMAF. However, TBAF is considerably more difficult to handle and prepare in anhydrous form than TMAF due to its poor thermal stability and tendency to undergo Hoffmann elimination—eight β-hydrogen atoms (Scheme 3d)—at temperatures as low as 0 °C (Section 2.1).
3.2. C(sp2)-F Bond Formation with TMAF: Nucleophilic Aromatic Substitution
During recent decades, the development of general, selective, and cost-efficient C(sp2)-F bond-forming methods has remained a tantalizingly elusive goal in synthetic organofluorine chemistry due to the troublesome access to competent sources of nucleophilic fluoride, the hitherto limited understanding of ion-pairing processes governing their reactivity, and the exceptional strength of the C(sp2)-F bond (126 kcal/mol) [49]. In this section, we will cover the mechanistic and synthetic aspects, as well as the historical evolution in the utilization of TMAF to conduct this key class of fluorination reactions.
3.2.1. Nucleophilic Aromatic Substitution. General Mechanistic Overview
Gaining fundamental mechanistic insight has direct practical implications to predict key aspects of a given chemical process such as the selectivity, robustness to medium effects and product distribution.
Most of the reactions that employ TMAF as a fluorinating agent for the formation of C(sp2)-F bonds are nucleophilic aromatic substitutions (SNAr) [37]. Physical organic chemistry defines SNAr reactions as the substitution of a leaving group with a nucleophile on a (hetero)aromatic system [103] and establishes two major categories according to their mechanism: Reactions proceeding via (i) a stepwise addition–elimination mechanism, which is largely the most common type—especially amongst reported TMAF-mediated SNAr reactions—and (ii) stepwise elimination–addition SNAr reactions.
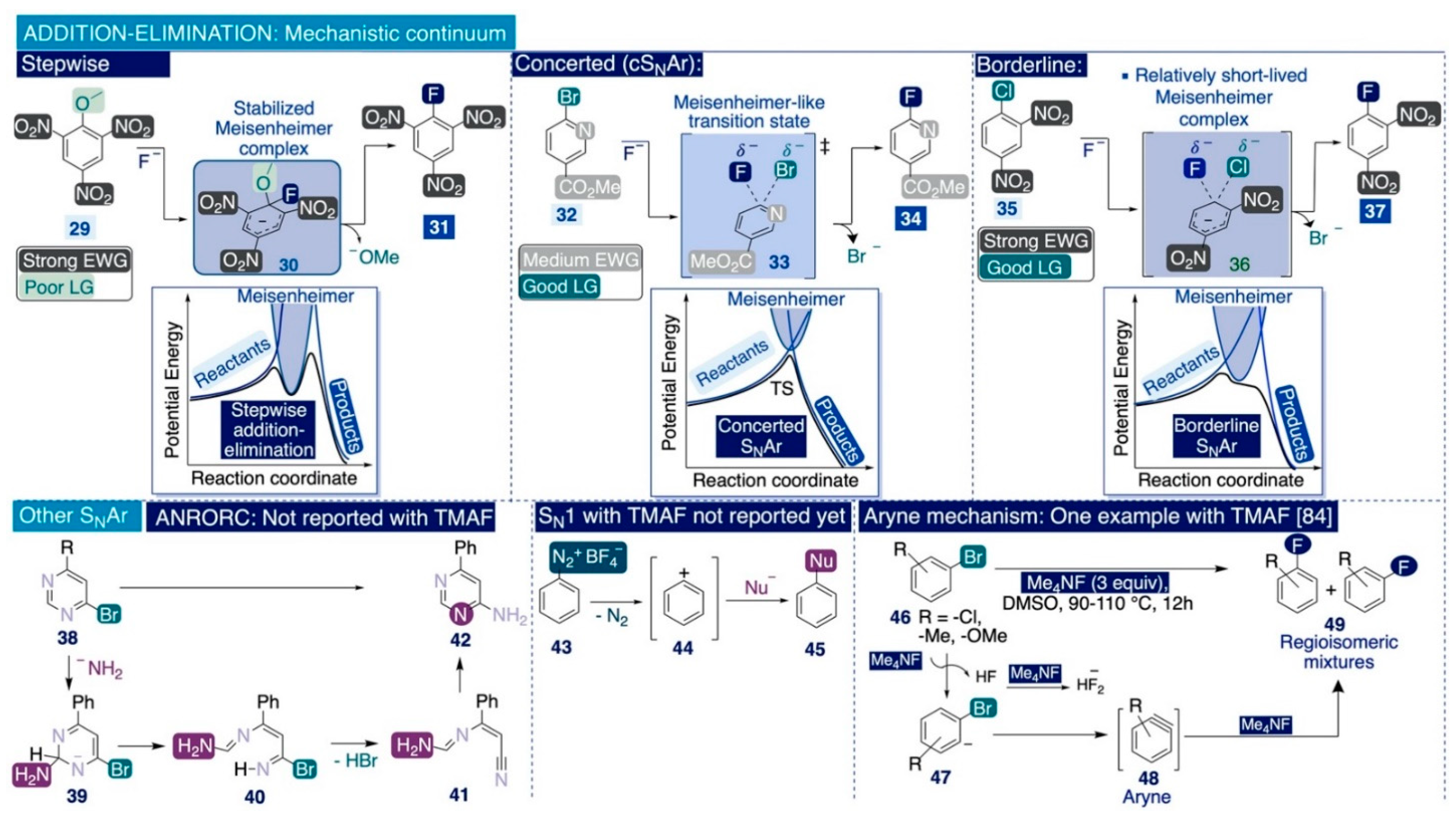
In general, the nature of the substituents on the aryl ring of the substrate is the main factor dictating the operative mechanism in SNAr transformations and TMAF-mediated fluorinations of C(sp2) bonds. Indeed, stepwise addition–elimination reactions involve arene substrates with strong electron-withdrawing substituents (e.g. CN, NO2, or SO2R) and poor leaving groups (e.g. F, OMe, NR2, H) that stabilize anionic σ-complexes such as 30—known as Meisenheimer complexes (Scheme 7). The formation of these complexes takes place immediately after the initial nucleophilic attack at the ipso-carbon and is usually the rate-limiting step of the whole process due to the elevated activation energy required to break the aromaticity of the initial substrate [104,105,106]. It is worth noting that Meisenheimer complexes are not merely putative transient species since their existence was unequivocally proven by NMR spectroscopy in the 1960s [107]. Then, the Meisenheimer complex collapses, releasing the leaving group and yielding the energetically favored neutral products with the aromaticity reinstalled [103]. A representative example of this mechanistic type would be the fluorination of 2-methoxy-1,3,5-trinitrobenzene (29) (Scheme 7, left).
Furthermore, the groups of Jacobsen [108], Ritter [109,110], Sanford [111], and others [112] have postulated, in recent years, single-step concerted mechanisms (cSNAr) to explain the occurrence of SNAr reactions defying the hitherto widely accepted stepwise mechanism. This type of reaction occurs on arene substrates with relatively electron-rich substituents and a good leaving group (e.g. Br). A representative example would be the reaction of a nitrogen-containing heterocycle equipped with an ester substituent and a good leaving group (e.g. Br), such as 32, with TMAF or TBAF in a polar aprotic solvent such as DMF or DMSO (Scheme 7, center). Hammett studies revealed that variations of the electronic effects associated with changes to the aryl substituents have little impact on the rate-limiting step [109], a result incompatible with the intermediacy of Meisenheimer complexes [113].
Furthermore, Jacobsen and co-workers demonstrated that the evaluation of kinetic isotope effects (KIEs) can be utilized to elucidate the operative pathway on a given SNAr reaction [108]. In that study, DFT analyses of the calculated transition states of several SNAr processes allowed for postulating the existence of a mechanistic continuum, which may be visualized by representing the computed potential energy against the progress of the reaction (Scheme 7).
Scheme 7.
Possible reaction mechanisms for SNAr fluorination reactions with TMAF.

In such representations, the presence of a distinctive minimum of potential energy along the reaction coordinate corresponds to discrete, stabilized Meisenheimer complexes and a reaction proceeding via a conventional stepwise pathway. Conversely, the absence of that energy minimum may be associated with a single-step cSNAr reaction. These studies also underlined the existence of borderline substrates that do not react through wither of those two extremes in the mechanistic continuum. For instance, Jacobsen considered substrates combining strong electron-withdrawing substituents, such as 35, that stabilize the potential Meisenheimer complex, with a relatively good leaving group (e.g. Cl) that destabilizes it (Scheme 7, right) [108]. In those cases, the reaction transition state corresponds to a small minimum in the representation of potential energy vs. the progress of the reaction.
Some authors have suggested that such mechanistic scenarios may be rationalized either as a concerted pathway with a relatively long-lived transition state (in relation to archetypical cSNAr reactions), or through a stepwise mechanism with a relatively short-lived transition state (in relation to classical stepwise mechanisms) [104]. However, the widely accepted definition for “transition state” usually refers to the maximum energy alongside the reaction coordinate, which does not correspond to discrete intermediate species [114]. Consequently, it may be sensible to avoid using the concept of a “long-lived transition state” which may foster unnecessary confusion.
In general, SNAr reactions may proceed through other mechanistic types, which are either fundamentally incompatible with TMAF-mediated SNAr fluorination reactions or there are no examples reported of them (Scheme 7, bottom). For instance, the vicarious SNAr mechanism is not operative on TMAF-mediated SNAr reactions because it involves carbanions as nucleophilic species. Moreover, the strong reactivity of TMAF may favor an ANRORC SNAr pathway (the addition of a nucleophile, ring-opening, ring-closure) in some heterocyclic systems [115]. However, to the best of our knowledge, there are no reported examples of ANRORC SNAr fluorinations using TMAF as the reagent.
Furthermore, SN1 elimination–addition pathways constitute another major type of SNAr reactions. This type of mechanism involves the formation of very reactive aromatic carbocations after the initial elimination event. The classic substrates fluorinated through this type of mechanism are tetrafluoroborate diazonium salts such as 43 [103], which undergo thermal or photolytic decomposition in what is known as the Balz–Schiemann reaction [116]. Nevertheless, to the best of our knowledge, there are no examples reported of TMAF-mediated fluorinations proceeding through a SN1 pathway.
Despite that, Grushin and Marshall reported in 2008 that the SNAr of substituted bromoarenes, such as 46, with TMAF in DMSO at elevated temperatures yields regioisomeric mixtures of the corresponding fluorinated arenes (Scheme 7, bottom-left) [84]. To explain these results, the authors proposed an elimination–addition mechanism mediated by extremely reactive aryne species 48, which requires the consumption of three equivalents of TMAF. Whilst the first equivalent of TMAF would deprotonate the bromoarene and generate the reactive aryne species 48, a second equivalent of TMAF would capture the HF formed after the initial deprotonation step, and a third equivalent of TMAF would ultimately yield the nucleophilic addition across the aryne triple bond leading to the fluorinated end-products. To the best of our knowledge, there are no more reports of such TMAF-mediated fluorination reactions proceeding via an aryne mechanism.
3.2.2. Seminal Nucleophilic Aromatic Substitution Reactions on Nitro- and Halo-Arenes
The discovery of the Balz–Schiemann reaction in 1927 marks the advent of the first fluorination transformations on arene systems [116]. In practice, this process comprises the treatment of aniline substrates (50) with HBF4 upon diazotation conditions to generate relatively stable diazonium salts (51) that may only undergo decomposition at elevated temperatures to yield the corresponding fluorinated products (52) (Scheme 8, up-left). The major drawbacks of this reaction are the considerable danger of explosion when the thermal decomposition is carried out at a large scale, and the dramatic drops in yield associated with the presence of electron-withdrawing groups in the substrates. In fact, the reaction has been mainly useful for the fluorination of electron-rich arene after some improvements [117].
Complementing the Balz–Schiemann reaction, Gottlieb reported in 1936 the direct conversion of 1-chloro-2,4-dinitrobenzene (53) into 1-fluoro-2,4-dinitrobenzene (54) using an excess of anhydrous KF at 200 °C (Scheme 8, up-right) [118]. This process, later known as “Halex”—halogen exchange—constitutes the seminal example of SNAr on electron-deficient arenes, which had enormous implications for the development of important applications such as the preparation of [18F]-labelled radiopharmaceuticals [2,3]. Two decades later, Finger and Kruse systematically studied and successfully expanded the Halex process to the fluorination of an amplified set of aryl chloride substrates, and furthermore, also described the first examples for the fluorodenitration of various nitro- and chloro-nitroarenes (Scheme 8, up-right). The latter reaction involves the nucleophilic attack of fluoride into the carbon atom that bears the nitro group and takes place using a large excess of anhydrous KF at elevated temperatures [119].
It was not until the 1980s when Clark and colleagues described further improved versions of Halex and fluorodenitration SNAr processes. First, they reported that tetraphenylphosphonium bromide can significantly accelerate SNAr fluorinations with KF on polar aprotic solvents at lower temperatures [120]. Next, it was in 1993 when they published the first use of TMAF in a SNAr process, such as the fluorodenitration of several nitroarenes (e.g. 55) [121] (Scheme 8, bottom-left). Two years later, they expanded the synthetic scope of this reaction to diphenyl sulfone substrates [122]. In both cases, they found that a slight excess of azeotropically dried TMAF was highly competent to minimize the considerable amounts of phenolic and diaryl ether side products (usually associated with fluorodenitration processes) and substantially reduce the required reaction times and temperatures in DMSO (Scheme 8). A few years later, a re-examination of those fluorodenitration processes enabled the Clark group to infer that the basicity of TMAF is the key parameter to further optimize the reaction yields (Scheme 8). Indeed, the use of slightly less basic TMAF (TMAF·4/3H2O) in N,N’-dimethylacetamide (DMA) led to improved yields [86] that were further improved by later switching to the even less basic tetramethylammonium hydrogen difluoride (TMAHF2) in DMA, which in turn required using larger stoichiometries and longer reaction times [123].
Scheme 8.
Seminal SNAr fluorination reactions of (hetero)arenes.

Along those lines, the Clark group also realized that the pronounced basicity of TMAF could be successfully exploited to render one-pot transformations where a base-promoted step adds to the fluorination reaction. For instance, the reaction of 57 with TMAF yielded 59 via benzylic oxidation followed by difluorodenitration (Scheme 8, bottom) [124]. The authors found that this process was especially sensitive to variations in the solvent and temperature. Indeed, whilst 59 was obtained in low yields after just 1 h of reaction with an excess of pre-dried TMAF in DMSO at 100 °C, only the non-fluorinated oxidized product 58 was quantitatively formed by conducting the reaction under the same conditions but instead using a positive atmosphere of oxygen at room temperature.
Furthermore, the authors indicated that such benzylic oxidation proceeded even using catalytic amounts of TMAF and aptly tolerated moisture and variable levels of hydration on the TMAF reagent. Moreover, DMA was found again to be the most effective media to sequentially carry out the one-pot process, obtaining 59 in 59% yield by first conducting the benzylic oxidation with a catalytic amount of TMAF at room temperature, and subsequently adding 3 equiv more of TMAF to the reaction mixture and raising the temperature to 100 °C to accomplish the fluorodenitration reaction.
As it was previously mentioned (Section 2.1, Scheme 3), it was not until 2005 that DiMagno and co-workers documented an elegant approach to tame the enormous instability that the presence of β-hydrogen atoms imparts to anhydrous TBAF, and thus exploit its outstanding reactivity as an ionic source of “naked” fluoride in a diverse set of SNAr processes [68]. This method superseded all the previously reported procedures for the nucleophilic fluorination of (hetero)arenes and constitutes a suitable golden standard to compare and fully understand the progress made, in terms of performance, in subsequent SNAr fluorination methodologies with TMAF. The key finding in this approach was identifying the cyanide anion as a potent and weakly basic nucleophile capable of substituting and releasing fluoride from hexafluorobenzene by forming another strong C(sp2) bond (BDE (C(sp2)-CN) = 133 kcal/mol). In practice, anhydrous TBAF was generated in nearly quantitative yields by treating hexafluorobenzene with tetrabutylammonium cyanide (TBACN) (1:1 to 1:6 molar ratio) in a polar aprotic solvent such as THF, MeCN, or DMSO at sub-zero temperatures (Scheme 9a). Remarkably, when the synthesis was conducted in THF at −65 °C, TBAF was isolated in an anhydrous form (70% yield) that is stable for weeks under nitrogen at temperatures below −35 °C, for hours in CD3CN solutions, and for more than one day in DMSO solutions at 25 °C. According to 19F NMR spectroscopy, such anhydrous TBAF contains only 2% of tetrabutylammonium hydrogendifluoride as impurity.
The authors ascribed this unexpected stability of TBAF in solution to the dehydrating properties of the reaction byproducts, cyanoarenes (60), which scavenge any traces of water that may be present in the solution during the reaction.
Scheme 9.
(a) DiMagno synthesis of anhydrous TBAF, [68], (b) Preliminary study for SNAr fluorinations with anhydrous TBAF, [68], and (c) Sun-DiMagno SNAr fluorination of heteroaromatics with anhydrous TBAF, [125].

With a solid method in hand to efficiently prepare solutions of anhydrous TBAF, the authors preliminarily compared its performance with other sources of nucleophilic fluorine, such as “vacuum-dried” TBAF, CoCp2F (24), or TBAT (9), in SNAr reactions such as the fluorination of benzylic and primary alkyl bromides, the fluorodenitration of nitroarenes, or the direct fluorination of alkyl tosylates. In this task, the use of anhydrous TBAF displayed superior performance to afford the fluorinated end-products (60–67) in nearly quantitative yields and dramatically reduced the reaction times from hours to a few minutes (Scheme 9b).
In this connection, Sun and DiMagno reported in 2006 the implementation of this anhydrous TBAF-based SNAr methodology to the fluorination of a diverse set of chloropyridines (68–73) in combination with theoretical calculations of the thermodynamic activation parameters involved (Scheme 9c) [125]. These are relevant scaffolds in the agrochemical industry [126,127,128]. Experimentally, the standard optimized conditions involved mixing pre-isolated solid anhydrous TBAF (1.3 equiv) with the corresponding chloropyridine substrate in DMSO at room temperature. The authors observed a clear pattern of reactivity in those processes that directly depended on the substrate substitution. For instance, in the absence of other electron-withdrawing groups in the pyridine ring, the reaction proceeded sluggishly when the leaving chlorine group was located at an ortho-position (i.e. 4 equiv of TBAF and 14 days were needed to convert 69 into the fluorinated end-product in 80% yield) and did not occur at all when the chlorine was placed in a meta-position as in 68. However, those SNAr fluorinations went significantly faster and smoother for chloropyridine substrates equipped with a second electron-withdrawing substituent in the ring such as chlorine in 70 and 71, trifluoromethyl in 72, or a carbonyl group (i.e. >95% yield obtained in 1 h or less for chloropyridine 73 with 1.3 equiv of TBAF). Moreover, other electron-poor heterocyclic substrates, such as chloropyrazine 74 and chloropyridazine 75, also underwent swift fluorination in nearly quantitative yields at room temperature (Scheme 9c). Remarkably, N-protected 6-chloropurine 76 and chlorobenzimidazole 77 could also be fluorinated with excellent yields in less than 1 h at room temperature.
Additionally, the authors further evaluated the ability of their method to fluorinate other electron-deficient arene systems such as chlorinated benzonitriles (78–83), which in general proved to be more reactive than the chlorinated pyridine analogues yet with a similar relationship between their ease to undergo fluorination and their substitution pattern. Furthermore, difluorination only took place in dichloropyridines, such as 71, and dichlorinated benzonitriles, such as 83, with both chlorine groups in ortho- and para-positions in relation to the activating group, since meta-positions are barely activated for SNAr reactions. To conclude, Sun and DiMagno revisited the same type of fluorodenitration reactions on nitroarene substrates, such as 84–90, that the Clark group had studied a decade earlier [86,121,123]. Effectively, they found that such substrates are fluorinated in nearly quantitative yields and significantly faster than their chloropyridine and chlorinated benzonitrile analogues (i.e. monofluorination of 1,4-dinitrobenzene 85 in 95% yield using 1.3 equiv of TBAF during less than 5 min), with the exception of 84 and its analogues containing weakly activating substituents in the para-position, such as fluorine and methyl groups, that did not react at all. However, meta-substituted nitrobenzenes (e.g. 90) could be fluorinated on such conditions.
In general, the impressive reaction rates and mild conditions of the DiMagno TBAF-based fluorination methods made possible a hitherto unusual functional group tolerance in traditional SNAr fluorinations, and aryl esters, ethers, aldehydes, and N-benzyl protecting groups were shown to be compatible with those reaction conditions.
3.2.3. Recent SNAr Fluorination Methodologies with TMAF
In DiMagno’s TBAF-based method, the mandatory use of sub-zero temperatures and at least one equivalent of a potentially highly toxic reagent such as TBACN per equivalent of anhydrous TBAF prepared—and ultimately per atom of fluorine introduced in the corresponding heteroaromatic substrate—constitutes a drastic restraint that is very detrimental to its utilization at the multi-kilogram scales associated with industrial processes. Therefore, subsequent initiatives—such as the research program launched during the last decade by the Sanford group [37]—focused on developing alternative methods for the fluorination of heteroaromatics with viable industrial applicability.
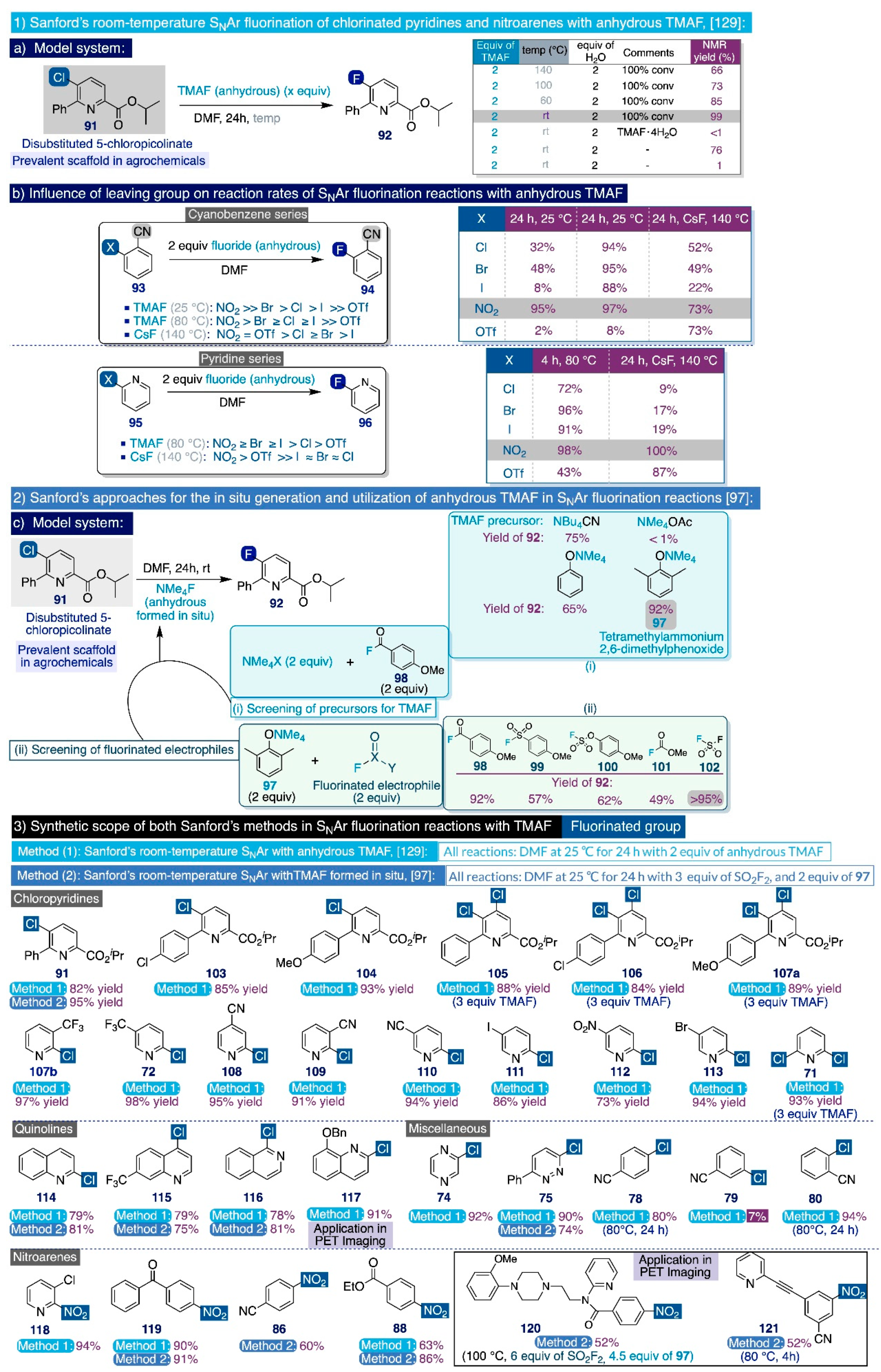
In that context, the Sanford group documented in 2015 a systematic study on the utilization of anhydrous TMAF for the room-temperature SNAr fluorination of chlorinated pyridines and nitroarene substrates (Scheme 10, up) [129]. The first task in that study was identifying a suitable model system, such as the 5-chloropicolinate substrate 91, which is substituted at the ortho positions with phenyl and isopropyl ester groups. Such a scaffold is frequently encountered in agrochemicals and possesses a meta-relationship between the pyridine nitrogen and chlorine reacting group, which constitutes a daunting barrier for aromatic fluorinations, although the simultaneous presence of an ester group para- to the reaction site activates the substrate. In practice, treatment of 91 with 2 equiv of anhydrous TMAF in DMF at room temperature quantitatively rendered the desired fluorinated end-product 92 (Scheme 10a). Conversely, conducting that model reaction at elevated temperatures (100–140 °C) promoted undesired competing side reactions such as the formation of the fluorinated picolinic acid analogue. Additionally, the authors also inspected the effect of adding stoichiometric amounts of water to the reaction media, finding that the fluorination reaction is completely suppressed after merely adding just two equivalents of H2O.
Furthermore, the authors decided to evaluate the influence that the nature of the leaving group imparts on the relative fluorination rates in DMF of a series of ortho-substituted cyanobenzene substrates (93) (Scheme 10b). In that task, they also compared the performance of anhydrous TMAF (2 equiv at 25 °C and 80 °C) with another industrially viable source of nucleophilic fluorine such as CsF (2 equiv at 140 °C). First, the rate of the SNAr fluorinations with anhydrous TMAF at room temperature (for 24 h) significantly varied with the nature of the leaving group in the order of NO2 (95% NMR yield) >> Br (48%) > Cl (32%) > I (8%) >> OTf (2%). Secondly, that variation was drastically reduced by conducting the same reactions at 80 °C for 3h: NO2 (95% NMR yield after just 5 min, 97% after 3h) > Br (95%) ≥ Cl (94%) ≥ I (88%) >> OTf (8%). Furthermore, CsF proved to be a significantly inferior reagent, and only rendered those SNAr fluorinations in moderate to good yields after 24h at 140 °C with substantial changes in the following order of relative rates: NO2 (73% NMR yield) = OTf (73%) > Cl (52%) ≥ Br (49%) > I (22%). A similar evaluation was carried out on a series of ortho-substituted pyridine substrates (95) using anhydrous TMAF (2 equiv) in DMF at 80 °C for 4 h, and CsF (2 equiv) also in DMF at 80 °C for 24 h (Scheme 10b). Experimentally, the relationship between the relative SNAr fluorination rates and the nature of the leaving group is fundamentally the same for both series of substrates (93 and 95) reacting under the same conditions. Additionally, anhydrous TMAF continued to substantially supersede CsF as a fluoride source in these SNAr reactions.
The substrate scope of this synthetic method was next explored utilizing two equivalents of anhydrous TMAF in DMF at room temperature for 24h (Scheme 10, bottom). Effectively, a diverse set of industrially relevant monochloro- and dichloro-picolinates (91, 103–107a), chloro-pyridines (71–72 and 107b–113), chloro-(iso)quinolines (114–117), and other heteroaromatics (74–75) were successfully fluorinated under those general reaction conditions in isolated yields close to those afforded by the DiMagno fluorination method. Moreover, those transformations with dichloropicolinate substrates (105–107a) only required 1.5 equivalents of anhydrous TMAF per chloride. Special mention deserves the fluorination of (benzyloxy)-2-chloroquinoline 117, which afforded the desired fluorinated product in 91% yield, whose [18F]-version has been used in the PET imaging of neurodegenerative processes [130].
Remarkably, this fluorination procedure could also be applied at the multigram scale without significant drops in reaction yield. Additionally, the method was demonstrated to possess comparable, if not slightly superior, functional group substrate tolerance to the DiMagno method, and it allowed fluorinating substrates containing methoxy (104, 107a, 120), cyano (e.g. 108–110), trifluoromethyl (72, 107, 115), ester (e.g. 103–107a), keto (119), iodo (111), and bromo (113) substituents in high to excellent yields. As in previous SNAr fluorination reports, only meta-substituted substrates with electron-withdrawing groups, such as 3-fluorobenzonitrile (79), were not activated enough to undergo SNAr fluorination through this methodology.
Finally, the authors run a comparative cost analysis between the use of anhydrous TMAF and CsF as reagents in this type of SNAr fluorinations at process scale, which concluded that anhydrous TMAF is substantially more economically competitive than CsF since the use of the latter ultimately implies a 3- to 10-fold increase in the final costs. This superior cost-efficiency of TMAF directly increases with the value of the fluorinated end-products and their difficulty to be synthesized.
The complications associated with the direct preparation of large amounts of dry anhydrous TMAF remained perhaps one of the greatest hurdles for the use of this reagent in industrial SNAr fluorinations. As such, the Sanford group deemed it highly convenient to next settle the development of more practical synthetic approaches that could simultaneously generate in situ anhydrous TMAF and render the SNAr fluorinations upon the same reaction conditions, thus avoiding the lengthy drying steps required to prepare anhydrous TMAF. In that context, in 2017, they described several elegant synthetic methods able to expeditiously provide anhydrous TMAF in DMF solution from relatively inexpensive reagents (Section 2.3) [97].
As in their previous work [129], to begin their study the authors selected the same substrate 91 as model system. Next, they screened a series of tetraalkylammonium salts (2 equiv) that could react as nucleophiles with an activated acid fluoride 98 (2 equiv). In this manner, the authors devised that fluoride anions could be rapidly generated, which would be stabilized with the free tetramethylammonium cation, and thus promote the SNAr fluorination of the model substrate in situ (Scheme 10c). In practice, tetramethylammonium acetate proved to be ineffective to yield any fluorinated end-product (92). However, the tetramethylammonium salt of 2,6-dimethylphenoxide (97) exhibited superior performance compared to tetrabutylammonium cyanide or tetramethylammonium phenoxide to afford 92 in an excellent 92% of yield after 24h of reaction in DMF at room temperature. That yield was further elevated to >95% by increasing the ratio of acid fluoride to tetramethylammonium salt from 1:1 to 1.3:1. Furthermore, the SNAr fluorination of a small series of chloro- and nitro-arenes was chosen to compare the efficiency of this method with the use of anhydrous TMAF or acyl azolium fluoride 7 [66]. To that end, all three methods exhibited similar performances.
Next, the authors directed their attention to the screening of other fluorinated electrophiles that, combined with tetramethylammonium 2,6-dimethylphenoxide (97), could generate TMAF in DMF in situ after a short reaction time of 15 min, with the subsequent addition of the initial model substrate. In this task, the relatively inexpensive sulfuryl fluoride (SO2F2) was slightly superior (>95% of SNAr fluorination after 24h of reaction at room temperature) to the previously used acid fluoride (98) (92% of SNAr fluorination), and the rest of surveyed fluorine-containing electrophiles (99–101) (Scheme 10c).
Scheme 10.
Sanford’s SNAr fluorination reactions with: (1) anhydrous TMAF [129], (2) TMAF generated in situ [97], and (3) their synthetic substrate scope.

With the optimized combination of reagents in hand to generate TMAF in situ, the authors next explored the synthetic scope of this method by conducting the SNAr fluorinations as a one-pot reaction where an excess of both precursors—2 equiv of 97 and 3 equiv of SO2F2—could be directly mixed with the corresponding substrate and allowed to react for 24h in DMF at room temperature (Scheme 10 bottom). Upon those conditions, the SNAr fluorination of a varied set of (hetero)aryl chlorides (e.g. 75, 91), including quinolines (114–117) and activated nitroarenes (86, 88, 119–121), proceeded smoothly and delivered the corresponding fluorinated end-products with minor differences of functional group tolerance and yields in relation to the previous protocol based on anhydrous TMAF. Ultimately, this method was competent to render the fluorination of two complex nitroarene heteroaromatic substrates (120, 121), used in PET imaging, in slightly more severe conditions in relation to the use of TMAF anhydrous.
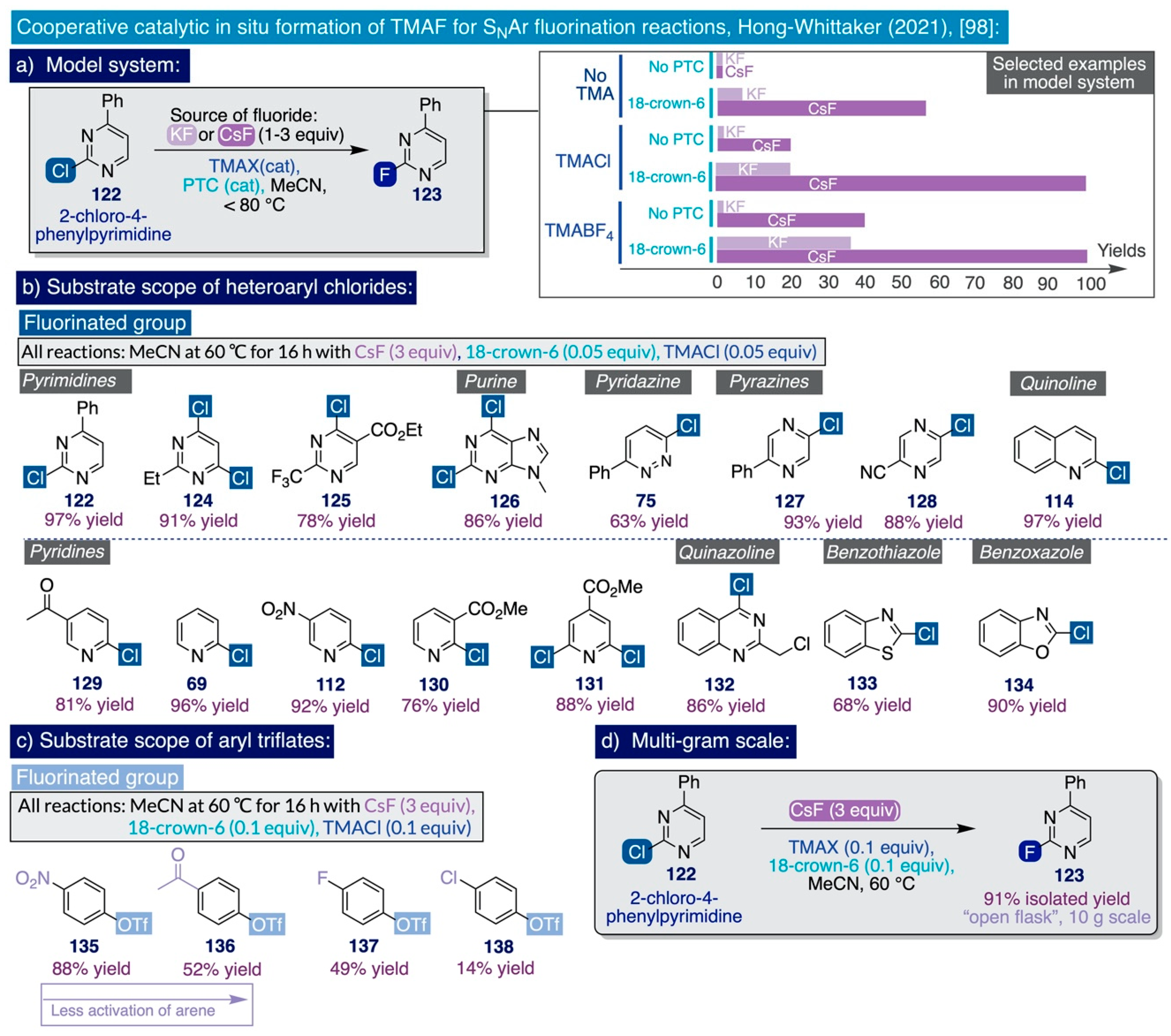
Despite the relevance and short time that has elapsed since the publication of the two initial reports by Sanford on the use of TMAF in SNAr fluorination reactions with amplified industrial applicability, very recently, the significance of this field has propelled the advent of further synthetic methodologies. Indeed, a team of process chemists at Merck & Co reported in early 2021 an expeditive method to generate and use in situ TMAF for the SNAr fluorination of a diverse set of electron-deficient heteroaryl chlorides and aryl triflates (Scheme 11) [98]. Building on Sanford’s protocol, this methodology constitutes a substantial conceptual improvement since it involves the use of catalytic amounts of TMAF generated in situ rather than the use of a stoichiometric excess. Additionally, it involves the cost-efficient use of stoichiometric amounts of alkali salts, such as CsF or KF, as viable and operationally simple ionic sources of nucleophilic fluoride amenable for facile implementation at large process scale, and precluding the drastic measures to handle a highly hygroscopic reagent such as TMAF. The key finding of this methodology lies in the realization that the poor solubility and reactivity of alkali fluorides in organic solvents may be circumvented by a catalytic ion-pairing approach comprising the cooperative action of a phase-transfer catalyst (PTC) such as a crown ether—a competent chelator of alkali cations—with catalytic amounts of a tetramethylammonium salt, which traps the released fluoride anion in situ. In this manner, catalytic amounts of TMAF are rendered available in solution that effectively drive the desired SNAr fluorinations.
The initial catalytic screening was conducted in a high-throughput regime using an electron-deficient chloropyrimidine substrate (122), which is suitably activated to undergo SNAr fluorination (Scheme 11a). Strongly polar aprotic solvents like DMF and DMSO, which are in the ideal solvation window to ensure optimum reactivities for ionic sources of nucleophilic fluorine such as TMAF (Section 2.2.3), were avoided in these studies due to the potential risks of explosion associated with their use in the presence of strong bases and temperatures as low as 50 °C [87,88,131]. To this end, MeCN offered the best compromise as a solvent between polarity, safety profile, and operational simplicity, allowing the removal of salt byproducts by filtration. Furthermore, CsF clearly outperformed KF and managed to fluorinate the model substrate 122 under a variety of combinations of the phase-transfer catalyst and tetramethylammonium salt, both of which were shown in control experiments to be mandatory for the progress of the reaction.
In practice, optimization of the reaction conditions demonstrated that TMACl and 18-crown-6 ether as the phase-transfer catalyst—both added at 5–10 mol% and operating in MeCN at 60 °C—was the best possible binomial out of the catalysts surveyed, as a result of their ‘broader substrate applicability’ (sic), catalytic performance, cost-efficiency, and easy handling. Moreover, inspection of the synthetic substrate scope revealed that such optimized reaction conditions were able to effectively fluorinate relevant chlorinated heteroaromatic scaffolds such as pyrimidines (122–125), N-methylpurine 126, pyridazine 75, pyridines (69, 112, 129–130), pyrazines (127–128), quinoline 114, quinazoline 132, benzothiazole 133, and benzoxazole 134 (Scheme 11b).
Scheme 11.
SNAr fluorination of heteroaryl chlorides and aryl triflates via the cooperative catalytic in situ generation of TMAF: (a) Model system, (b) Substrate scope of heteroaryl chlorides, (c) Substrate scope of aryl triflates, and (d) Multi-gram scale.
Scheme 11.
SNAr fluorination of heteroaryl chlorides and aryl triflates via the cooperative catalytic in situ generation of TMAF: (a) Model system, (b) Substrate scope of heteroaryl chlorides, (c) Substrate scope of aryl triflates, and (d) Multi-gram scale.

Remarkably, the reaction exhibited excellent functional group tolerance, and substrates containing ketones, esters, nitro, cyano, and even a benzyl chloride group were all selectively fluorinated. Conversely, substrates containing nucleophilic functionalities dimerized under such reaction conditions, namely electron-rich furans and thiophenes, did not react, and the presence of highly acidic functional groups made the substrates less reactive, likely as a result of their hydrogen bonding to the fluoride anions in solution. Furthermore, this methodology was applied—as a proof-of-principle—to the SNAr fluorination of four para-substituted aryl triflates (135–138) (Scheme 11c). In that task, the yield of the reaction increased in a linear fashion with the electron-withdrawing character of the functional group at the para-position (NO2: 88%, methyl ketone: 52%, F: 49% and Cl: 14%).
This constitutes a particularly useful extension of the substrate scope of this methodology since aryl triflates are easily accessed from readily available phenolic substrates. Ultimately, the authors proved the viability of this method at process scale by conducting the SNAr fluorination of the initial model substrate, 5-phenyl-2-chloropyrimidine 122, at a 10 g scale without any rigorous exclusion of air and moisture (Scheme 11d). At such scale, the corresponding fluorinated end-product 123 was obtained in excellent isolated yield and purity by merely filtering off the salts generated at the end of the reaction, followed by concentration in vacuo and recrystallization from dichloromethane. Experimental evidence for the origin of the observed reactivity was gathered by 19F NMR spectroscopy, which indicated the generation of catalytic amounts of TMAF in solution upon the reaction conditions. Indeed, the diagnostic resonance that TMAF exhibits in MeCN-d3 (apparent triplet 1:1:1 around −151 ppm) was only observed when CsF, TMACl, and 18-crown-6-ether were mixed in that deuterated solvent.
Up to date, the most recent report on the utilization of TMAF for SNAr fluorinations was published in May 2021 by the Sanford group [132]. This report directly builds on the work of Kim et al. [133], who discovered that the combination of t-BuOH with TBAF forms the crystalline adduct (Bu4N+F–·(t-BuOH)4 with attenuated basicity compared to anhydrous TBAF but rather less hygroscopic, and thus operationally simpler to handle (Scheme 12a). The attenuated basicity of this adduct has been synthetically exploited to fluorinate alkyl halides [133] and organophosphorus compounds [134], with lower amounts of the E2 side products that frequently accompany this type of SN2 reactions. However, the Sanford group pinpointed two important factors obstructing the use of this adduct in SNAr fluorinations.
First, Bu4N+F−·(t-BuOH)4) is not only less basic but also possesses reduced fluoride nucleophilicity due to the intermolecular hydrogen bonding interaction between the alcohol proton and fluoride. This is problematic in SNAr fluorination processes, which are typically slower than SN2 reactions. Second, the aryl fluoride end-products in SNAr reactions are substantially more electrophilic than alkyl fluoride SN2 products, and may react in basic media with alcohols to furnish significant amounts of undesired aryl ethers. To supersede these obstacles, the Sanford group decided to investigate TMAF·R(OH)x adducts that could maintain the high reactivity and selectivity of TMAF for SNAr fluorinations, and simultaneously be less hygroscopic, thus precluding the need for strictly excluding moisture and ambient air from the reaction media.
To begin their studies, the authors selected the SNAr fluorination of 2-chloroquinoline as the model system (Scheme 12b). This process was previously shown to deliver 2-fluoroquinoline (139) in 79% and 81% yields in Sanford’s two initial synthetic methods (Scheme 10, [97,129]), and in 97% yield via the cooperative catalytic generation of TMAF in situ (Scheme 11, [98]). Furthermore, a key intermediate in the preparation of anhydrous TMAF, such as Me4N+F−·MeOH, was selected as the fluoride source (1 equiv), which was allowed to react in DMF for 24h in room temperature and benchtop conditions (without prior drying of reagents or solvents). Upon such initial conditions, no conversion was observed. This result clearly underscores the difference of reactivity between the Me4N+F−·MeOH adduct and anhydrous TMAF, which in turn, had previously afforded 2-fluoroquinoline (139) in 99% yield under exactly the same reaction conditions. In practice, 139 was only sluggishly formed (10% yield) when the reaction temperature was raised to 60 °C, but along with 19% of the SNAr etherification product 2-methoxyquinoline (140). Further optimization identified DMSO and 80 °C as the most suitable reaction solvent and temperature, respectively.
Next, a series of Me4N+F−·(ROH)x (x = 1−1.4) adducts were synthesized in benchtop conditions by dissolving Me4N+F−·MeOH in the corresponding ROH solvent (Scheme 12b). Inspection of the performance of those TMAF alcohol adducts upon the optimized conditions for the model system allowed to clearly establish that the more sterically hindered the alcohol (MeOH < EtOH < i-PrOH < t-BuOH ≤ t-amylOH), the lower the extent of the competing SNAr etherification, where it acts as a nucleophile, and the more the fluoride nucleophilicity of the adduct, and thus the final yield, increases. Indeed, 2-chloroquinoline (114) was quantitatively fluorinated in benchtop conditions by reacting it with 1.5 equiv of Me4N+F−·t-amylOH in DMSO after 24 h at 80 °C. Remarkably, the process showcased high reproducibility, and minor variations in yield were observed using different batches of DMSO or with changes in the degree of ambient humidity.
Scheme 12.
Sanford’s SNAr fluorination of halo- and nitro- (hetero)arenes with alcohol adducts of TMAF: (a) Precedent: Preparation of TBAF alcohol adducts [133,134], (b) Sanford’s preparation of non-hygroscopic TMAF alcohol adducts, and initial screening [132], and (c) Substrate scope.

Next, the authors proceeded to investigate the substrate scope for the SNAr fluorination in the optimized benchtop conditions (DMSO, 80 °C, 24 h, Scheme 12c). Notably, this synthetic survey comprised a large and diverse set of halo- and nitro-(hetero)arene substrates (more than 50 compounds) including electron-deficient heterocyclic substrates that had not been tested before in SNAr processes. In practice, all the reactions were conducted with small variations in the amounts of Me4N+F−·t-amylOH adduct used (1.1 –2.0 equiv per atom of fluorine introduced), providing comparable reaction yields on scales ranging from 40 mg to 1 g of substrate, although in some cases, with significant disparities between the recorded NMR and isolated yields (e.g. 145, 149, 162, 183).
This synthetic methodology displayed an exceptionally broad functional group tolerance—for a SNAr process—and substrates featuring halogen (F, Cl, Br, I), nitrile (e.g. 108–110, 165–169), nitro (112, 156), trifluoromethyl (107), ether (117), ester (103, 154, 157), sulfonamide (170–171) and tertiary nitrogen (174–175) functional groups were all compatible with the reactions conditions, some of which may provide a synthetic handle for further functionalization. Moreover, the relatively mild reactivity of the Me4N+F−·t-amylOH adduct not only made this broad functional group tolerance possible, but also allowed achieving remarkable site-selectivity for SNAr fluorination to exclusively occur at the most reactive site in substrates with several feasible leaving groups (e.g. 112, 143–146, 156, 158–160).
Furthermore, this synthetic methodology followed reactivity patterns hitherto well-documented in SNAr fluorination processes such as (i) the reactivity and reaction yields increases with the electronic nature of the leaving group according to the order NO2 > Cl ≥ Br > I (e.g. series of substituted pyridines 113, 158–160), or ortho- and para-substituted benzonitriles (e.g. 80, 89, 168–169 and 78, 86, 165–166, respectively); and (ii) ortho-substituted substrates are generally more reactive than their corresponding para-substituted isomers, and meta-substituted isomers are not activated enough to undergo SNAr fluorination upon these conditions (e.g. nitro-substituted benzonitrile 89 vs 86 vs 90, or bromo-substituted benzonitrile 169 vs 166 vs 167). Finally, this synthetic methodology enabled the fluorination in moderate to excellent yields of heterocyclic scaffolds relevant in medicinal chemistry and drug discovery such as pyridazines (e.g. 75, 172), pyrazines (74, 173), indole (176), pyrimidines (122, 174–178), purines (76, 184–187), and the 1,8-naphthyridine-containing antibiotic 154.
Despite the remarkable progress in the development of practical, efficient, and industrially viable SNAr fluorination methods that has been described in this section, the importance of fluorinated (hetero)arene building blocks in several key areas combined with an understanding of gaps and limitations underlying the current methods certainly warrants further research in this area.
3.3. Deoxyfluorination of Phenols and Aldehydes with TMAF
3.3.1. Seminal Examples for the Nucleophilic Deoxyfluorination of Phenolic Compounds
Phenolic compounds are the most abundant category of secondary metabolites in the plant kingdom, thus constituting readily available, versatile, and increasingly important synthetic building blocks amidst the imperative transit to economic and industrial models based on renewable natural resources such as biomass [135]. In terms of synthetic fluorine chemistry, the substitution of the hydroxy group in phenols by fluoride—namely, the deoxyfluorination reaction—has recently received increasing attention [28,36].
In 2011, Akai and co-workers reported that one of the hydroxyl groups in catechol substrates can be replaced by fluorine by means of an umpolung strategy involving one-pot oxidation–fluorination with the hypervalent iodine oxidant PhI(OAc)2 and nucleophilic fluorinating reagent Deoxofluor (16), followed by reductive rearomatization of the resulting fluorinated diketones (190) to afford regioisomeric mixtures of the corresponding ortho-fluorinated phenols (191) in moderate yields (Scheme 13a) [136].
Previously, Hayashi and co-workers had described the use of 2,2-difluoro-1,3-dimethylimidazoline (DFI) as a new nucleophilic fluorinating agent that could directly deoxyfluorinate para-nitrophenol in relatively mild conditions (2 equiv, MeCN, 85 °C, 15 h) and moderate yield (62%) (Scheme 13b) [137].
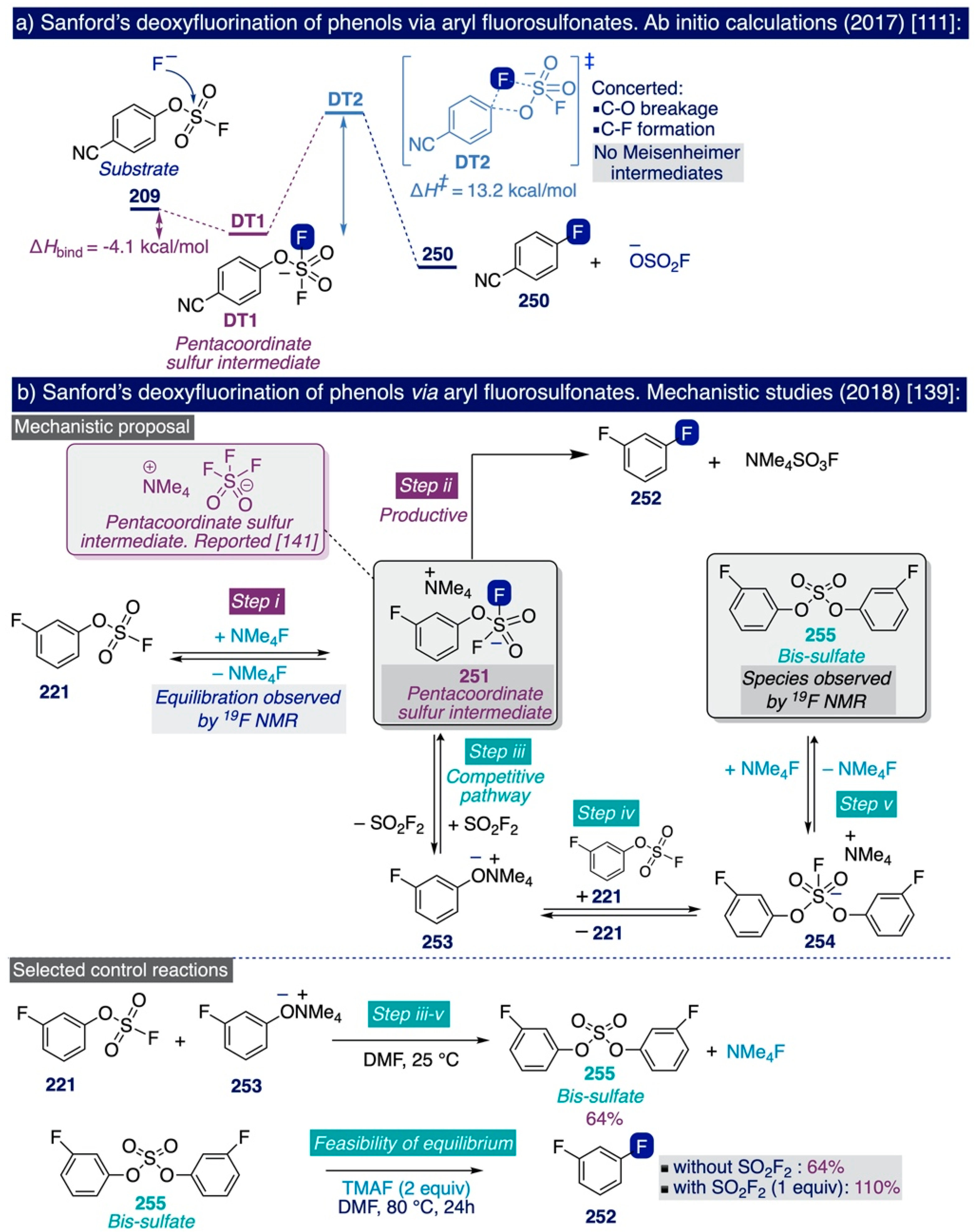
These seminal precedents inspired the Ritter group to develop an elegant and general method for the nucleophilic ipso-deoxyfluorination of phenols (Scheme 13c) [138]. This reaction takes place through an umpolung pathway whereby the phenol substrates react first with PhenoFluor to form an intermediate 2-phenoxyimidazolium bifluoride salt (206), which then can undergo the nucleophilic attack of fluoride (from CsF) due to the intramolecular activation by hydrogen bonding created by the presence of the bifluoride anion in the salt (Scheme 13c, right). The remarkable synthetic scope of this method encompasses a diverse set of electron-deficient, -neutral, and -rich (hetero)aryl fluorides—including challenging meta-substituted aryl fluorides (200–201), which can be obtained in good yields by directly treating the corresponding phenolic substrate with a slight excess (1.2 equiv) of PhenoFluor and CsF (3 equiv) upon relatively mild conditions such as allowing the mixture to react overnight in toluene at 80–110 °C.
3.3.2. Usage of TMAF for the Deoxyfluorination of Phenols
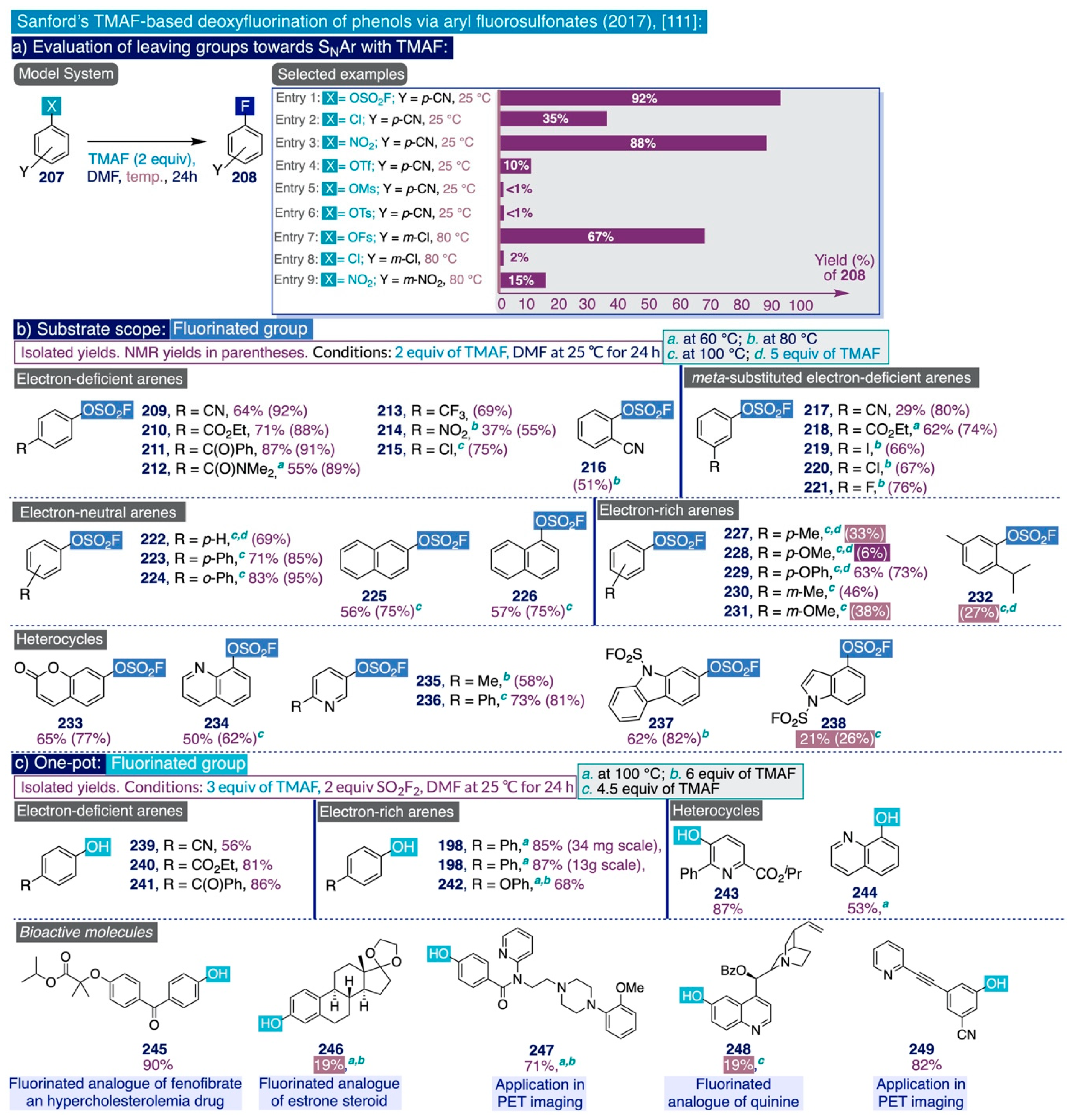
The pioneering Ritter deoxyfluorination approach avoids the need to prefunctionalize the corresponding phenolic substrate, which can be very diverse in electronic nature. However, it relies on the use of PhenoFluor, which can only be accessed after treatment of the dichloroimidazolium chloride precursor 205 with four equivalents of CsF [138]. Additionally, the preparation of that precursor comprises three synthetic steps from 2,6-diisopropylaniline (Scheme 13). That is, the deoxyfluorination of every single hydroxyl group requires at least 7 equivalents of CsF (3 equiv for the deoxyfluorination reaction and 4 equiv per equiv of PhenoFluor consumed).
In the context of their research program to develop effective, selective, and cost-efficient fluorinating methods with potential viability at industrial level—largely based on the use of TMAF as nucleophilic source of fluoride—the Sanford group disclosed a novel synthetic approach to deoxyfluorinate phenolic compounds in 2017 [111]. This transformation involves the prefunctionalization of the corresponding phenol substrate as an aryl fluorosulfonate (ArOFs) electrophile with the relatively inexpensive sulfuryl fluoride (SO2F2) gas. Then, the resulting ArOFs undergoes clean SNAr fluorination by treatment with TMAF in mild conditions without the need for employing additional stoichiometric fluorinating reagents or costly transition metal catalysts (sic).
To begin the study, an initial screening of the reactivity of several substituents as prospective leaving groups towards the SNAr with TMAF (2 equiv TMAF, DMF, rt, 24 h) on a series of para-substituted benzonitrile substrates (207) (Entries 1–6, Scheme 14a), allowed the authors to clearly establish that the fluorosulfonate leaving group (OFs) is more electrophilic and compares favorably to other standard leaving groups in SNAr, such as chloride, nitro, triflate, mesylate, or tosylate, rendering the corresponding fluorinated end-product 208 faster and in higher yield. Only para-nitrobenzonitrile reacts slightly faster at 80 °C than the para-fluorosulfonate benzonitrile, yet in inferior yield, as a result of side reactions that arise over time on the nitro leaving group. This trend also manifested when the reactivity of a series of meta-substituted chlorophenyl substrates was compared (Entries 7–9, Scheme 14a), which are usually poorly activated for SNAr fluorination reactions. In this task, only the meta-substituted aryl fluorosulfonate could effectively undergo SNAr fluorination after 24h of reaction with 2 equiv of TMAF in DMF at 80 °C. Conversely, the corresponding meta-substituted aryl chloride and nitroaryl analogues barely reacted (entries 8–9, Scheme 14a).
The importance of conducting this deoxyfluorination process in strictly anhydrous conditions is clearly illustrated by the reaction of aryl fluorosulfonate 209, whose NMR yield dramatically decreased from 92% to 1% after merely replacing the use of anhydrous TMAF (2 equiv) by TMAF hydrate (2 equiv).
Next, the authors proceeded to comprehensively assess the substrate scope and limitations of this method (Scheme 14b). Following usual patterns in SNAr fluorination processes, aryl fluorosulfonate substrates bearing electron-deficient para- and ortho-substituents (209–216) generally proved to be more reactive and could be converted into the corresponding fluorinated end-products upon relatively mild conditions (in some cases, at room temperature) with moderate to high yields. Remarkably, aryl fluorosulfonate substrates containing other potential leaving groups (i.e. Cl, 214 and NO2, 215) were converted with high site-selectivity to the corresponding aryl fluorides even at relatively high temperatures (80–100 °C). In line with the reactivity patterns observed in Ritter’s approach and during the initial screening in this work, the scope of this method also successfully covered electronically diverse substrates that typically are not accessible through classical SNAr fluorination methods. In this sense, aryl fluorosulfonate substrates equipped with electron-withdrawing groups at the meta-position (217–221) reacted selectively with TMAF at 25–100 °C to afford the corresponding aryl fluorides in moderate to good yields. Even more, this transformation was also competent to fluorinate electron-neutral aryl fluorosulfonates (222–226) in moderate to good yield at 100 °C.
Scheme 14.
Sanford’s deoxyfluorination of phenols via aryl fluorosulfonates: (a) Evaluation of leaving groups towards SNAr with TMAF, (b) Substrate scope, and (c) One-pot SNAr with TMAF.
Scheme 14.
Sanford’s deoxyfluorination of phenols via aryl fluorosulfonates: (a) Evaluation of leaving groups towards SNAr with TMAF, (b) Substrate scope, and (c) One-pot SNAr with TMAF.

However, as soon as the degree of electron donation on the aryl fluorosulfonate substrates increased (i.e. 227–232), the fluorination slowed down and afforded the corresponding fluoroarene products in lower yield, even at 100 °C, by adding higher amounts of TMAF. In this sense, it is especially illustrative the sluggish reactivity of aryl fluorosulfonate 228 (bearing a para-methoxy group) even upon forcing reaction conditions. Additionally, the method was competent to fluorinate heterocyclic aryl fluorosulfonate substrates (i.e. 233–238) in moderate to good yields. Importantly, this transformation could be conducted as a one-pot process where the phenolic substrates 198, 239–244 are transformed directly into the corresponding fluorinated end-products without significant drops in yield in relation to the sequential two-step procedure (Scheme 14c).
Such one-pot process involved simultaneously treating the corresponding phenol substrate with SO2F2 (2 equiv) and TMAF (3 equiv) at a temperature that depended on the electronic nature of the substrate (e.g. 25 °C or 100 °C), and it was successfully scaled up to 10 g for phenol 198, as a proof-of-principle of its industrial applicability. Ultimately, the phenol groups of a series of bioactive molecules (245–249) were deoxyfluorinated in one pot with low to high yields, to further illustrate the potential of this method in pharmaceutical and drug discovery settings as well as its compatibility with common functional groups such as alkenes (e.g. 248), non-enolizable esters and ketones (e.g. 245), amides and amines (e.g. 247), and some nitrogen-based heterocycles (e.g. 247–249). However, protection was needed for base-sensitive functional groups such as an enolizable ketone (i.e. 246) and a secondary alcohol (i.e. 248), which did not prevent significant erosion in their deoxyfluorination yields.
Aiming to gain mechanistic insight into this process, the authors next carried out ab initio calculations on the deoxyfluorination of the model aryl fluorosulfonate substrate (209) with TMAF [111], which suggested that the fluoride anion initially attacks the sulfur atom in the fluorosulfonate group exothermically, giving rise to the pentacoordinate sulfur intermediate DT1 (ΔH = −4.1 kcal/mol) (Scheme 15a). Then, the rupture of the C(sp2)-O bond and the formation of the C(sp2)-F bond occur in concerted fashion with a relatively low activation enthalpy (ΔH‡ = 13.2. kcal/mol), which is consistent with the fast reaction rates observed experimentally. Attempts to computationally identify possible Meisenheimer-type stable intermediates along the reaction coordinate failed (sic). This mechanistic scenario was consistent with those mechanistic observations in Ritter’s deoxyfluorination method, yet at that moment seemingly contravened the SNAr fluorination mechanisms classically accepted that usually involve the intermediacy of Meisenheimer complexes (Section 3.2.1).
Intrigued by these results, the Sanford group reported detailed mechanistic studies a year later on their deoxyfluorination method [139] (Scheme 15b). To begin, the Hammett plot of the initial rates for the deoxyfluorination reaction of a series of electronically different aryl fluorosulfonates with TMAF provided a value of the sensitivity constant (ρ) of +6.26, which is substantially superior to the value reported for Ritter’s deoxyfluorination reaction (ρ = +1.79) and in the range of typical values observed in classical SNAr reactions (ρ (SNAr) ≈ 3–8) [140]. This constant ρ in the Hammett equation describes the susceptibility of the reaction to the substituents of the aryl ring in relation to the ionization of benzoic acid. Typically, the larger is the value of ρ, the larger is the dependence of the rate-determining step with the electronic effects imparted by the aryl substituents [113]. Consequently, the larger value of ρ observed in Sanford’s deoxyfluorination reaction is consistent with the stronger dependence of its rate with the electronic nature of the aryl substituents (e.g. reacting slowly and with low yields with electron-rich substrates, such as 228, Scheme 14), compared to the rate of Ritter’s deoxyfluorination reactions [138].
Furthermore, monitoring the reaction of aryl fluorosulfonate substrate 221 with TMAF, via 19F NMR spectroscopy, indicated possible equilibration with the tetramethylammonium salt of the pentacoordinate sulfur intermediate 251 (occurrence of broad resonance in the 19F NMR spectrum), an observation in line with the detection of analogous pentacoordinate sulfur species after the treatment of sulfuryl fluoride with TMAF [141]. More importantly, careful analysis of that 19F NMR data also revealed the formation of a bis-sulfate species 255. Subsequently, the resonances of both intermediates 251 and 255 faded away over time leaving the fluorinated end-product 252 as the only species present in the mixture. Based on the combined theoretical and spectroscopic information gathered, Sanford proposed a mechanistic scenario (Scheme 15b), where the aryl fluorosulfonate substrate 221 reacts first with TMAF to form the pentacoordinate sulfur salt intermediate 251 (step i), which may either directly afford the fluorinated end-product 252 (step ii) or form the phenoxide intermediate 253 after extrusion of SO2F2. Then, the phenoxide species are able to attack a second molecule of the starting material 221 to generate a new pentacoordinate sulfur intermediate 254 (step iv) that finally dissociates into the spectroscopically observed bis-sulfate species 255 and TMAF (step v).
Scheme 15.
Sanford’s deoxyfluorination of phenols via aryl fluorosulfonates: (a) Ab initio calculations (2017) [111], and (b) Mechanistic studies (2018) [139].

Independent syntheses of these postulated intermediates allowed to corroborate the feasibility of the individual steps leading to the formation of bis-sulfate species 255 (Step iii-v, Scheme 15b) and the reverse reactions from 255 to the fluorinated end-product 252. Additional proof was obtained by adding external SO2F2 to the reaction mixture that eventually drove the equilibrium between bis-sulfate 255 and 251 towards the formation of 251, thus increasing the yield of the fluorinated end-product 252. Overall, the existence of a competing equilibrium between 255 and 251 was proven, which eroded the reaction yields of the main deoxyfluorination process. The detrimental effect of this equilibrium was particularly clear in electron-neutral and electron-rich substrates. In an attempt to address this, electron-neutral and electron-rich aryl triflates and nonaflates, that are not able to engage in such equilibria due to their inability to generate bis-sulfate species, were proven to be ineffective substrates to improve the deoxyfluorination yields, except for electron-deficient aryl triflates, which underwent deoxyfluorination in slightly higher yields than their aryl fluorosulfonate analogues.
3.3.3. Usage of TMAF for the Deoxyfluorination of Aldehydes and Ketoesters
In the aftermath of their report on the deoxyfluorination of phenols via aryl fluorosulfonates, the Sanford group swiftly described two approaches to transform (hetero)aryl aldehydes and α-ketoester substrates into the corresponding difluoromethylated (hetero)arene products (Scheme 16) [142,143]. The deoxyfluorination of aldehydes was already known for other reagents, such as DAST [73] or Deoxofluor [144] but is relatively less studied than the deoxyfluorination of other types of substrates. The first method relies on a two-chamber system whereby SO2F2 gas is generated ex situ in one chamber via the reaction of commercially available 1,1′-sulfonyldiimidazole (SDI), KF, and formic acid—a procedure that was previously developed to safely prepare aryl fluorosulfonates from phenols [145]. Subsequently, the SO2F2 gas is allowed to mix in a separate chamber with TMAF and the corresponding aromatic aldehyde substrate in DMF at room temperature (Scheme 16a, method A).
To begin their optimization studies, the authors selected the deoxyfluorination reaction of 4-bromobenzaldehyde (256) as a model system using their previous conditions to deoxyfluorinate phenols (1.5 equiv of SO2F2, 3 equiv of TMAF in DMF at 25 °C for 24 h) (Scheme 16b). In this task, the authors initially observed that the TMAF-SO2F2 two-chamber-based protocol rendered comparable or substantially increased deoxyfluorination yields (68% NMR yield for the model system) compared to their initial one-pot deoxyfluorination protocol (56%) that mixed directly TMAF and SO2F2. Additionally, an increase in the initial amounts of TMAF and SO2F2 used (from 3 equiv of TMAF and 1.5 equiv of SO2F2 to 4 equiv and 2 equiv, respectively), elevated the deoxyfluorination NMR yield to 91%, which could be maintained (90%) when the reaction time was reduced from 24 h to just 4 h.
Importantly, the authors demonstrated that this transformation must be conducted in strictly anhydrous conditions to preserve the reactivity of TMAF (e.g. the NMR yield drops from 90% to 42% if the reaction is set up in benchtop conditions instead of in the glovebox). Moreover, the difluoromethylated end-products can be easily purified by flash column chromatography without significant drops in yield (e.g. 80% isolated yield for the model substrate at 1 mmol scale).
Next, the authors investigated the synthetic scope of their optimized method for the deoxyfluorination reaction of aldehydes (Scheme 16c). In general, ortho- (e.g. products 261–264) and para-substituted (e.g. products 257–260, 269–272) aromatic aldehydes underwent deoxyfluorination in moderate to excellent yields. However, it is not possible to establish a direct formal comparison between the relationship of both patterns of aryl substitution and the reaction yield by merely looking at the reported examples, except for the phenyl-substituted benzaldehyde (forming 260 para-: 66% vs. forming 263 meta-: 72%).
Notably, the steric hindrance inherent to a large group, such as isopropyl, placed at the ortho-position in substrate 262 was not deleterious for the deoxyfluorination yield. Moreover, two meta-substituted aromatic aldehyde substrates (forming 273, 281) underwent deoxyfluorination in acceptable to good yields. Furthermore, bromide, iodide, and chloride substituents in the aromatic ring, which may provide a synthetic handle for further derivatization, were all compatible with the deoxyfluorination conditions and furnished the corresponding difluoromethylated end-products (257–259) in acceptable to good yields. Remarkably, aromatic aldehydes bearing strong electron-withdrawing substituents were those substrates providing better deoxyfluorination yields (e.g. products 266, 267), which in general slightly decreased for electron-neutral and moderately electron-rich substrates (e.g. forming 260–265). Importantly, carboxaldehydes of heterocycles such as pyridine (e.g. products 277–282) and (iso)quinoline (products 283–285), bearing either electron-donating or electron-withdrawing functional groups, also underwent smooth deoxyfluorination.
Scheme 16.
Sanford’s synthetic methodologies for the deoxyfluorination of aldehydes and α-ketoesters: (a) Methods, (b) Method A. Optimization [142], (c) Synthetic scope [142,143], and (d) Proposed mechanism [143]).

In aromatic substrates containing a second carbonyl-based functional group (e.g. those forming 267, 269), the deoxyfluorination reaction proceeded at the more electrophilic aldehyde group with high site-selectivity.
A brief exploration of other types of carbonyl groups for this reaction such as aromatic (e.g. acetophenone, benzophenone) or aliphatic ketones (i.e. cyclohexanone) was completely unsuccessful to provide rather than traces of the corresponding difluoromethylated products. The authors ascribed these results to either competitive enolate formation promoted by the high basicity of TMAF in the cases of acetophenone and cyclohexanone, or to the poor electrophilicity of the carbonyl group in benzophenone in relation to aromatic aldehydes.
Conversely, α-ketocarboxylic esters proved to be electrophilic enough to undergo the deoxyfluorination reaction under the optimized protocol to afford the corresponding α-gem-difluoro ester products, 286 and 287, in good yields. Finally, to further demonstrate the potential of this method in pharmaceutical and industrial settings, the proficiencies of this method and DAST were compared for the deoxyfluorination of several aromatic aldehydes and α-ketoesters. In that task, Sanford’s deoxyfluorination method provided comparable or substantially higher yields than the use of DAST, faring particularly well with ortho-substituted substrates (e.g. to form 261-264), pyridine and quinoline carboxaldehydes (e.g. to form 277, 281, and 285), and α-ketoesters (to give 286, 287).
The authors disclosed a simplified mechanistic scenario (Scheme 16d), where TMAF first attacks the aromatic aldehyde 288 giving rise to the tetramethylammonium salt of a fluorinated aromatic alkoxide 289, that then attacks SO2F2 to afford the corresponding fluorinated aryl fluorosulfonate intermediate 290. Then, 290 must undergo a second fluorination event with another equivalent of TMAF through an analogous pathway to the previously described fluorination of phenols via aryl fluorosulfonates [111,139].
The authors argued that their TMAF-SO2F2-based method has great potential for the deoxyfluorination of (hetero)aryl aldehydes on the process scale due to the relatively low cost of these reagents. However, they also inferred that the implementation of this method at laboratory scale may be limited by safety concerns related to the toxicity profile of SO2F2. Consequently, the second approach reported by the Sanford group on the deoxyfluorination of (hetero)aryl aldehydes dealt with the search of suitable liquid sulfur-based reagents that may replace SO2F2 gas as the electrophile that reacts with TMAF at room temperature, thus improving the possible applicability at the laboratory scale [143]. Based on their previous mechanistic proposal for this transformation, Sanford and co-workers devised that such liquid sulfur-based electrophiles should simultaneously contain a good leaving group and a strong electron-withdrawing group to enhance the electrophilicity—both attached to sulfur.
Two reagents were deemed suitable for this task, perfluorobutanesulfonyl fluoride (PBSF) (Scheme 16a, method B) and trifluoromethanesulfonic anhydride (Tf2O) (Scheme 16a, method C). In practice, neither of the two methods provided better yields for the deoxyfluorination of (hetero)aryl aldehydes at room temperature than the previous TMAF-SO2F2 two-chamber-based protocol even by extending the reaction time to 24h. However, the use of the TMAF–PBSF system provided nearly comparable yields to those obtained via TMAF–SO2F2 for the deoxyfluorination reaction of some electron-deficient aromatic aldehydes (e.g. to give 266, 268), and the TMAF-Tf2O system fared better with electron-neutral substrates (e.g. 265, 272, 275).
3.4. C(sp3)-F Bond Formation with TMAF
As it was previously discussed (Section 2.2), the basicity of TMAF is in the range of superbases (e.g. NaH or PhLi) in most organic solvents [74]—especially in those polar aprotic solvents such as DMSO and DMF, which are polar enough to solubilize significant amounts of TMAF and simultaneously lack the ability to establish strong hydrogen bonds with the fluoride anion that may attenuate its basicity. As a result, there are not many examples involving the use of TMAF in nucleophilic aliphatic substitution reactions leading to the formation of C(sp3)-F bonds, since in most cases, it may generate significant amounts of E2 side products.
However, the syntheses of [18F]-labeled radiopharmaceuticals constitutes a remarkable exception to this rule. In practice, the short life-time of [18F] isotopes (t1/2 = 109.8 min [2,3,4,5]) calls for the development of rapid fluorination methods enabling quick in situ access to the targeted [18F]-pharmaceuticals, which should ideally proceed at an ambient temperature to avoid the denaturalization or degradation of potentially desirable biomolecular targets such as peptides and proteins [4]. Therefore, the considerable fluoride nucleophilicity of TMAF seems to ideally fit that purpose, and thus unsurprisingly, it has been exploited to develop fast fluorination methods that can proceed upon lower temperatures than other fluorination methods commonly used in this field, such as those based on the use of K[1 8F] complexed with the aminopolyether cryptand Kryptofix 2.2.2 (K222).
The first use of TMAF in an aliphatic substitution fluorination reaction was reported in the context of the synthesis of 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG or 18F-FDG) by Tewson in 1983 [146] (Scheme 17a). This is the glucose analogue with the hydroxyl at the C-2 position replaced by the [18F]-radioisotope and is frequently used in positron emission tomography (PET) diagnostics to monitor the uptake and distribution of glucose in metabolically active tissue in the brain or cancer cells [147]. At that time, the usual synthetic protocol of [18F]FDG involved electrophilic fluorination of the so-called triacetal glucal with [18F]F2 gas [148], which was deemed inefficient with respect to fluorine due to hydrolysis of half of the obtained fluorinated products [146]. To overcome this issue, the authors devised that the development of a fluorination method based on the nucleophilic aliphatic substitution at the 2-position of hexoses may be highly convenient. For this purpose, choosing suitable substrates with the right leaving groups was critical to avoid side reactions and control the stereochemistry of the reaction. To that end, the author selected cyclic sulfur esters such as 2,3-O,O-sulfites and 2,3-O,O-sulfate as substrates, which were known to favor the desired attack with fluoride at the carbon rather than at the oxygen and sulfur atoms (e.g. with Grignard reagents). In mechanistic terms, such a process is related to the formation of aryl fluorides from bis-sulfates discussed years later in Sanford’s mechanistic studies for the deoxyfluorination of phenols (Section 3.3.2, [139]). Importantly, all fluorination reactions were conducted using TMAF tetrahydrate dried in a rotary evaporator with MeCN, which thus contained significant amounts of water. Such a protocol had been previously successfully implemented for the preparation of [18F]-tetraalkylammonium salts [149].
In practice, while the reaction of the selected β-D-mannopyranoside 2,3-O,O-sulfite substrate 293 (TMAF in MeCN under reflux for 1h) led to the exclusive formation of the corresponding diol 294 due to the water present in the reagent, the analogous β-D-mannopyranoside 2,3-O,O-sulfate substrate 295 underwent clean fluorination with full chirality transfer after just 5 min of reaction under reflux. This reaction was remarkably insensitive to the addition of external water. Subsequent hydrolysis followed by acylation provided the desired peracetylated 2-deoxy-2-fluoro-β-D-glucopyranoside 297 in 84% isolated yield over three steps.
A few years later, a more detailed study on the use of cyclic sulfates as substrates for nucleophilic aliphatic substitution reactions was also described by Tewson and co-workers [150] (Scheme 17a), which allowed extending this methodology to the fluorination and [18F]-radiolabelling of other substrates such as 5-, 6-, and 7-membered ring sultones (298–301) and cyclic sulfate derivatives of the estrogen hormone epiestriol (302–303) in excellent yields [150,151]. Notably, the presence of acetyl and allyl hydroxyl protecting groups in the epiestriol analogues was compatible with the reaction conditions. Afterwards, van Lier and colleagues proved that the methoxymethyl ether (MOM) protecting group is also compatible with Tewson’s reaction conditions [152]. However, attempts to fluorinate protected α- and β–D-glucopyranosides 304–305 under Tewson’s conditions failed to deliver any of the expected products [150].
Considering that the difference between the substrate previously successfully fluorinated (i.e. β-D-mannopyranoside 295) [146], and the substrate that did not react under the same conditions (i.e. β-D-glucopyranoside 305) [150], only lies in the relative stereochemistry of the leaving group at C-2, these results highlight the importance of stereoelectronic effects on nucleophilic fluorination reactions in complex substrates such as carbohydrates.
Scheme 17.
Radiopharmaceutical nucleophilic aliphatic fluorination reactions using TMAF: (a) Cyclic sulfates as leaving group, and (b) Triflate as leaving group.
Scheme 17.
Radiopharmaceutical nucleophilic aliphatic fluorination reactions using TMAF: (a) Cyclic sulfates as leaving group, and (b) Triflate as leaving group.

Finally, Kuwabara and co-workers reported that this protocol can also be applied for the preparation of 6-deoxy-6-[18F]fluoro-L-ascorbic acid ([18F]-DFA, 307) [153], although without indicating important experimental details such as the reaction yield and the precise water content and type of the employed [18F]-TMAF.
Along those lines, Kojima and co-workers described an improved TMAF-based protocol that could be adapted for the synthesis of [18F]-FDG (Scheme 17b) [154]. It involves replacing the cyclic sulfate by triflate as the leaving group at the C-2 position in a suitably protected β-D-mannopyranoside substrate (308). The authors ascribed this reaction improvement to the optimal trans-diequatorial relative disposition of the leaving group at C-2 and the vicinal hydrogen that precludes the possibility of an elimination side reaction. Effectively, treatment of 308 with TMAF in MeCN under reflux for 20 min afforded the corresponding fluorinated product 309 in 91% isolated yield. The same authors had reported that TMAF and tetraethylammonium fluoride (TEAF) are the most suitable sources of nucleophilic fluoride to convert protected glucopyranosides analogues (310–311) into the corresponding fluorinated mannopyranoside products (312–313) in good yields (Scheme 17b) [155]. Importantly, the authors found that acetyl protecting groups at the neighboring C-3 position are cleaved upon the reaction conditions due to TMAF basicity.
Conversely, other hydroxyl protecting groups at that position commonly used in carbohydrate chemistry (e.g. benzyl, methyl ether) may tolerate such reaction conditions. Considering the sensitivity of TMAF to water, and the lack of experimental details concerning the source of TMAF, its content of water, and the amounts used, it cannot be ruled out that further reaction optimization may provide higher yields for these transformations. However, these results already supersede those obtained by Tewson for the fluorination of glucopyranosides [146,150,151,152], and prove that triflate is a better leaving group than cyclic sulfates for the selective nucleophilic fluorination of carbohydrates.
Up to date, there are two reports outside the radiopharmaceutical field describing the use of TMAF in nucleophilic aliphatic fluorination reactions. First, Grygorenko and co-workers have recently described an isolated example for the use of mesylate as a leaving group for the TMAF-based nucleophilic fluorination of a 1,2-disubstituted cyclobutane (314) (Scheme 18) [156]. This transformation renders the desired fluorinated cyclobutane product (315) in 69% yield, and proceeds with just 1.3 equiv of TMAF under reflux for 18 h in t-BuOH. In this manner, the authors managed to attenuate the basicity of TMAF, and thus preclude side reactions such as elimination reactions, or the opening of the relatively strained cyclobutane ring.
Secondly, the only example of a general synthetic methodology based on the use of TMAF for the formation of C(sp3)-F bonds was reported in 2020 by Guo, Chen, and co-workers [157]. This transformation concerns the one-pot deoxyfluorination of aliphatic alcohols into alkyl fluorides via fluoroformate intermediates formed by the thermal decomposition of perfluoroalkyl ether esters (PFECAs) (Scheme 18). These substances have been identified as contaminants of emerging concern due to their toxicity and potential bioaccumulation in living organisms and are generated in relatively large amounts at the process scale as side products in the industrial production of hexafluoropropene oxide (HFPO)—a versatile building block for the synthesis of organofluorine compounds and polymers. On this basis, the authors argued that this may constitute a convenient methodology to upcycle a hazardous waste substance such as PFECAs (316) by transforming it into valuable fluorinated commodity chemicals such as aryl fluorides.
However, the thermal decomposition of PFECAs precursors generates at least one equivalent or more of carbon dioxide, a fact that obstructs the implementation of this synthetic methodology at process scale. Moreover, this synthetic method is inspired by the report of Olofson and colleagues concerning the reaction of aliphatic alcohols with the carbonyl fluoride (COF2) formed in situ via reaction of triphosgene and a large excess of KF in presence of catalytic amounts of 18-crown-6-ether at 0 °C. In this manner, fluoroformate products are afforded that then can be thermally converted at 120–125 °C into the corresponding alkyl fluorides with hexabutylguanidinium fluoride (HBGF) as the catalyst (Scheme 18) [158].
However, the fluoroformate intermediates in Olofson’s report require purification prior to further fluorination (Scheme 18). To address this, Guo and Chen conceived that alkyl alcohols could directly react with the COF2 generated by the thermal decomposition of PFECAs, and then the resulting fluoroformates may be further fluorinated in a one-pot manner with a nucleophilic source of fluoride. For this task, optimization studies found that the treatment of 4-phenyl-1-butanol with the combination of a standard PFECAs, namely N,N-dimethylpropyleneurea (DMPU), as solvent and substoichiometric amounts of TMAF (0.5 equiv) furnished the desired fluorinated product 317 in more than 90% yield after 5 h of reaction at 130 °C. This screening revealed that the obtained reaction yields are rather uniform, independently of the used PFECAs, and also that the formation of alkyl fluorides is accompanied by minimal amounts of trifluoromethoxylated products.
Scheme 18.
TMAF usage in traditional synthetic nucleophilic aliphatic fluorination reactions.

With the optimized conditions in hand, the authors proceeded to explore the synthetic scope of this method (Scheme 18). In this task, they found that a diverse group of primary alcohols (317–320), benzylic primary alcohols (322–328), and heterocyclic scaffolds with pendant alkylic primary hydroxyl groups (330–336) all underwent deoxyfluorination in good to excellent yields. Furthermore, the reaction conditions tolerated a broad series of aryl substituents such as cyano (321, 338), ester (337), methoxy (325), methylthio (326), sulfone (328), iodo (323), bromo (324), chloro (333), and vinyl (327) as well as an internal alkyne (329) and a sulfonamide group (335). Additionally, the deoxyfluorination of 4-iodobenzyl alcohol (323) was successfully conducted at gram scale in 81% yield, as a proof-of-principle of the potential viability of this transformation at process scale. Remarkably, a diol substrate could be efficiently deoxyfluorinated to afford the corresponding difluorinated end-product (336) in 79% yield.
Next, the authors examined the suitability of this method to deoxyfluorinate alkyl secondary and tertiary alcohols. First, although the reactivity slightly decreased for alkyl and benzylic secondary alcohols, the corresponding fluorinated end-products (337–342) were obtained in moderate to excellent yields, except for fluorinated steroid 343. On the contrary, the reactivity remarkably decreased for cyclic aliphatic secondary alcohols and alkyl tertiary alcohols which only furnished traces of the desired fluorinated end-products 345–347, with the exception of 1-adamantanol that remarkably was converted into 1-fluoroadamantane (344) in 93% yield. The authors attributed those negative results to the fact that the process may follow a typical SN2 mechanism. To confirm this mechanistic scenario, the authors evaluated the deoxyfluorination of a borderline secondary alkyl alcohol such as (S)-4-phenyl-2-butanol (348). Effectively, (R)-(3-fluorobutyl)benzene (341) was obtained in an isolated yield of 81% with full inversion of the configuration (>99% ee), indicating that, indeed, the process proceeds through a typical SN2 mechanism. Ultimately, the authors also demonstrated the generation of COF2 via the thermal decomposition of PFECAs precursors.
3.5. Other Types of C-F Bond Formation with TMAF
3.5.1. Nucleophilic Additions to C=X bonds with TMAF
Formally, Sanford’s method to deoxyfluorinate aromatic aldehydes and α-ketoesters substrates (Section 3.3.3, Scheme 16) [142,143] constitutes the only example in the literature involving the use of TMAF for the nucleophilic addition of fluorine to multiple C=X bonds in organic molecules. Conversely, there is a larger number of examples of such a transformation in inorganic molecules. For instance, the Seppelt group described in 1995 that anhydrous TMAF—and other ionic sources of “naked” fluoride, but not inorganic salts such as CsF or AgF—reacts spontaneously with carbon dioxide, both without solvent or dissolved in CH2F2 and CF3CHFCF3, to generate fluorocarbonate insoluble solid salts ([FCO2−]) 349 (Scheme 19a) [159].
Although the existence of the fluorocarbonate anion had only been previously postulated without conclusive experimental confirmation [160], the authors experimentally corroborated the identity of such salts by NMR, IR, and Raman spectroscopy. Furthermore, ab initio calculations indicated that the C-F bond in FCO2– is very weak (ΔH = −110 kJ/mol, d(C-F) = 144.6 pm for ([FCO2−]) (e.g. vs. 139.9 pm in CH3F or 134.6 pm for typical C(sp2)–F bonds), as a result it behaves more like a fluoride anion solvated in carbon dioxide rather than as an ionic salt (sic) [159]. Indeed, careful experimental observation confirmed that in the presence of traces of moisture these salts spontaneously decompose into hydrogen carbonate, hydrogen fluoride, and gaseous carbon dioxide. Furthermore, protic solvents such as alcohols also immediately cleave the C-F bond with the evolution of carbon dioxide and solvation of the fluoride anion. Additionally, MeCN solutions of fluorocarbonate slowly release the fluoride anion after prolonged times, which further causes slow deprotonation and dimerization of that solvent (Section 2.2.2, [55]).
Perhaps as a result of this instability, fluorocarbonate salts have not yet found application in synthesis or catalysis, with the exception of the isolated reaction reported by Rüdiger and Seppelt between anhydrous TMAF, carbon dioxide, and 1,1,1,2,3,3,3-heptafluoropropane, which, after several days at 80 °C, led to the formation of the tetramethylammonium salt of hexafluoroisobutyric acid 350 (Scheme 19a) [161].
Along those lines, the Seppelt group also studied the reaction of TMAF with other inorganic molecules bearing C=X bonds such as carbon disulfide (CS2) (Scheme 19b) [161,162]. Such process involves the nucleophilic addition of fluoride to the electrophilic carbon at CS2 to form the dithiofluorocarbonate anion ([FCS2−]) 351. Unfortunately, there are not reported details concerning the isolation and characterization of such anion in the literature. However, the Seppelt group described its generation in situ and reactivity towards other species during their studies. For instance, a mixture of TMAF, an excess of CS2, and a deactivated fluorinated secondary amine (i.e. bis(2,2,2-trifluoroethyl)amine) was allowed to warm up from –196 °C to room temperature during the course of 3 h, and then it was maintained at 60 °C for 1 h to yield the tetramethylammonium salt of the N,N’-bis(2,2,2-trifluoroethyl)dithiocarbamate anion 352. The authors did not mention the yield of the reaction. This salt only can be formed after in situ deprotonation of the secondary amine by the FCS2–anion [161].
Scheme 19.
TMAF usage in nucleophilic addition reactions to C=X bonds: (a) CO2, Seppelt, [159,161], (b) CS2, Seppelt, [161,162], and (c) Cl2C=S, Clark, [164,166].

The same year, Seppelt and co-workers also reported that the analogous reaction of TMAF with an excess of CS2, in the presence of pentafluorobenzonitrile (F5PhCN), with DMF as solvent, eventually delivers a mixture of trifluoromethylthiolated compounds in low yield (17%) after 3 days at room temperature (Scheme 19b). Despite the fact that this transformation requires considerable optimization to acquire any synthetic applicability, interestingly, the major component of the mixture is 353, where the fluoride in a meta-position in relation to the nitrile is the substituent that has actually been replaced by the trifluoromethylthiolate group (SCF3) [162]. The authors noted that this transformation also proceeds with only catalytic amounts of TMAF due to the fact that the starting material may also act as a source of fluoride anions.
Thiophosgene (Cl2C=S) is another inorganic molecule whose reactivity with ionic sources of fluoride was studied in the 1990s as a potential route to the trifluoromethylthiolate anion (SCF3−).
The incorporation of this fragment into organic molecules has a prime interest in medicinal chemistry due to the high lipophilicity and membrane permeability that is able to induce in potential drugs (e.g. the Hansch constant that quantifies lipophilicity is 1.44 for SCF3 vs. 0.88 for CF3) [163]. In this sense, the Clark group reported in 1997 that Cl2C=S reacts reversibly with an excess of KF (6 equiv) in MeCN at -15 °C to generate KSCF3 354 [164], which is intrinsically unstable at room temperature and exists in equilibrium with thiocarbonyl fluoride 355 and KF (Scheme 19c) [165].
However, the trifluoromethylthiolate anion can render in situ the nucleophilic aromatic substitution reaction of electron-deficient fluoro- and chloro-arene substrates (with two or more activating groups). Such processes proceed in moderate to good yields under reaction conditions mild enough to tolerate the presence of one additional substituent on the arene ring such as nitro (e.g. 357, 358), cyano (362), trifluoromethyl (360), and furazane (361). In particular, aryl fluoride substrates provided better results than more activated aryl chlorides, presumably because the halide groups may react with further thiophosgene molecules. Importantly, the authors observed a significant drop in the trifluoromethylthiolation yields when this process was started at temperatures higher than 0 °C, as a result of the formation of a series of SCF3-containing side products, which were also observed during the reaction of relatively unreactive substrates. The authors managed to overcome this limitation for the relatively unreactive 2-chloro-5-nitrobenzonitrile by increasing the amounts of KF used and raising the temperature to reflux during the reaction. These two improvements raised tenfold the yield of the corresponding trifluoromethylthiolated product 362.
Two years later, the Clark group also published the replacement of KF by TMAF for the generation of the trifluoromethylthiolate anion from thiophosgene, and its use on identical halex reactions (Scheme 19 c) [166]. To that end, the amounts of TMAF used and the temperature must be decreased (from 6 equiv to 3 equiv of anhydrous TMAF, and from -15 to -40 °C) to conduct the reaction with thiophosgene and generate the trifluoromethylthiolate anion without side products. Upon these reaction conditions, the authors demonstrated that the trifluoromethylthiolation of a small set of electron-deficient fluoro- and chloro-arene substrates proceeded in slightly higher yields than using KF (e.g. formation of 356, 357), with the exception of the furazane-containing substrate 361 and the relatively inactivated 2-chloro-5-nitrobenzonitrile 362. Finally, Clark’s TMAF-based method for the generation of the trifluoromethylthiolate anion has been superseded in recent years by other TMAF-based synthetic procedures that imply the use of less toxic, more atom-economical and sustainable starting materials than thiophosgene such as for instance elemental sulfur and the Ruppert-Prakash reagent [167,168].
3.5.2. Nucleophilic Addition to Diaryliodonium Salts with TMAF
As it was mentioned before (Section 3.2), the importance of fluorinated (hetero)arene scaffolds in drug discovery, medical imaging diagnostics, and agrochemical applications has propelled the investigation of novel synthetic methodologies for the formation of C(sp2)-F bonds that may address the limitations underlying the traditional procedures for nucleophilic aromatic fluorination reactions.
In this context, DiMagno and co-workers reported in 2010 that the similarities between the iodonium ion and late transition metal chemistries could be exploited to develop a rapid method providing access to otherwise unattainable electron-rich and meta-substituted inactivated (hetero)arene fluorides with prospective applicability as [18F]-radiolabelled imaging PET agents [169]. Such method involves the fluorination of diaryliodonium salts (363–371) with TMAF, which then may thermally undergo reductive elimination to generate the targeted aryl fluorides (373–381) (Scheme 20) [170].
However, the prospective radiopharmaceutical application of this method required the authors to conduct the reaction very rapidly at high temperatures. To that end, the selection of solvent was critical, especially when the authors had observed in previous studies that difluorinated phenyliodonium (Ph(I)F2) species undergo spontaneous reduction to PhI and HF2− in the presence of an excess of the fluoride anion in MeCN. The authors noted that this side reaction can be suppressed by using an aprotic non-polar solvent, such as benzene, to conduct the thermal decomposition of the intermediate fluorinated diaryliodonium salts 372.
However, benzene is not a suitable solvent for the initial fluorination of diaryliodonium salts since it barely dissolves the TMAF that is needed. Hence, the authors conducted the whole process sequentially (Scheme 20), first performing the fluorination with TMAF in deuterated MeCN at room temperature to minimize that side reaction. Then, upon completion of the fluorination reaction, the solvent was evaporated from the reaction mixture. Next, they dissolved the crude product in deuterated benzene and subsequently filtered off the precipitated inorganics. Finally, they reacted the resulting fluorinated diaryliodonium salt (372) in deuterated benzene at 140 °C for 15 min. Unfortunately, the authors did not provide information about the screening of other solvents (e.g. DMF) that perhaps may simultaneously solubilize TMAF, minimize the undesired reduction to iodoarene and the bifluoride anion (Section 2.2.2), and suitably allow for conducting the whole process in a more rapid one-pot manner at lower temperatures. However, at a later stage, the authors stated that benzene can be replaced by the less hazardous toluene as the solvent for the reaction.
With that experimental setting developed, the authors proved the feasibility of accessing several electron-rich aryl fluoride end-products (373–381) from the corresponding diaryliodonium salt precursors (363–371) in moderate to good yields, in a rapid and convenient manner for radiolabelling applications. That set of aryl fluorides included suitably protected precursors of actively investigated radiotracers such as [18F]-6-fluorodopamine 380 and [18F]-2-fluoroestradiol 381, although the latter was produced in low yield.
However, this methodology possesses several serious limitations that may encourage future research. First, diaryliodonium salts are electrophilic arylating reagents that are traditionally obtained via oxidation of the corresponding iodoarene precursor followed by ligand exchange (often in acidic conditions) with a second molecule of arene or an organometallic reagent (Scheme 20) [171]. This need to prefunctionalize first the iodoarene precursors as diaryliodonium salts (i.e. three steps in the case of DiMagno’s report) coupled with the sequential nature of the aforementioned protocol compromises the atom economy, practicality, and cost-effectiveness of the whole approach. Secondly, the reductive elimination of fluorinated diaryliodonium salts (372) is accompanied by regioselectivity issues that are often difficult to suppress. As such, those undesired regioisomeric fluorinated side products add as impurities to the equivalent of 4-iodoanisole (382) that is formed by default at the end of the reaction (i.e. per equivalent generated of the desired aryl fluoride). In practice, the purification of such mixtures may be laborious and time-consuming, making lengthier the whole method and compromising its realistic implementation in the radiopharmaceutical field.
4. Use of TMAF as a Base
The primary application of ionic sources of fluoride in synthesis was originally the fluorination of small organic molecules. Hence, the seminal reports describing their use as basic reagents or catalysts were often serendipitous discoveries made accidentally while attempting to fluorinate a given target molecule. However, there is relatively rich literature on the use of ionic sources of fluoride as bases in synthesis and catalysis. Extensive reviews on this topic were published elsewhere by Clark in 1980 [172], and subsequently by others [173].
In this context, the distinct physicochemical properties of TMAF have increasingly attracted attention in recent years to render a diverse set of chemical transformations where it participates—either stoichiometrically or catalytically—(i) as a base, most importantly (ii) with a dual role as a basic and methylating or fluorinating reagent, or (iii) triggering the formation of the active species in a given chemical reaction. Overall, the number of examples of such processes remains relatively low; however, the fact that many of them are challenging or essentially new chemical reactions, unattainable by other means, definitely warrants further synthetic and catalytic research in this area.
In this section, we will cover the most important synthetic and mechanistic aspects of this promising class of TMAF transformations.
4.1. TMAF Participation in Classic Acid-Base Transformations
Classic acid–base processes involve the use of Brønsted bases with the suitable pKb to deprotonate the most acidic protons of a given molecule. Considering the plethora of relatively simple and readily available inorganic and organic substances that are amenable to cover this role in the entire spectrum of basicity, TMAF is a relatively expensive base that has traditionally been primed for the syntheses of high added-value fluorinated molecules, and its use as a Brønsted base is drastically limited to a few examples. Such reports do not necessarily always justify the reasons underlying the choice of TMAF as base, or accurately describe its preparation and degree of hydration. The omission of such key experimental details may seriously compromise the reproducibility of those transformations given the great variability on the reactivity of TMAF that is imparted by its degree of hydration.
To the best of our knowledge, the first report explicitly describing the use of TMAF as a basic reagent dates back to 2005 [174]. In that work, Ménand and Dalla demonstrated that catalytic amounts of TMAF promote the nucleophilic conjugate addition of N-based nucleophiles (e.g. oxazolidinone 384) to a small set of Michael acceptors featuring electron-withdrawing functional groups such as esters, aldehydes, and ketones (Scheme 21a).
This type of reactions, that is, the conjugate addition of a carbamate nucleophile to an unsaturated carboxylic acid derivative, provides a convenient entry to biologically relevant β-amino acid scaffolds, but is challenging due to the weak nucleophilicity of carbamates. In practice, this method involves very mild reaction conditions (0.2 equiv of TMAF, excess of the oxazolidinone nucleophile 384, MeCN, rt, 72-96h). However, the reaction yields dramatically dropped on a series of Michael acceptors such as crotonates and enoates when the electron-donating character of their substituents increased (e.g. the yield went from 90% for methyl crotonate to no reaction with ethyl crotonate). Conversely, the yield of the reaction did not experience the same variability when a series of chalcones—typically unreactive substrates towards the conjugate addition of carbamates—and enones (e.g. cyclohexanone) were evaluated as Michael acceptors, and the desired products (385) were afforded in moderate to good yields (55–82%).
Subsequently, the authors observed the same reactivity trend when better nucleophiles (i.e. thiols 387) were subjected to this transformation. In practice, thiols and thiophenols reacted smoothly, without the requirement of being added in excess, with a broader variety of Michael acceptors (386) containing electron-poor groups such as esters, ketones, aldehydes, and nitro moieties. These processes took place in substantially shorter times (1h instead of 96h) and excellent yields (60–100%), which even allowed for the reduction of the catalytic loading of TMAF (from 0.2 equiv to 0.02 equiv) without substantial losses in the reaction yield (Scheme 21a).
Acting as a base, TMAF is capable of promoting many other nucleophilic conjugate addition reactions. However, it is rather unusual to observe, in this class of reactions, TMAF being selected as the base of choice amongst the vast number of simpler and milder options available. The enantioselective organocatalytic conjugate addition of nitrophenylacetonitriles to enals and enones—reported by Ruano and co-workers in 2010 [175]—provides a good example of this principle. This process takes place in mild conditions, and several organocatalysts were able to promote moderate enantioselectivities. Furthermore, a weak inorganic base, such as LiOAc, was able to outperform TMAF in terms of yield, although the choice of base did not affect the enantioselectivities.
A specific example in which TMAF was the base of choice in a synthetic methodology was published by Behr and co-workers in 2019 (Scheme 21b) [176]. This report concerns the aza-Henry nitromethylation reaction of nitrones (389) to obtain β-nitrohydroxylamine products (390) that can be further converted into scaffolds containing valuable moieties such as vicinal diamines. To begin their studies, the authors selected nitromethane and the nitrone derivative 392 as the model reaction, by which they devised the feasibility of preparing the drug Praziquantel 404 (antiparasitic in the WHO list of essential medicines [177]). After a comprehensive screening of reaction conditions, the authors met three problems: (i) The bases typically used in previous reports failed to promote any reactivity at room temperature (e.g. NaOEt, NaOMe K2CO3), (ii) Henry and Aza–Henry reactions are known to be reversible [178], and (iii) the hydroxylamine moiety in the desired product is prone to undergo oxidation in basic conditions.
The authors addressed these issues by conducting the reaction in an inert atmosphere and utilizing nitromethane as a solvent to drive the equilibrium forward. Upon those conditions, the addition of either TMAF or the bicyclic guanidine base 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) in slight excess (1.6 equiv) to 392 at room temperature provided the best conversions and yields. However, the use of TMAF tetrahydrate failed to deliver any product, likely due to its poor solubility in neat nitromethane.
Scheme 21.
TMAF acting as a classic base: (a) Nucleophilic conjugate additions of carbamates and thiols to Michael acceptors [174], and (b) Aza-henry nitromethylation of nitrones [176]).

In the model substrate (392), the use of TMAF exhibited three advantages over TBD. Firstly, the reaction proceeded twice faster (i.e. 6 h were required with TBD to achieve 68% yield, compared to the 2.5 h required with TMAF to reach 74% yield). Secondly, the extension of the undesired retro-Aza–Henry side reaction increased after prolonged reaction times, and was more pronounced using TBD as the base. Finally, the use of TMAF in substoichiometric amounts (0.25 equiv) promoted twice the conversion than the use of TBD in a given reaction time.
In addition to the model tetrahydroisoquinoline nitrone (392), the synthetic scope of this process comprised acyclic nitrones (393–395), glyceraldehyde-derived nitrone 396, and cyclic nitrones (392, 397–400) that include carbohydrate-derived ones (i.e. 399 and 400). The use of TMAF and TBD delivered comparable results affording the desired products in moderate to good yields, except for acyclic nitrones (393-395), in which the competing formation of (bis)nitromethyl alkanes (391) significantly eroded the yields. This issue was partially minimized using TBD instead of TMAF. Remarkably, the reaction proceeded with excellent diastereoselectivities for the chiral carbohydrate-derived nitrones (399, 400). This result was consistent with previous studies and may be attributed to the stereoinduction imparted by the steric hindrance at the carbon adjacent to the reaction site. Finally, the authors implemented this Aza–Henry reaction in a synthetic route that allowed the synthesis of praziquantel (404) from the model substrate (392). Such synthetic sequence also involved the reduction of the nitro group in 401 followed by two acylation steps and proceeded in a competitive overall yield of 21%. Unfortunately, the authors did not provide further mechanistic insight into this methodology.
4.2. Novel Uses of TMAF as a Base
In the last two years, a couple of important synthetic methodologies have been reported that epitomize the full potential of harnessing the unique properties of TMAF to render novel chemical transformations. To the best of our knowledge, both processes are the first of their kind.
The first methodology was published in early 2020 by the Schoenebeck group and constitutes the first method where the prominent basic character of TMAF is merged with a special secondary role as a methylating reagent. In this unprecedented manner, the authors chemoselectively methylated several protic functional groups such as amides, alcohols, and thiols. Remarkably, this method was also competent to render the chemoselective methylation of some N-based heterocycles with widespread applicability in the medicinal chemistry arena (e.g. indoles, pyrroles, imidazoles) [179]. In that field, the methylation of protic functional groups is a pivotal strategy to tune the physicochemical, conformational, and protein-binding properties of therapeutically relevant molecules. As a result of this “magic methyl” effect, methyl groups attached to other carbon or protic functional groups are ubiquitous in approved and candidate drug molecules [180].
To begin their studies, Schoenebeck decided to focus first on secondary amides amongst all the functional groups amenable to be methylated, due to their prominent biological profile and pervasive presence in drug molecules. Additionally, the current synthetic methods to synthesize secondary amides either use toxic agents (e.g. MeI, MeOTf) or proceed in harsh reaction conditions with relatively low yields that often require a sophisticated catalyst limiting their implementation at process scale (e.g. methylation using sustainable feedstocks as sources of methyl such as formaldehyde or methanol). Conceptually, this work is based on previous mechanistic observations that recognized the ability of tetramethylammonium trifluoromethanethiolate (Me4N-SCF3) to transfer relatively rapidly methyl groups to aniline substrates in processes with a low activation barrier [181] (Scheme 22a).
The authors selected N-phenylacetamide (405) as model substrate for the initial evaluation of reaction conditions, and found that its methylation is optimal after 12 h of reaction with TMAF as a methyl source (2.5 equiv) in toluene at 100 °C (95% yield of 406, entry 1, Scheme 22b). Importantly, the use of TMAF as a methylating source is essential for the reactivity since other tetramethylammonium salts (e.g. TMACl and TMABr) failed to deliver any product (entries 7,8). This screening also underlined the great sensitivity of the reaction to the temperature, for instance, twice the time, 24 h, was needed to reach 93% yield at 80 °C, and no conversion was observed at room temperature (entries 2 and 3).
Scheme 22.
Schoenebeck’s chemoselective methylation [182]: (a) Mechanistic observation [181], (b) Model system for reaction development [179], (c) Synthetic scope [179], (d) Chemoselectivity experiments [179] and (e) Mechanistic DFT studies [179]).

Additionally, other solvents were not as optimal as toluene (e.g. 70% yield with DMF and 57% with MeCN, entries 5 and 6) with the exception of the polar solvent N-Methyl-2-Pyrrolidone (NMP), in which the methylated model amide 406 was obtained in 95% (entry 4). However, toluene was deemed as the solvent of choice due to the fact that the main byproduct of the reaction—Me3NHF—precipitates in toluene at room temperature, which facilitates straightforward purification of the crude reaction mixture by filtration through a small pad of silica.
Next, the authors demonstrated that this methodology is highly efficient to methylate a broad scope of secondary amide substrates upon the optimized conditions, although no mention was made of the possibility of methylating primary amide functional groups (Scheme 22c). In practice, aromatic amides (407–415) including pyridine amides (410, 411), aliphatic cyclic amides (416–418), and a substrate featuring a pharmaceutically relevant sulfonamide moiety (419), all could be methylated in moderate to excellent yields. Remarkably, 3-methylindolin-2-one (417) underwent methylation at both N-1 and C-3 positions, a strong indication that this method can directly methylate acidic enough C-H bonds. Furthermore, as a proof of concept of the proficiency of the method to potentially render late-stage methylation reactions in drug discovery programs, substrates with an important pharmaceutical role were methylated, such as the anesthetic lidocaine (420), thalidomide (421), and the antitumoral enzalutamide (422), in moderate to excellent yields and site-selectivities.
Additionally, the scalability of the reaction was demonstrated by conducting the methylation of the secondary amide 408 at 7.0 mmol scale without significant losses of yield (88% at 7.0 mmol scale vs 94% yield at 0.4 mmol scale). Remarkably, the authors further demonstrated the generality of this method by methylating an impressive and diverse set of functional groups other than secondary amides such as indoles (at the N-1 position) (423–434), pyrroles (435, 436), imidazoles (437), alcohols (438–440), and thiols (441, 442) in excellent yields. In relation to indole substrates, a significant variety of functional groups (e.g. ester, aldehyde, cyano, bromo, iodo, fluoro)—placed at most positions of their structure—tolerated the methylation conditions without noticeable drops in yield.
Foremost, perhaps the most impressive attribute of this method is the chemoselectivities in which secondary amides (443, 444) and indoles (445, 446) can be methylated with excellent yields in the presence of other potentially reactive groups such as primary anilines and alkyl amines. This site-selectivity was also displayed for alcohol substrates (447, 448) with aromatic and aliphatic primary amines. In comparison, the site-selective methylation of a secondary amide group in a substrate such as 444 would require a three-step sequence by known synthetic methods due to the presence of the aromatic amine moiety [183].
Finally, the authors carried out DFT studies to shed light on the mechanism of this novel transformation (Scheme 22e). These calculations clearly excluded a mechanistic scenario analogous to the methylation of aromatic amines with Me4NSCF3 where the methyl group is directly transferred from the tetramethylammonium cation to the secondary amide without participation of the counterion—the free energy activation barrier for that process was estimated to exceed 45 kcal/mol.
Considering the basicity of TMAF and the fact that the initial screening clearly demonstrated its key role in the reaction, the authors postulated a concerted pathway where the amide is concomitantly deprotonated by the fluoride and the methyl is transferred from the tetramethylammonium cation—the computed free energy activation barrier for that process was 30.9 kcal/mol (TS1, Scheme 22e). Additionally, the authors also considered that the secondary amide could tautomerize to the corresponding imidic acid (only 3.4 kcal/mol higher in energy than the amide), finding that the free energy activation associated with that process was 25.8 kcal/mol (TS2). That is, the concerted methylation/deprotonation pathway is favored to proceed via the imidic acid tautomer in 5.1 kcal/mol over the pathway taking place directly from the amide.
Finally, these calculations also revealed that the exquisite chemoselectivity in which this method methylates secondary amides in the presence of primary amine sites (e.g. in N-(4-aminophenyl)acetamide) is due to the fact that the free energy activation barrier for the methylation of amines (TS3, Scheme 22e) is 8 kcal/mol higher than the one to methylate secondary amides (TS2, Scheme 22e).
During the preparation of this review, König’s group published a second report where TMAF displays a critical role as a base beyond the classical definition of the term [184]. This work details the unprecedented photo-catalyzed radical ipso-borylation of relatively stable aromatic bonds such as C(sp2)-F, C(sp2)-S, C(sp2)-O, and C(sp2)-N. The generated C(sp2)-B bonds constitute a quintessential functional group in organic synthesis to swiftly increase molecular complexity through cross-coupling C-C bond-forming reactions [185], and have also revealed promising potential in drug discovery and material sciences [186,187,188].
In recent years, photocatalytic methods have emerged as a powerful tool that complements and further expands the capabilities of traditional metal-based methodologies to access C(sp2)-B bonds [189]. However, the majority of these photocatalyzed borylation methods rely on substrates with low reduction potentials and/or featuring labile redox-active bonds, such as aryldiazonium salts, arylammonium salts, aryl (pseudo)halides, or redox-active esters. While the use of this type of prefunctionalized substrates facilitates generating the active radical species on the reaction media upon mild visible-light irradiation, high redox potentials and more forcing conditions are required for radical generation from substrates containing the relatively chemically stable C(sp2)-F, C(sp2)-S, C(sp2)-O, or C(sp2)-N bonds covered in this study. In this work, the authors addressed that challenge by harnessing the high reducing potentials arising from the photoexcitation of anionic species [190].
To begin their studies, the authors selected the defluoroborylation of fluorobenzene (449) with bis(catecholato)diboron (B2pin2) (2.0 equiv) as the model system with a series of readily available sulfur-based anions as prospective photocatalysts (0.3 equiv), an approach that they had already followed in previous studies [191,192]. Indeed, the authors found in this screening that light irradiation and a photocatalyst are mandatory for the reaction to proceed, achieving the optimum conditions with pyridine-2-thiolate (2-PySNa) operating at 385–390 nm (Scheme 23). Moreover, the authors noticed that the use of a slight excess of base (2.0 equiv) is also key for the reaction to progress. In this role, ionic sources of fluoride superseded inorganic bases, such as cesium carbonate or cesium acetate, and amongst them, TMAF provided the best results by far, a fact attributed to its superior solubility in the reaction solvent—MeCN (e.g. 19% NMR yield with KF, 56% with CsF, and 74% with TMAF).
With suitable working conditions in hand, the authors next investigated the range of functional groups in the arene substrates that could act as suitable radical precursors (Scheme 23a). To that end, the photocatalytic borylation of chlorobenzene (450) and bromobenzene (451) proceeded in good yields (74% and 78%, respectively), both of which with high redox potentials. Then, König and co-workers set their attention on extending their method to different phenolic derivatives due to the increasing importance of these compounds as versatile and sustainable building blocks directly obtainable from biomass [135]. Moreover, up to date only a few synthetic methods are capable of effecting the radical borylation of non-activated aryl esters via C-O bond cleavage [193,194], apart from reactive aryl sulfonate substrates (e.g. aryl triflates, aryl mesylates). In this task, O-Boc (452, 65%), sulfamate (453, 51%), triflate (454, 60%), phosphate (455, 48%), and carbamate (456, 46%) phenolic derivatives all underwent radical borylation smoothly. Additionally, aryltrimethylammonium halide salts were also successfully borylated (e.g. 458, 76%). However, this photocatalytic borylation method provided variable results on aryl substrates with sulfur-based functionalities as leaving groups.
Scheme 23.
König’s photocatalytic ipso-borylation of relatively stable (hetero)arenes with the key participation of TMAF: (a) Scope of leaving groups for the generation of phenyl radicals, (b) Synthetic scope, (c) Selected control experiments, and (d) Mechanistic proposal.
Scheme 23.
König’s photocatalytic ipso-borylation of relatively stable (hetero)arenes with the key participation of TMAF: (a) Scope of leaving groups for the generation of phenyl radicals, (b) Synthetic scope, (c) Selected control experiments, and (d) Mechanistic proposal.

Whilst diphenyl sulfone (460), which has a relatively high redox potential, provided the desired borylated product in excellent yield (96%), the reaction yields dropped for alkyl phenyl sulfones (461 and 462, 38% and 32%, respectively) and vinyl phenyl sulfone (463, 30%).
The same reactivity pattern was observed for the borylation of aryl sulfoxides and aryl sulfides, that is the borylation proceeds with lowest efficiencies when there is one alkyl group attached to the sulfur center (e.g. diphenyl sulfoxide 464 and diphenyl sulfide 466 were borylated in 61% and 70% yields, respectively, but their methyl phenyl counterparts 465 and 468 were borylated in 23% and 13% yields, respectively). The authors attributed that trend to the increased negative reduction potentials imparted by the augmented electron donation caused by the alkyl groups.
Further examination of the scope of this photocatalytic reaction revealed that a broad set of para-substituted (e.g. 469–479, 34–80% yield) aryl fluorides could be smoothly borylated to furnish the desired boronic ester products in moderate to good yields (Scheme 23b). Remarkably, that set included substrates with para-substituting electron-donating groups (e.g. OMe, 471, 80%, and piperidinyl, 475, 62%), electron-neutral (e.g. ethyl, 469, 68%, and benzyl, 470, 62%), functionalities with acidic protons (e.g. NH2, 474, 52%), or heteroaromatic substituents (e.g. pyrrole, 476, 52%, and pyrazole, 477, 49%). The authors stated that the yield dropped for para-substituted aryl fluorides with electron-withdrawing functional groups (i.e. CN, 479, 56%). Concerning ortho-substituted aryl fluorides, the authors found that the reaction yield notably decreases by increasing the steric hindrance posed by the neighboring group (e.g. Me, 480, 57% vs. Ph, 481, <20%). Additionally, the borylation reaction proceeded in moderate yields for meta-substituted aryl fluorides (e.g. Me, 482, 58% and OPh, 483, 48%). The authors stated that, in most cases, hydrogen atom transfer (HAT) was the main side reaction, which led to the formation of variable amounts of the corresponding dehydrofluorinated side products.
To further illustrate the generality of their method, the authors applied their photocatalytic borylation to several para- and meta-substituted Boc-protected phenols (484–492), aryl sulfones (493–495), the aryl sulfoxide 496, and the aryl sulfide 497. Such processes rendered the desired borylated products in slightly lower yields than aryl fluoride substrates and tolerated a broad diversity of functional groups (e.g. alkyl, phenyl, phenoxy, ester, amide, etc.). Remarkably, the utility of this method was ultimately demonstrated by the direct ipso-borylation of the Boc-O-protected δ-tocopherol (491) and Boc-O-protected estrone (492) derivatives in 65% and 45% yields, respectively.
To gain insight into the mechanism of this photocatalyzed process (Scheme 23c), first the authors conducted control experiments with radical scavengers that confirmed the intermediacy in the process of phenyl radicals (505). Then, UV-visible spectroscopic measurements of the reaction components indicated the formation of electron donor–acceptor charge-transfer (EDA) complexes during the progress of the reaction. Further 11B NMR spectroscopic analysis allowed postulating that the photocatalyst 2-PySNa forms an adduct with a borylating reagent B2pin2 such as 501, which is responsible for one of the observed redshifts in the UV-vis measurements that indicate the formation of EDA complexes. In parallel, 19F NMR spectroscopic studies allowed to observe the generation of a second adduct between TMAF and B2pin2. Considering the key impact that the use of TMAF had in the reactivity compared to other ionic sources of fluoride, the authors deemed that such adduct may give rise to boryl anionic species [B2pin2-F–] (506) that are nucleophilic enough to interact with the leaving group of the substrate and activate it.
Based on all this experimental evidence, the authors proposed a mechanism in which the association of such activated species 500 with the adduct PySNa-B2pin2 (501) gives rise to the EDA complex 502 observed in the UV-vis measurements (Scheme 23d). Upon photoexcitation, it is in such EDA complexes where an inner-sphere electron-transfer event may occur that leads to the generation of a thiyl radical (503) and the arene radical anion 504 from which the leaving group easily cleaves. As a result, the active phenyl radical species 505 are formed, that then may directly react with the boryl anionic species [B2pin2-F]− 506 and yield the desired borylated product 507. Finally, a boryl radical anion 508 is simultaneously formed to 507, which is reduced in situ by the thiyl radical 503 to regenerate the anion of the photocatalyst 2-PySNa and close the catalytic cycle.
Although the authors stated the need to conduct future studies to provide a more detailed mechanistic rationale for this reaction, this work constitutes the first precedent where TMAF is key to generate anionic species such as [B2pin2-F−] 506 with a key dual role in: (i) the formation upon irradiation of the EDA complexes that facilitate the inner-sphere transfer event leading to the active phenyl radical intermediates 505 (even from arene substrates with high reduction potentials) and in (ii) their reaction with those phenyl radicals 505 to provide the targeted borylated end-products 507. This precedent may open new avenues in reaction development for the utilization of TMAF to generate fluorinated excited species with untapped potential to participate in unprecedented chemical transformations.
5. Conclusions and Outlook
Synthetic organofluorine chemistry has experienced burgeoning progress in recent decades to meet the urge-pressing need of fluorinated molecules in critical areas for human development such as pharmaceutical drug discovery where the installation of fluorine-based functional groups is often instrumental to impart intriguing properties to small-molecule drug candidates [28,29,30,31,32,33]. These new methodologies strive to amplify the structural diversity of the potentially accessed fluorinated molecules by developing new chemical reactions, adding new reagents to the arsenal of fluorinating reagents, or improving the selectivity, functional group tolerance, and substrate scope of the known fluorinating methods (Section 1). However, it is mandatory that these fluorination methodologies are highly efficient, atom-economical, and do not generate toxic waste, thus enabling their implementation in large industrial processes where sustainability and safety are often the decisive factors.
In this review, we have illustrated that TMAF is a nucleophilic ionic source of fluoride with all the features and physicochemical attributes needed to develop new or improve existing fluorinating methodologies with the realistic aim of industrial implementation. For decades, a scientific milieu has evolved that encompasses an increasing understanding of the distinctive physicochemical properties of TMAF, the intermolecular interactions and solvation effects governing its reactivity in solution, and the progress of the synthetic methods for its in preparation in an increasingly more efficient and less time-consuming manner, even at bulk scale (Section 2). Nurturing from that scientific milieu, the use of TMAF in C(sp2)-F bond-forming reactions has experienced a vertiginous progression that has culminated in the emergence of synthetic methods capable of fluorinating a wide range of halide and pseudohalide (hetero)arene systems with unparalleled efficiency (even compared to anhydrous TBAF), atom-economy, and often upon reactions conditions mild enough to enable high site-selectivities that are unprecedented in nucleophilic SNAr fluorinations. Moreover, the refinement in the methods to generate in situ TMAF [97], even catalytically [98], and the development of non-hygroscopic TMAF adducts [132], allows tuning the reactivity of the reagent in solution, and further expands its synthetic utility and versatility (Section 3.2). More recently, TMAF has been also shown capable of promoting other C(sp2)-F bond-forming reactions on important building blocks such as the deoxyfluorination of phenols via aryl fluorosulfonates with a performance comparable to the better existing methods for this task [138], and improved atom-economy [111,139] (Section 3.3). To that end, TMAF has also been utilized for the relatively understudied deoxyfluorination reaction of (hetero)aromatic aldehydes that yields the corresponding difluoromethylated products [142,143], with better results than DAST—the only reagent hitherto known to render this transformation.
Concerning C(sp3)-F bond formation, the use of TMAF has been mainly limited to the synthesis of [18F]-labeled radiopharmaceuticals via nucleophilic substitution reactions on manno- and gluco-pyranoside carbohydrates, estrogen hormones, or L-ascorbic acid, with cyclic sulfate or triflate leaving groups (Section 3.4).
Although those processes seem to fulfill their initial role, granting rapid access to those molecules relevant for medical imaging, the therapeutical potential of this field warrants further research to develop selective methods proceeding under milder reaction conditions (e.g. lower reaction temperatures), that may allow the rapid last-stage [18F]-fluorination of a broader set of (bio)molecules with untapped potential in medical imaging. We propose that the prominent fluorination properties of TMAF endows it with the potential to play a prime role in this area.
Additionally, TMAF has been demonstrated to react with important molecules such as carbon dioxide (Section 3.5) [159], with which it spontaneously generates fluorocarbonate salts that may have considerable potential as sustainable fluorinated synthetic building blocks. This is of great importance amidst the current global warming crisis and the impact underlying the search of high added-value applications to upcycle this greenhouse gas. Furthermore, TMAF has also been shown to activate other relevant inorganic molecules such as carbon disulfide [161,162], thiophosgene [166], and it is also known to act as a promising non-toxic source of the pharmaceutically valuable trifluoromethylthiolate anion [167,168].
Regarding the reactive profile of TMAF as a base (Section 4), its use was traditionally seldom in classic Brønsted base-mediated reactions due to its relatively high cost in comparison to a vast number of other bases, the gaps of knowledge on its properties, and the fact that it was unclear what could specifically provide in relation to those other bases. It is only been in the last two years when two pioneering works have showcased the intriguing potential of TMAF to participate as a base in new chemical transformations. First, Schoenebeck elegantly described that TMAF enables the chemoselective methylation of protic groups that are pervasive in organic chemistry and pharmaceutical sciences such as secondary amides, nitrogenated heterocyclic scaffolds (e.g. indoles, pyrroles, and imidazoles) alcohols, thiols, and sulfonamides in a highly efficient manner and often with remarkable site-selectivities (e.g. methylation of amides, indoles, and alcohols in the presence of anilines and primary alkyl amines) [179]. Moreover, Schoenebeck and co-workers proved in that work that TMAF possesses an unprecedented dual role acting as a base and a methyl source in a concerted manner. Finally, König’s group reported the photocatalyzed ipso-borylation of relatively stable aromatic bonds such as C(sp2)-F, C(sp2)-S, C(sp2)-O, and C(sp2)-N [184]. Although the authors stated that more studies are needed to provide a concise mechanistic rationale for this process, the experimental evidence gathered allowed them to suggest that TMAF also plays a unique role in this transformation to generate upon photoexcitation the anionic borylating species [B2pin2-F–], and simultaneously activate the substrates facilitating the generation of phenyl radicals via electron–donor complexes (EDA).
Most of the physicochemical properties of TMAF in solid state and solution were studied and described in the 1980s and 1990s, combining experimental methods with theoretical models that have been largely superseded ever since. In this sense, we devise that the application of current methods coupled with the implementation of the latest theoretical and computational models (e.g. activation strain model and molecular theory [195]), could provide a more comprehensive and accurate description of those physicochemical properties, and definitely assist to gain further fundamental understanding of the mechanistic pathways operating in TMAF-mediated transformations. We further devise that this knowledge combined with the large amount of experimental data already available (e.g. TMAF-mediated SNAr fluorinations) and the implementation of artificial intelligence could realistically facilitate the digitalization of TMAF-mediated industrial processes [196], so the average benchtop or process chemist may predict the optimal experimental conditions required for a given TMAF-mediated process on a particular substrate with minimal experimentation.
We also devise the intriguing potential of merging the use of TMAF with the latest trends in transition metal catalysis, photocatalysis (e.g. [184]), or synthetic electrochemistry as a promising approach to generate excited fluorinated species that could facilitate overcoming the significant limitations associated with the current TMAF-based synthetic methodologies (e.g. SNAr fluorination of meta-substituted (hetero)aryl (pseudo)halides, and/or to expand the set of functional groups amenable to undergo SNAr fluorination reactions). Additionally, we also anticipate that such hybrid approaches may facilitate transformations that are currently unattainable by other means, such as the direct nucleophilic fluorination of C(sp3)-H bonds and novel difunctionalization reactions.
Author Contributions
V.I. and J.E.P.-B. conceived and conceptualized this work, reviewed the literature, and wrote the advance draft of this manuscript. T.W. contributed to the proofreading and critical analysis of the advanced draft of this manuscript, V.I. and J.E.P.-B. edited and wrote the final and subsequent corrected version of the manuscript. J.E.P.-B. wrote the conclusions of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was enabled by the financial support received from the Academy of Finland (decision no. 321778). We are also grateful for the support from the Faculty of Science of the University of Helsinki.
Acknowledgments
We gratefully acknowledge Israel Fernández (Universidad Complutense de Madrid) for insightful discussions during the preparation of this manuscript and Karin Seppä Pavón for valuable assistance with the design of the graphics.
Conflicts of Interest
The authors declare no competing interests.
References
- Swallow, S. Fluorine in Pharmaceutical and Medicinal Chemistry. From biophysical Aspects to Clinical Applications, 1st ed.; Gouverneur, V., Müller, K., Eds.; ICP: Oxford, UK, 2012; Volume 6, pp. 141–174. [Google Scholar]
- Ametamey, S.M.; Honer, M.; Shubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Tredwell, M.; Gouverneur, V. 18F Labeling of Arenes. Angew. Chem. Int. Ed. 2012, 51, 11426–11437. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [18F]fluorination: New solutions to old problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [Green Version]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Hiyama, T.; Yamamoto, H. Fluorine-Containing Materials. In Organofluorine Compounds; Yamamoto, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Babudri, F.; Farinola, G.M.; Naso, F.; Ragni, R. Fluorinated organic materials for electronic and optoelectronic applications: The role of fluorine atom. Chem. Commun. 2007, 1003–1022. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in Health Care: Organofluorine Containing Blockbuster Drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2018 FDA drug approvals. Nat. Rev. Drug. Discov. 2019, 18, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug. Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amines Basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry, J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Thiehoff, C.; Rey, Y.P.; Gilmour, R. The Fluorine Gauche Effect: A Brief History. Isr. J. Chem. 2017, 57, 92–100. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Gribble, G.W. Naturally Occurring Organofluorines. In Organofluorines. The Handbook of Environmental Chemistry; (Volume 3 Series: Anthropogenic Compounds), vol 3N; Neilson, A.H., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 3. [Google Scholar] [CrossRef]
- Gribble, G.W. A recent survey of naturally occurring organohalogen compounds. Environ. Chem. 2015, 12, 396–405. [Google Scholar] [CrossRef]
- Höfler, G.T.; But, A.; Hollmann, F. Haloperoxidases as catalysts in organic synthesis. Org. Biomol. Chem. 2019, 17, 9267–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.; Huang, Q.; Li, M.; Wang, J. Enzyme-catalyzed C–F bond formation and cleavage. Bioresour. Bioprocess. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, D.; Schaffrath, C.; Cobb, S.L.; Hamilton, J.T.G.; Murphy, C.D. Biosynthesis of an organofluorine molecule. Nature 2002, 416, 279. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Hammond, C. Catalytic C(sp3)-F bond formation: Recent achievements and pertaining challenges. Green Chem. 2020, 22, 5195–5209. [Google Scholar] [CrossRef]
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorination of C−H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [Green Version]
- Fustero, S.; Sedgwick, D.M.; Román, R.; Barrio, P. Recent advances in the synthesis of functionalized monofluorinated compounds. Chem. Commun. 2018, 54, 9706–9725. [Google Scholar] [CrossRef]
- Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef]
- Pan, Y. The Dark Side of Fluorine. ACS Med. Chem. Lett. 2019, 10, 1016–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.J.; Shu, Y.-Z.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compound. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Ritter, T. Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev. 2015, 115, 612–633. [Google Scholar] [CrossRef] [PubMed]
- See, Y.Y.; Morales-Colón, M.T.; Bland, D.C.; Sanford, M.S. Development of SNAr Nucleophilic Fluorination: A Fruitful Academia-Industry Collaboration. Acc. Chem. Res. 2020, 53, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.S.; Pez, G.P.; Syvret, R.G. Electrophilic NF Fluorinating Agents. Chem. Rev. 1996, 96, 1737–1755. [Google Scholar] [CrossRef]
- Murphy, G.K.; Gulder, T. Hypervalent Iodine Fluorination for Preparing Alkyl Fluorides (Stoichiometrically and Catalytically). In Fluorination. Synthetic Organofluorine Chemistry; Hu, J., Umemoto, T., Eds.; Springer: Singapore, 2018. [Google Scholar] [CrossRef]
- Rozatian, N.; Ashworth, I.W.; Sandford, G.; Hodgson, D.R.W. A quantitative reactivity scale for electrophilic fluorinating reagents. Chem. Sci. 2018, 9, 8692–8702. [Google Scholar] [CrossRef] [Green Version]
- Sibi, M.P.; Landais, Y. Csp3-F Bond Formation: A Free-Radical Approach. Angew. Chem. Int. Ed. 2013, 52, 3570–3572. [Google Scholar] [CrossRef]
- Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F. Recent advances in radical fluorination. Synthesis 2015, 47, 2554–2569. [Google Scholar] [CrossRef]
- Lantaño, B.; Postigo, A. Radical fluorination reactions by thermal and photoinduced methods. Org. Biomol. Chem. 2017, 15, 9954–9973. [Google Scholar] [CrossRef]
- Koike, T.; Akita, M. Photocatalytic Introduction of Fluorinated Groups. In Science of Synthesis; König, B., Ed.; Thieme Verlagsgruppe: Stuttgart, Germany, 2019; pp. 559–574. [Google Scholar]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of Solvation of Ions. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Lemal, D.M. Perspective on Fluorocarbon Chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Liotta, C.L.; Harris, H.P. Chemistry of Naked Anions. I. Reactions of the 18-Crown-6 Complex of Potassium Fluoride with Organic Substrates in Aprotic Organic Solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Emsley, J. Very Strong Hydrogen Bonding. Chem. Soc. Rev. 1980, 9, 91–124. [Google Scholar] [CrossRef]
- Seppelt, K. Does the Naked Fluoride Ion Exist? Angew. Chem. Int. Ed. 1992, 31, 292–293. [Google Scholar] [CrossRef]
- Christe, K.O.; Jenkins, H.D.B. Quantitative Measure for the “Nakedness” of Fluoride Ion Sources. J. Am. Chem. Soc. 2003, 125, 9457–9461. [Google Scholar] [CrossRef]
- Borrmann, T.; Lork, E.; Mews, R.; Stohrer, W.-D. Fluoride Ion Transfer and Stabilisation of Reactive Ions. J. Fluor. Chem. 2004, 125, 903–916. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Wilson, R.D.; Bau, R.; Feng, J.A. Syntheses, Properties, and Structures of Anhydrous Tetramethylammonium Fluoride and Its 1:1 Adduct with Trans-3-Amino-2-Butenenitrile. J. Am. Chem. Soc. 1990, 112, 7619–7625. [Google Scholar] [CrossRef]
- Harmon, K.M.; Southworth, B.A.; Harmon, J. Hydrogen Bonding Part 46. Thermodynamic and IR Study of Stability and Structure of Tetraethylammonium Fluoride·2.75H2O, Tetraethylammonium Fluoride·2.00H2O, Tetramethylammonium Fluoride·3.00H2O, and Tetraethylammonium Fluoride·5.00H2O. J. Mol. Struct. 1993, 300, 339–349. [Google Scholar] [CrossRef]
- Harmon, K.M.; LaFave, N.J. Hydrogen Bonding Part 66. Further Studies of the Fluoride Ion Assisted Dissolution of 1-Methyl-4,5-Dicarboxyimidazole: Absence of Cation Participation and Stoichiometric Considerations. J. Mol. Struct. 1997, 404, 297–306. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W. Nuclear Magnetic Resonance Spectrum of the Fluoride Anion. J. Fluor. Chem. 1990, 46, 339–342. [Google Scholar] [CrossRef]
- Gerken, M.; Boatz, J.A.; Kornath, A.; Haiges, R.; Schneider, S.; Schroer, T.; Christe, K.O. The 19F NMR Shifts Are Not a Measure for the Nakedness of the Fluoride Anion. J. Fluor. Chem. 2002, 116, 49–58. [Google Scholar] [CrossRef]
- Shodai, Y.; Kohara, S.; Ohishi, Y.; Inaba, M.; Tasaka, A. Anionic Species (FH) x F—In Room-Temperature Molten Fluorides (CH 3) 4 NF· m HF. J. Phys. Chem. A 2004, 108, 1127–1132. [Google Scholar] [CrossRef]
- Harmon, K.M.; Southworth, B.A.; Wilson, K.E.; Keefer, P.K. N,N,N-Trimethyl-1-Adamantylammonium Fluoride, a Completely Anhydrous Quaternary Ammonium Fluoride Salt. J. Org. Chem. 1993, 58, 7294–7295. [Google Scholar] [CrossRef]
- Mahjoub, A.R.; Zhang, X.; Seppelt, K. Reactions of the “Naked” Fluoride Ion: Syntheses and Structures of SeF 62− and BrF 6−. Chem. Eur. J. 1995, 1, 261–265. [Google Scholar] [CrossRef]
- Gnann, R.Z.; Wagner, R.I.; Christe, K.O.; Bau, R.; Olah, G.A.; Wilson, W.W. Naked Fluoride Ion Sources: Synthesis, Characterization, and Coupling Reaction of 1-Methylhexamethylenetetramine Fluoride. J. Am. Chem. Soc. 1997, 119, 112–115. [Google Scholar] [CrossRef]
- Kornath, A.; Neumann, F.; Oberhammer, H. Tetramethylphosphonium Fluoride: “Naked” Fluoride and Phosphorane. Inorg. Chem. 2003, 42, 2894–2901. [Google Scholar] [CrossRef]
- Schwesinger, R.; Link, R.; Wenzl, P.; Kossek, S. Anhydrous Phosphazenium Fluorides as Sources for Extremely Reactive Fluoride Ions in Solution. Chem. Eur. J. 2005, 12, 438–445. [Google Scholar] [CrossRef]
- Ryan, S.J.; Schimler, S.D.; Bland, D.C.; Sanford, M.S. Acyl Azolium Fluorides for Room Temperature Nucleophilic Aromatic Fluorination of Chloro- and Nitroarenes. Org. Lett. 2015, 17, 1866–1869. [Google Scholar] [CrossRef]
- Elias, S.; Karton-Lifshin, N.; Yehezkel, L.; Ashkenazi, N.; Columbus, I.; Zafrani, Y. Synthesis, Characterization, and Reactivity of Thermally Stable Anhydrous Quaternary Ammonium Fluorides. Org. Lett. 2017, 19, 3039–3042. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; DiMagno, S.G. Anhydrous Tetrabutylammonium Fluoride. J. Am. Chem. Soc. 2005, 127, 2050–2051. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.K.; Harrison, R.G.; Richmond, T.G. Cobaltocenium Fluoride: A Novel Source of “Naked” Fluoride Formed by Carbon-Fluorine Bond Activation in a Saturated Perfluorocarbon. J. Am. Chem. Soc. 1994, 116, 11165–11166. [Google Scholar] [CrossRef]
- Grushin, V.V. Generation of “Naked” Fluoride Ions in Unprecedentedly High Concentrations from a Fluoropalladium Complex. Angew. Chem. Int. Ed. 1998, 37, 994–996. [Google Scholar] [CrossRef]
- Chen, Z.; Tonouchi, Y.; Matsumoto, K.; Saimura, M.; Atkin, R.; Nagata, T.; Katahira, M.; Hagiwara, R. Partially Naked Fluoride in Solvate Ionic Liquids. J. Phys. Chem. Lett. 2018, 9, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilcher, A.S.; DeShong, P. Utilization of Tetrabutylammonium Triphenyldifluorosilicate as a Fluoride Source for Silicon−Carbon Bond Cleavage †. J. Org. Chem. 1996, 61, 6901–6905. [Google Scholar] [CrossRef]
- Middleton, W.J. New Fluorinating Reagents. Dialkylaminosulfur Fluorides. J. Org. Chem. 1975, 40, 574–578. [Google Scholar] [CrossRef]
- Behr, J.-B.; Massicot, F. Tetramethylammonium Fluoride. Encycl. Reag. Org. Synth. 2001, 1–5. [Google Scholar] [CrossRef]
- Aue, D.H.; Webb, H.M.; Bowers, M.T. A Thermodynamic Analysis of Solvation Effects on the Basicities of Alkylamines. An Electrostatic Analysis of Substituent Effects. J. Am. Chem. Soc. 1976, 98, 318–329. [Google Scholar] [CrossRef]
- McLean, W.J.; Jeffrey, G.A. Crystal Structure of Tetramethylammonium Fluoride Tetrahydrate. J. Chem. Phys. 1967, 47, 414–417. [Google Scholar] [CrossRef]
- Stäben, D.; Mootz, D. Die Kristallinen Hydrate von Tetramethylammoniumfluorid. Bildung, Struktur, Wasserstoffbrückenbindung [1]/The Crystalline Hydrates of Tetramethylammonium Fluoride. Formation, Structure, Hydrogen Bonding. Z. Naturforsch. B 1993, 48, 1057–1064. [Google Scholar] [CrossRef]
- Harmon, K.M.; Madeira, S.L. Hydrogen Bonding. Part 77. Molecular Orbital Study of C–H⋯F Hydrogen Bonds in Tetramethylammonium Fluoride; Potential Effects on Solid State Structure. J. Mol. Struct. 2001, 560, 179–188. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables; 4th ed.; Monograph 9 (Part I and Part II) (1998); 1963. Available online: https://janaf.nist.gov/ (accessed on 13 January 2022).
- Atkins, P.; Overton, T.; Rourke, J.; Weller, M.; Armstrong, F. Inorganic Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2010; p. 94. [Google Scholar]
- Pliego, J.R., Jr.; Piló-Veloso, D. Effects of Ion-Pairing and Hydration on the SNAr Reaction of the F − with p-Chlorobenzonitrile in Aprotic Solvents. Phys. Chem. Chem. Phys. 2008, 10, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Dalessandro, E.V.; Pliego, J.R., Jr. Reactivity and Stability of Ion Pairs, Dimers and Tetramers versus Solvent Polarity: SNAr Fluorination of 2-Bromobenzonitrile with Tetramethylammonium Fluoride. Theor. Chem. Acc. 2020, 139, 27. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W. Reaction of the Fluoride Anion with Acetonitrile. Chloroform and Methylene Chloride. J. Fluor. Chem. 1990, 47, 117–120. [Google Scholar] [CrossRef]
- Grushin, V.V.; Marshall, W.J. Fluorination of Nonactivated Haloarenes via Arynes under Mild Conditions, Resulting from Further Studies toward Ar−F Reductive Elimination from Palladium(II). Organometallics 2008, 27, 4825–4828. [Google Scholar] [CrossRef]
- Bastos, M.M.; Barbosa, J.P.; Pinto, A.C.; Kover, W.B.; Takeuchi, Y.; Boechat, N. Reductive Debromination of 1-Methyl-2,4,5-Tribromoimidazole Mediated by Dry Tetramethylammonium Fluoride in Aprotic Solvents. J. Braz. Chem. Soc. 2001, 12, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.J.; Clark, J.H.; Nightingale, D.J. The Effect of Basicity on Fluorodenitration Reactions Using Tetramethylammonium Salts. Tetrahedron 1999, 55, 7725–7738. [Google Scholar] [CrossRef]
- Yang, Q.; Sheng, M.; Henkelis, J.J.; Tu, S.; Wiensch, E.; Zhang, H.; Zhang, Y.; Tucker, C.; Ejeh, D.E. Explosion Hazards of Sodium Hydride in Dimethyl Sulfoxide, N,N-Dimethylformamide, and N,N-Dimethylacetamide. Org. Process Res. Dev. 2019, 23, 2210–2217. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Sheng, M.; Huang, Y. Potential Safety Hazards Associated with Using N,N-Dimethylformamide in Chemical Reactions. Org. Process Res. Dev. 2020, 24, 1586–1601. [Google Scholar] [CrossRef]
- Wilson, J.W. Standard Enthalpy of Solution and Formation of Tetramethylammonium Fluoride. J. Chem. Thermodyn. 1976, 8, 1107–1108. [Google Scholar] [CrossRef]
- Lawson, A.T.; Collie, N. XLVII.—The action of heat on the salts of tetramethylammonium. J. Chem. Soc. Trans. 1888, 53, 624–636. [Google Scholar] [CrossRef] [Green Version]
- Harmon, K.M.; Gennick, I. Hydrogen Bonding. V. Possible Existence of Strongly Hydrogen-Bonded Water-Fluoride and Water-Hydroxide Complex Anions, (F-.H2O)22- and (OH-.H2O)22-, in Tetramethylammonium Ion Salt Hydrates. Inorg. Chem. 1975, 14, 1840–1845. [Google Scholar] [CrossRef]
- Hawk, M.K.; Ryan, S.J.; Zhang, X.; Huang, P.; Chen, J.; Liu, C.; Chen, J.; Lindsay-Scott, P.J.; Burnett, J.; White, C.; et al. Tetramethylammonium Fluoride Tetrahydrate for SNAr Fluorination of 4-Chlorothiazoles at a Production Scale. Org. Process Res. Dev. 2021, 25, 1167–1175. [Google Scholar] [CrossRef]
- Tunder, R.; Siegel, B. The SF5 Anion. J. Inorg. Nucl. Chem. 1963, 25, 1097–1098. [Google Scholar] [CrossRef]
- Klanberg, F.; Muetterties, E.L. Nuclear Magnetic Resonance Studies on Pentacoordinate Silicon Fluorides. Inorg. Chem. 1968, 7, 155–160. [Google Scholar] [CrossRef]
- Kolomeitsev, A.A.; Seifert, F.U.; Röschenthaler, G.-V. Simple Preparation of Difluorophosphoranes Using Anhydrous Zinc and Tetramethylammonium Fluorides. J. Fluor. Chem. 1995, 71, 47–49. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, T.; Zhou, X. The Synthesis of Quaternary Ammonium Salts from Ammonium Salts and Dialkyl Carbonate. Chem. Commun. 2006, 1864–1865. [Google Scholar] [CrossRef]
- Cismesia, M.A.; Ryan, S.J.; Bland, D.C.; Sanford, M.S. Multiple Approaches to the In Situ Generation of Anhydrous Tetraalkylammonium Fluoride Salts for SNAr Fluorination Reactions. J. Org. Chem. 2017, 82, 5020–5026. [Google Scholar] [CrossRef]
- Hong, C.M.; Whittaker, A.M.; Schultz, D.M. Nucleophilic Fluorination of Heteroaryl Chlorides and Aryl Triflates Enabled by Cooperative Catalysis. J. Org. Chem. 2021, 86, 3999–4006. [Google Scholar] [CrossRef]
- Lee, S.J.; Morales-Colón, M.T.; Brooks, A.F.; Wright, J.S.; Makaravage, K.J.; Scott, P.J.H.; Sanford, M.S. SNAr Radiofluorination with In Situ Generated [18F]Tetramethylammonium Fluoride. J. Org. Chem. 2021, 86, 14121–14130. [Google Scholar] [CrossRef]
- Bloom, S.; Pitts, C.R.; Miller, D.C.; Haselton, N.; Holl, M.G.; Urheim, E.; Lectka, T. A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination. Angew. Chem. Int. Ed. 2012, 51, 10580–10583. [Google Scholar] [CrossRef]
- Guo, R.; Huang, J.; Zhao, X. Organoselenium-Catalyzed Oxidative Allylic Fluorination with Electrophilic N–F Reagent. ACS Catal. 2018, 8, 926–930. [Google Scholar] [CrossRef]
- Sorlin, A.M.; Usman, F.O.; English, C.K.; Nguyen, H.M. Advances in Nucleophilic Allylic Fluorination. ACS Catal. 2020, 10, 11980–12010. [Google Scholar] [CrossRef]
- Terrier, F. Modern Nucleophilic Aromatic Substitution; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–94. [Google Scholar] [CrossRef]
- Lennox, A.J.J. Meisenheimer Complexes in SNAr Reactions: Intermediates or Transition States? Angew. Chem. Int. Ed. 2018, 57, 14686–14688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artamkina, G.A.; Egorov, M.P.; Beletskaya, I.P. Some Aspects of Anionic. Sigma.-Complexes. Chem. Rev. 1982, 82, 427–459. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G.; Uggerud, E. Rate-Determining Factors in Nucleophilic Aromatic Substitution Reactions. J. Org. Chem. 2010, 75, 2971–2980. [Google Scholar] [CrossRef]
- Cramption, M.R.; Gold, V. 824. Reactions of Aromatic Nitro-Compounds in Alkaline Media. Part IX. Nuclear Magnetic Resonance Spectra of Meisenheimer Complexes. J. Chem. Soc. 1964, 4293–4295. [Google Scholar] [CrossRef]
- Kwan, E.E.; Zeng, Y.; Besser, H.A.; Jacobsen, E.N. Concerted Nucleophilic Aromatic Substitutions. Nat. Chem. 2018, 10, 917–923. [Google Scholar] [CrossRef]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted Nucleophilic Aromatic Substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.N.; Ritter, T. Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res. 2017, 50, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Schimler, S.D.; Cismesia, M.A.; Hanley, P.S.; Froese, R.D.J.; Jansma, M.J.; Bland, D.C.; Sanford, M.S. Nucleophilic Deoxyfluorination of Phenols via Aryl Fluorosulfonate Intermediates. J. Am. Chem. Soc. 2017, 139, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Ba-Saif, S.; Luthra, A.K.; Williams, A. Concertedness in Acyl Group Transfer in Solution: A Single Transition State in Acetyl Group Transfer between Phenolate Ion Nucleophiles. J. Am. Chem. Soc. 1987, 109, 6362–6368. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- der Plas, H.C.V. The SN(ANRORC) Mechanism: A New Mechanism for Nucleophilic Substitution. Acc. Chem. Res. 1978, 11, 462–468. [Google Scholar] [CrossRef]
- Balz, G.; Schiemann, G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Ber. Dtsch. Chem. Ges. B 1927, 60, 1186–1190. [Google Scholar] [CrossRef]
- Cresswell, A.J.; Davies, S.G.; Roberts, P.M.; Thomson, J.E. Beyond the Balz–Schiemann Reaction: The Utility of Tetrafluoroborates and Boron Trifluoride as Nucleophilic Fluoride Sources. Chem. Rev. 2014, 115, 566–611. [Google Scholar] [CrossRef]
- Gottlieb, H.B. The Replacement of Chlorine by Fluorine in Organic Compounds. J. Am. Chem. Soc. 1936, 58, 532–533. [Google Scholar] [CrossRef]
- Finger, G.C.; Kruse, C.W. Aromatic Fluorine Compounds. VII. Replacement of Aromatic -Cl and -NO2 Groups by -F 1,2. J. Am. Chem. Soc. 1956, 78, 6034–6037. [Google Scholar] [CrossRef]
- Clark, J.H.; Macquarrie, D.J. The Synthesis of Organofluorine Compounds Using Potassium Fluoride-Tetraphenylphosphonium Bromide Systems. Tetrahedron Lett. 1987, 28, 111–114. [Google Scholar] [CrossRef]
- Boechat, N.; Clark, J.H. Fluorodenitrations using tetramethylammonium fluoride. J. Chem. Soc. Chem. Commun. 1993, 921–922. [Google Scholar] [CrossRef]
- Clark, J.H.; Wails, D. Fluorodenitration of Activated Diphenyl Sulphones Using Tetramethylammonium Fluoride. J. Fluor. Chem. 1995, 70, 201–205. [Google Scholar] [CrossRef]
- Adams, D.J.; Clark, J.H.; Nightingale, D.J. Tetramethylammonium Hydrogendifluoride: A Convenient Source of Nucleophilic Fluoride. Synth. Commun. 1998, 28, 4295–4301. [Google Scholar] [CrossRef]
- Adams, D.J.; Clark, J.H.; McFarland, H. The Formation of 4,4′-Difluorobenzophenone from 4,4′-Dinitrodiphenylmethane. J. Fluor. Chem. 1998, 92, 127–129. [Google Scholar] [CrossRef]
- Sun, H.; DiMagno, S.G. Room-Temperature Nucleophilic Aromatic Fluorination: Experimental and Theoretical Studies. Angew. Chem. Int. Ed. 2006, 45, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Eckelbarger, J.D.; Epp, J.B.; Schmitzer, P.R.; Siddall, T.L. 3-Alkenyl-6-halo-4-aminopicolinates and Their Use as Herbicides. U.S. Patent 20120190548A1, 26 July 2012. [Google Scholar]
- Whiteker, G.T.; Arndt, K.E.; Renga, J.M.; Yuanming, Z.; Lowe, C.T.; Siddall, T.L.; Podhorez, D.E.; Roth, G.A.; West, S.P.; Arndt, C. Process for the preparation of 4-amino-5-fluoro-3-halo-6-(substituted)picolinates. U.S. Patent 20120190860, 26 July 2012. [Google Scholar]
- Yerkes, C.N.; Lowe, C.T.; Eckelbarger, J.D.; Epp, J.B.; Guenthenspberger, K.A.; Siddall, T.L.; Schmitzer, P.R. Arylalkyl Esters of 4-amino-6-(substitutedphenyl)picolinates and 6-amino-2-(substitutedphenyl)-4-pyrimidinecarboxylates and Their Use as Selective Herbicides for Crops. U.S. Patent 20150025238A1, 22 January 2015. [Google Scholar]
- Schimler, S.D.; Ryan, S.J.; Bland, D.C.; Anderson, J.E.; Sanford, M.S. Anhydrous Tetramethylammonium Fluoride for Room-Temperature SNAr Fluorination. J. Org. Chem. 2015, 80, 12137–12145. [Google Scholar] [CrossRef] [Green Version]
- Hicken, E.J.; Marmsater, F.P.; Munson, M.C.; Schlachter, S.T.; Robinson, J.E.; Allen, S.; Burgess, L.E.; DeLisle, R.K.; Rizzi, J.P.; Topalov, G.T.; et al. Discovery of a Novel Class of Imidazo[1,2- a ]Pyridines with Potent PDGFR Activity and Oral Bioavailability. ACS Med. Chem. Lett. 2013, 5, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, R.; Ironside, M.D.; Vogt, P.F. Safety Notables: Information from the Literature. Org. Process Res. Dev. 2010, 14, 1480–1484. [Google Scholar] [CrossRef]
- Morales-Colón, M.T.; See, Y.Y.; Lee, S.J.; Scott, P.J.H.; Bland, D.C.; Sanford, M.S. Tetramethylammonium Fluoride Alcohol Adducts for SNAr Fluorination. Org. Lett. 2021, 23, 4493–4498. [Google Scholar] [CrossRef]
- Kim, D.W.; Jeong, H.; Lim, S.T.; Sohn, M. Tetrabutylammonium Tetra(Tert-Butyl Alcohol)-Coordinated Fluoride as a Facile Fluoride Source. Angew. Chem. Int. Ed. 2008, 47, 8404–8406. [Google Scholar] [CrossRef]
- Gupta, H.K.; Pardasani, D.; Mazumder, A.; Purohit, A.K.; Dubey, D.K. Tetrabutylammonium Tetra (Tert-Butyl Alcohol) Coordinated Fluoride-an Efficient Reagent for the Synthesis of Fluorine Derivatives of Phosphorus(V) Compounds. Tetrahedron Lett. 2009, 50, 2697–2699. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Kryachko, E.S.; Vanquickenborne, L.G. General and Theoretical Aspects of Phenols. In The Chemistry of Phenols; Rappoport, Z., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2003. [Google Scholar] [CrossRef]
- Nemoto, H.; Nishiyama, T.; Akai, S. Nucleophilic Deoxyfluorination of Catechols. Org. Lett. 2011, 13, 2714–2717. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sonoda, H.; Fukumura, K.; Nagata, T. 2,2-Difluoro-1,3-Dimethylimidazolidine (DFI). A New Fluorinating Agent. Chem. Commun. 2002, 1618–1619. [Google Scholar] [CrossRef]
- Tang, P.; Wang, W.; Ritter, T. Deoxyfluorination of Phenols. J. Am. Chem. Soc. 2011, 133, 11482–11484. [Google Scholar] [CrossRef] [Green Version]
- Schimler, S.D.; Froese, R.D.J.; Bland, D.C.; Sanford, M.S. Reactions of Arylsulfonate Electrophiles with NMe4F: Mechanistic Insight, Reactivity, and Scope. J. Org. Chem. 2018, 83, 11178–11190. [Google Scholar] [CrossRef]
- Fry, S.E.; Pienta, N.J. Effects of Molten Salts on Reactions. Nucleophilic Aromatic Substitution by Halide Ions in Molten Dodecyltributylphosphonium Salts. J. Am. Chem. Soc. 1985, 107, 6399–6400. [Google Scholar] [CrossRef]
- Hohenstein, C.; Kadzimirsz, D.; Ludwig, R.; Kornath, A. Synthesis and Characterization of Tetramethylammonium Trifluorosulfate. Chem. Eur. J. 2011, 17, 925–929. [Google Scholar] [CrossRef]
- Melvin, P.R.; Ferguson, D.M.; Schimler, S.D.; Bland, D.C.; Sanford, M.S. Room Temperature Deoxyfluorination of Benzaldehydes and α-Ketoesters with Sulfuryl Fluoride and Tetramethylammonium Fluoride. Org. Lett. 2019, 21, 1350–1353. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Melvin, P.R.; Sanford, M.S. Deoxyfluorination of (Hetero)Aryl Aldehydes Using Tetramethylammonium Fluoride and Perfluorobutanesulfonyl Fluoride or Trifluoromethanesulfonic Anhydride. Isr. J. Chem. 2020, 60, 398–401. [Google Scholar] [CrossRef]
- Lal, G.S.; Pez, G.P.; Pesaresi, R.J.; Prozonic, F.M.; Cheng, H. Bis(2-Methoxyethyl)Aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability. J. Org. Chem. 1999, 64, 7048–7054. [Google Scholar] [CrossRef]
- Veryser, C.; Demaerel, J.; Bieliūnas, V.; Gilles, P.; Borggraeve, W.M.D. Ex Situ Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org. Lett. 2017, 19, 5244–5247. [Google Scholar] [CrossRef] [PubMed]
- Tewson, T.J. Cyclic Sulfur Esters as Substrates for Nucleophilic Substitution. A New Synthesis of 2-Deoxy-2-Fluoro-D-Glucose. J. Org. Chem. 1983, 48, 3507–3510. [Google Scholar] [CrossRef]
- Fatangare, A.; Svatoš, A. Applications of 2-Deoxy-2-Fluoro-D-Glucose (FDG) in Plant Imaging: Past, Present, and Future. Front. Plant. Sci. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido, T.; Wan, C.; Casella, V.; Fowler, J.S.; Wolf, A.P.; Reivich, M.; Kuhl, D.E. Labeled 2-deoxy-D-glucose Analogs. 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose. J. Labelled Compd. Radiopharm. 1978, 14, 175–183. [Google Scholar] [CrossRef]
- Gatley, S.J.; Shaughnessy, W.J. Synthesis of 18F-3-Deoxy-3-Fluoro-D-Glucose with Reactor-Produced 18F. Int. J. Appl. Radiat. Isot. 1980, 31, 339–341. [Google Scholar] [CrossRef]
- Berridge, M.S.; Franceschini, M.P.; Rosenfeld, E.; Tewson, T.J. Cyclic Sulfates: Useful Substrates for Selective Nucleophilic Substitution. J. Org. Chem. 1990, 55, 1211–1217. [Google Scholar] [CrossRef]
- Lim, J.L.; Zheng, L.; Berridge, M.S.; Tewson, T.J. The Use of 3-Methoxymethyl-16β, 17β-Epiestriol-O-Cyclic Sulfone as the Precursor in the Synthesis of F-18 16α-Fluoroestradiol. Nucl. Med. Biol. 1996, 23, 911–915. [Google Scholar] [CrossRef]
- Seimbille, Y.; Ali, H.; Lier, J.E. van Synthesis of 2,16α- and 4,16α-Difluoroestradiols and Their 11β-Methoxy Derivatives as Potential Estrogen Receptor-Binding Radiopharmaceuticals. J. Chem. Soc. Perkin Trans. 2002, 1, 657–663. [Google Scholar] [CrossRef]
- Kim, J.; Yamamoto, F.; Kuwabara, Y.; Honda, H.; Mukai, T.; Maeda, M. 6-[18F]Fluoro-Dehydroascorbic Acid:Synthesis and Tissue Distribution in Mice. Radioisotopes 2009, 58, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Haradahira, T.; Maeda, M.; Kai, Y.; Kojima, M. A New, High Yield Synthesis of 2-Deoxy-2-Fluoro- D -Glucose. J. Chem. Soc. Chem. Commun. 1985, 364–365. [Google Scholar] [CrossRef]
- Haradahira, T.; Maeda, M.; Omae, H.; Yano, Y.; Kojima, M. Synthesis of 2-Deoxy-2-Fluoro-D-Mannose Using Fluoride Ion. Chem. Pharm. Bull. 1984, 32, 4758–4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchuk, O.P.; Hryshchuk, O.V.; Vashchenko, B.V.; Trofymchuk, S.A.; Melnykov, K.P.; Skreminskiy, A.; Volochnyuk, D.M.; Grygorenko, O.O. Fluoroalkyl-Containing 1,2-Disubstituted Cyclobutanes: Advanced Building Blocks for Medicinal Chemistry. Eur. J. Org. Chem. 2021, 2021, 87–95. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Su, Z.; Cao, W.; Wu, C.; Chen, Q.-Y. A Series of Deoxyfluorination Reagents Featuring OCF2 Functional Groups. Org. Lett. 2020, 22, 8634–8637. [Google Scholar] [CrossRef] [PubMed]
- Flosser, D.A.; Olofson, R.A. A Useful Conversion of Alcohols to Alkyl Fluorides. Tetrahedron Lett. 2002, 43, 4275–4279. [Google Scholar] [CrossRef]
- Zhang, X.; Gross, U.; Seppelt, K. Fluorocarbonate, [FCO2]−: Preparation and Structure. Angew. Chem. Int. Ed. 1995, 34, 1858–1860. [Google Scholar] [CrossRef]
- Lawlor, L.; Passmore, J. Existence of Cesium Salts of CO2F-, CO2F22- and NO2F2-. Inorg. Chem. 1979, 18, 2923–2924. [Google Scholar] [CrossRef]
- Rüdiger, S.; Seppelt, K. On Reactions of Carbon Disulphide Induced by ‘Naked’ Fluoride Part 2: Reactions with 2-H-Heptafluoropropane, Hexafluoropropene, and Bis (2,2,2-Trifluoroethyl) Amine. J. Fluor. Chem. 1997, 82, 29–32. [Google Scholar] [CrossRef]
- Rüdiger, S.; Seppelt, K. On Reactions of Carbon Disulphide Induced by ‘Naked’ Fluoride Part 1: Reactions with Fluoroaromatics. J Fluor. Chem. 1997, 82, 25–28. [Google Scholar] [CrossRef]
- Kovács, S.; Bayarmagnai, B.; Goossen, L.J. Preparation of Electrophilic Trifluromethylthio Reagents from Nucleophilic Tetramethylammonium Trifluoromethylthiolate. Adv. Synth. Catal. 2017, 359, 250–254. [Google Scholar] [CrossRef]
- Clark, J.H.; Tavener, S.J. The Preparation of Trifluoromethyl Aryl Sulfides Using KF and Thiophosgene. J. Fluor. Chem. 1997, 85, 169–172. [Google Scholar] [CrossRef]
- Dmowski, W.; Haas, A. Trifluoromethanethiolate Ion. Part 2. Nucleophilic Substitution in Pentafluoropyridine. Synthesis and Characteristics of Trifluoromethylthio and Trifluoromethylsulphonyl Derivatives. J. Chem. Soc. Perkin Trans. 1987, 1, 2119–2124. [Google Scholar] [CrossRef]
- Tavener, S.J.; Adams, D.J.; Clark, J.H. Trifluoromethylthiolation of Aromatic Substrates Using Thiophosgene—Fluoride Salt Reagents, and Formation of Byproducts with Multi-Carbon Chains. J Fluor. Chem. 1999, 95, 171–176. [Google Scholar] [CrossRef]
- Tyrra, W.; Naumann, D.; Hoge, B.; Yagupolskii, Y.L. A New Synthesis of Trifluoromethanethiolates—Characterization and Properties of Tetramethylammonium, Cesium and Di(Benzo-15-Crown-5)Cesium Trifluoromethanethiolates. J. Fluor. Chem. 2003, 119, 101–107. [Google Scholar] [CrossRef]
- Yin, G.; Kalvet, I.; Schoenebeck, F. Trifluoromethylthiolation of Aryl Iodides and Bromides Enabled by a Bench-Stable and Easy-To-Recover Dinuclear Palladium(I) Catalyst. Angew. Chem. Int. Ed. 2015, 54, 6809–6813. [Google Scholar] [CrossRef]
- Wang, B.; Qin, L.; Neumann, K.D.; Uppaluri, S.; Cerny, R.L.; DiMagno, S.G. Improved Arene Fluorination Methodology for I(III) Salts. Org. Lett. 2010, 12, 3352–3355. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, B.; DiMagno, S.G. A Method for Detecting Water in Organic Solvents. Org. Lett. 2008, 10, 4413–4416. [Google Scholar] [CrossRef] [Green Version]
- Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef]
- Clark, J.H. Fluoride Ion as a Base in Organic Synthesis. Chem. Rev. 1980, 80, 429–452. [Google Scholar] [CrossRef]
- Hameed, A.; Alharthy, R.D.; Iqbal, J.; Langer, P. The Role of Naked Fluoride Ion as Base or Catalyst in Organic Synthesis. Tetrahedron 2016, 72, 2763–2812. [Google Scholar] [CrossRef]
- Ménand, M.; Dalla, V. TMAF-Catalyzed Conjugate Addition of Oxazolidinone and Thiols. Synlett 2004, 2005, 95–98. [Google Scholar] [CrossRef]
- Cid, M.B.; Duce, S.; Morales, S.; Rodrigo, E.; Ruano, J.L.G. Nitrophenylacetonitriles as Versatile Nucleophiles in Enantioselective Organocatalytic Conjugate Additions. Org. Lett. 2010, 12, 3586–3589. [Google Scholar] [CrossRef] [PubMed]
- Messire, G.; Massicot, F.; Vallée, A.; Vasse, J.; Behr, J. Aza-Henry Reaction with Nitrones, an Under-Explored Transformation. Eur. J. Org. Chem. 2019, 2019, 1659–1668. [Google Scholar] [CrossRef]
- da Silva, V.B.; Campos, B.R.; de Oliveira, J.F.; Decout, J.L.; de Lima, M.D. Medicinal Chemistry of Antischistosomal Drugs: Praziquantel and Oxamniquine. Bioorg. Med. Chem. 2017, 25, 3259–3277. [Google Scholar] [CrossRef] [PubMed]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic Studies on the Michael Addition of Amines and Hydrazines To Nitrostyrenes: Nitroalkane Elimination via a Retro-Aza-Henry-Type Process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-G.; Pu, M.; Kundu, G.; Schoenebeck, F. Selective Methylation of Amides, N-Heterocycles, Thiols, and Alcohols with TMAF. Org. Lett. 2020, 22, 331–334. [Google Scholar] [CrossRef]
- Schönherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. [Google Scholar] [CrossRef]
- Scattolin, T.; Pu, M.; Schoenebeck, F. Investigation of (Me4N)SCF3 as a Stable, Solid and Safe Reservoir for S=CF2 as a Surrogate for Thiophosgene. Chem. Eur. J. 2018, 24, 567–571. [Google Scholar] [CrossRef]
- For full details of Schoenebeck’s Mechanistic DFT Studies for the Chemoselective Methylation of Secondary Amides and Alcohols, See pp. S14–S21 in the Supporting Information of That Work.
- Wang, X.; Xu, Y.; Mo, F.; Ji, G.; Qiu, D.; Feng, J.; Ye, Y.; Zhang, S.; Zhang, Y.; Wang, J. Silver-Mediated Trifluoromethylation of Aryldiazonium Salts: Conversion of Amino Group into Trifluoromethyl Group. J. Am. Chem. Soc. 2013, 135, 10330–10333. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; König, B. Photo-Induced Thiolate Catalytic Activation of Inert Caryl-Hetero Bonds for Radical Borylation. Chem 2021, 7, 1653–1665. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, G.F.S.; Denny, W.A.; Santos, J.L.D. Boron in Drug Design: Recent Advances in the Development of New Therapeutic Agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Plescia, J.; Moitessier, N. Design and Discovery of Boronic Acid Drugs. Eur. J. Med. Chem. 2020, 195, 112270. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Wrigstedt, P.; Moslova, K.; Iashin, V.; Mäkkylä, H.; Ghemtio, L.; Heikkinen, S.; Tammela, P.; Perea-Buceta, J.E. Installation of an Aryl Boronic Acid Function into the External Section of N-Aryl-Oxazolidinones: Synthesis and Antimicrobial Evaluation. Eur. J. Med. Chem. 2021, 211, 113002. [Google Scholar] [CrossRef]
- Tian, Y.-M.; Guo, X.-N.; Braunschweig, H.; Radius, U.; Marder, T.B. Photoinduced Borylation for the Synthesis of Organoboron Compounds. Chem. Rev. 2021, 121, 3561–3597. [Google Scholar] [CrossRef] [PubMed]
- Schmalzbauer, M.; Marcon, M.; König, B. Excited State Anions in Organic Transformations. Angew. Chem. Int. Ed. 2021, 60, 6270–6292. [Google Scholar] [CrossRef]
- Wang, S.; Lokesh, N.; Hioe, J.; Gschwind, R.M.; König, B. Photoinitiated Carbonyl-Metathesis: Deoxygenative Reductive Olefination of Aromatic Aldehydes via Photoredox Catalysis. Chem. Sci. 2019, 10, 4580–4587. [Google Scholar] [CrossRef] [Green Version]
- Schmalzbauer, M.; Svejstrup, T.D.; Fricke, F.; Brandt, P.; Johansson, M.J.; Bergonzini, G.; König, B. Redox-Neutral Photocatalytic C−H Carboxylation of Arenes and Styrenes with CO2. Chem 2020, 6, 2658–2672. [Google Scholar] [CrossRef]
- Wang, M.; Shi, Z. Methodologies and Strategies for Selective Borylation of C–Het and C–C Bonds. Chem. Rev. 2020, 120, 7348–7398. [Google Scholar] [CrossRef]
- Jin, S.; Dang, H.T.; Haug, G.C.; He, R.; Nguyen, V.D.; Nguyen, V.T.; Arman, H.D.; Schanze, K.S.; Larionov, O.V. Visible Light-Induced Borylation of C–O, C–N, and C–X Bonds. J. Am. Chem. Soc. 2020, 142, 1603–1613. [Google Scholar] [CrossRef]
- Fernández, I.; Bickelhaupt, F.M. The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Davies, I.W. The Digitization of Organic Synthesis. Nature 2019, 570, 175–181. [Google Scholar] [CrossRef] [PubMed]
Scheme 2.
Outline of this review.

Scheme 13.
Initial reports describing the deoxyfluorination of phenols: (a) The deoxyfluorination of catechols [136], (b) The deoxyfluorination of p-nitrophenol [137], and (c) General method for the deoxyfluorination of phenolic substrates [138]).

Scheme 20.
Syntheses of electron-rich aryl fluorides from diaryliodonium salts.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Iashin, V.; Wirtanen, T.; Perea-Buceta, J.E. Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base. Catalysts 2022, 12, 233. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020233
AMA Style
Iashin V, Wirtanen T, Perea-Buceta JE. Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base. Catalysts. 2022; 12(2):233. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020233
Chicago/Turabian StyleIashin, Vladimir, Tom Wirtanen, and Jesus E. Perea-Buceta. 2022. "Tetramethylammonium Fluoride: Fundamental Properties and Applications in C-F Bond-Forming Reactions and as a Base" Catalysts 12, no. 2: 233. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020233
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.