Oxygen Reduction Reaction Electrocatalysis in Alkaline Electrolyte on Glassy-Carbon-Supported Nanostructured Pr6O11 Thin-Films


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
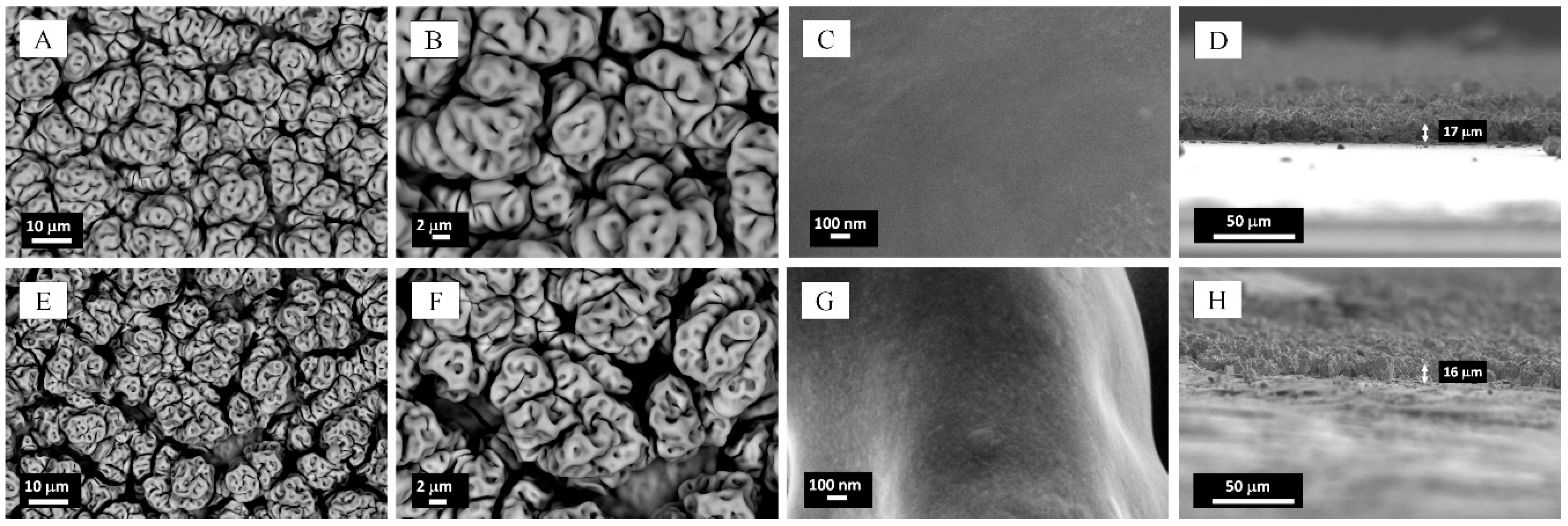
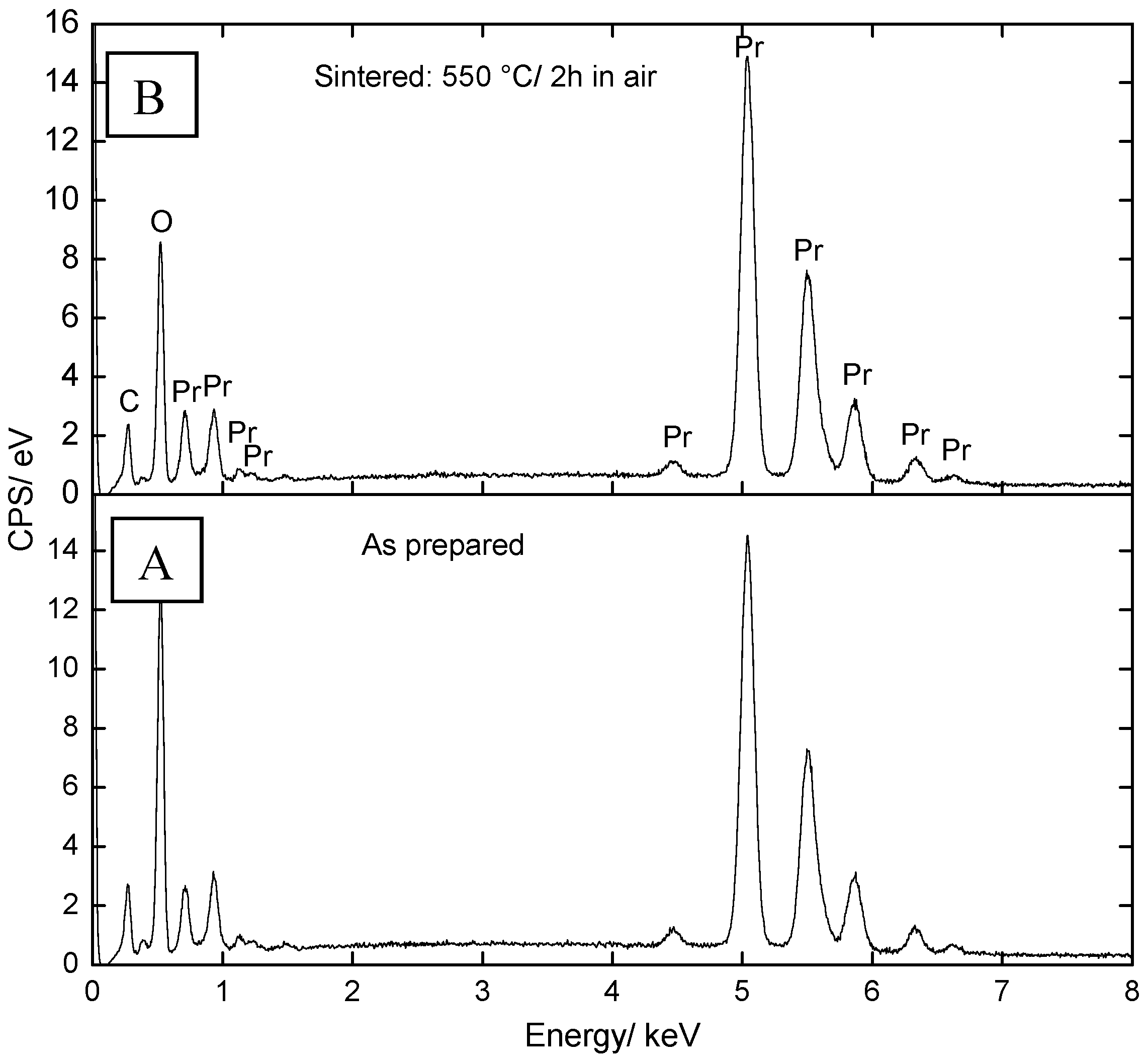
2.1. Microstructural Characterization and Elemental Analysis
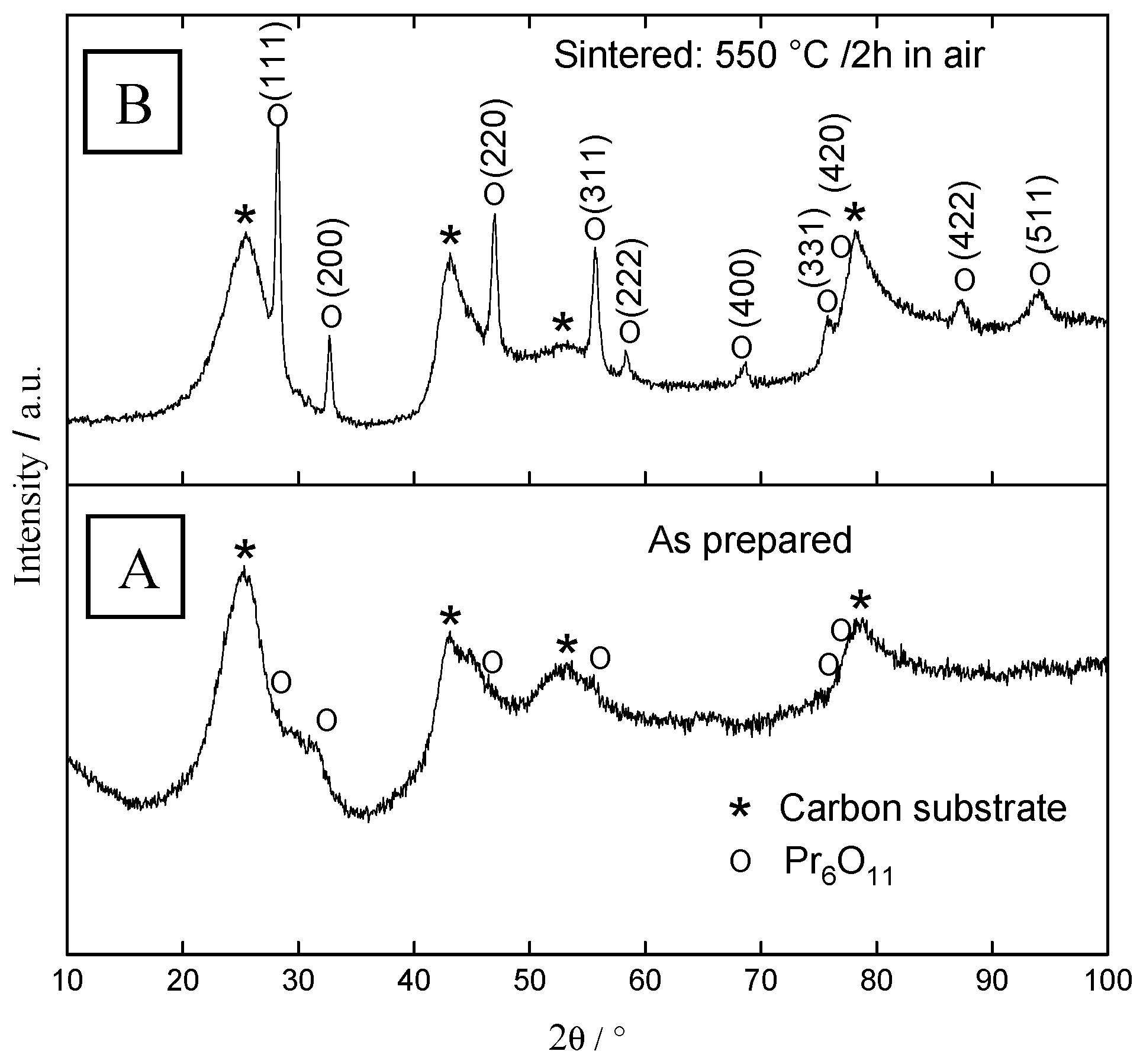
2.2. Structural Characterization
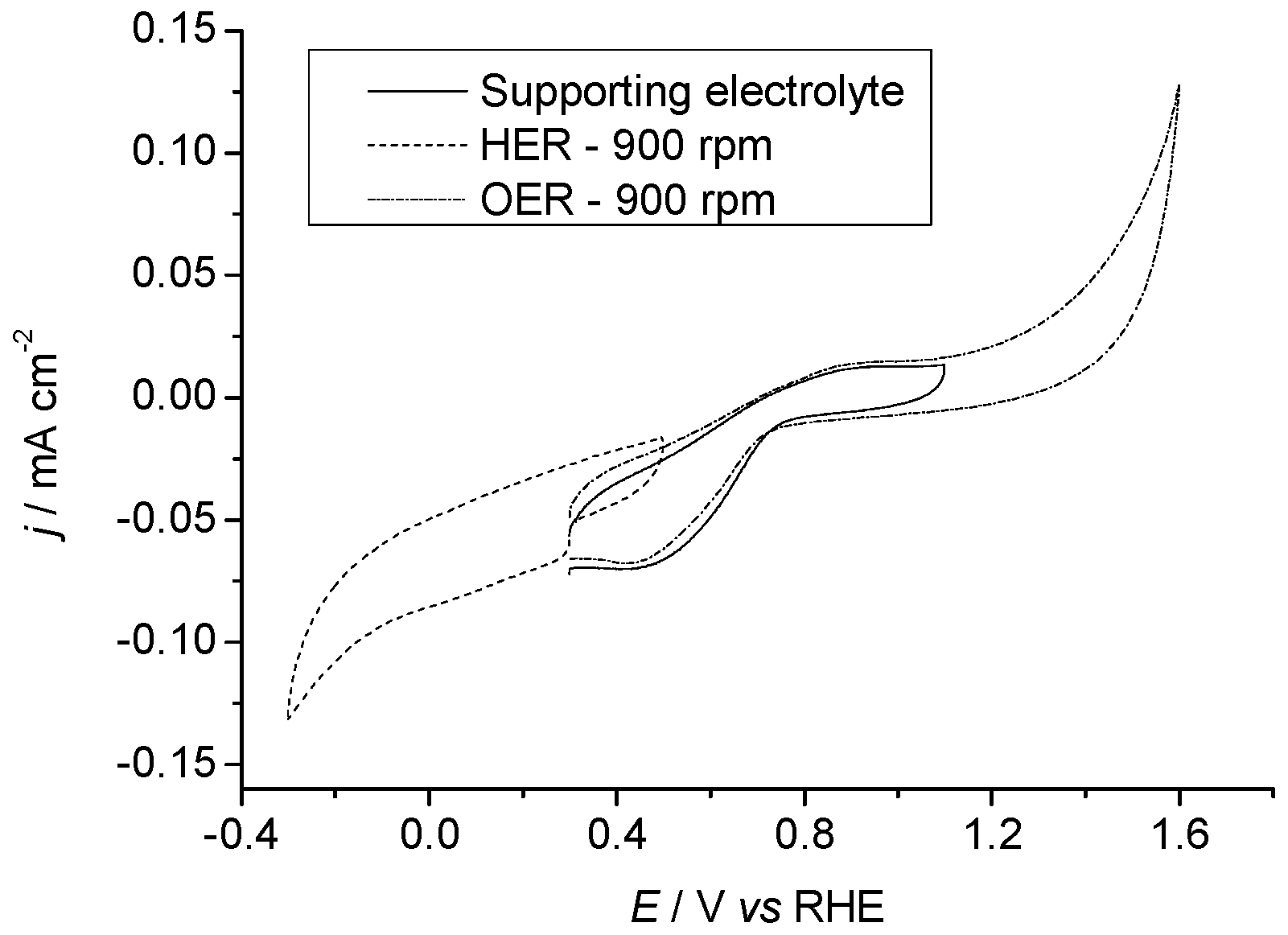
2.3. Electrochemical Properties
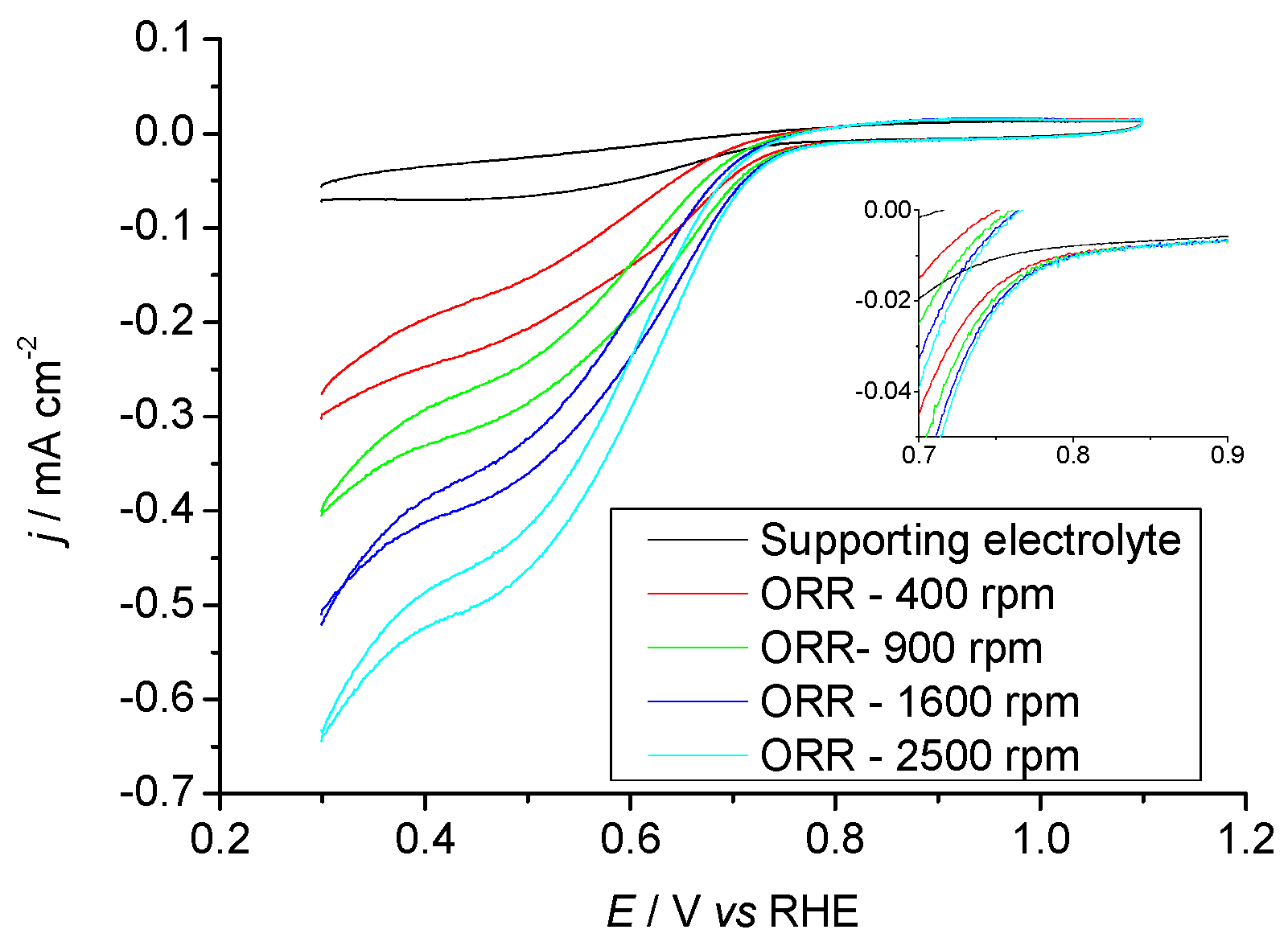
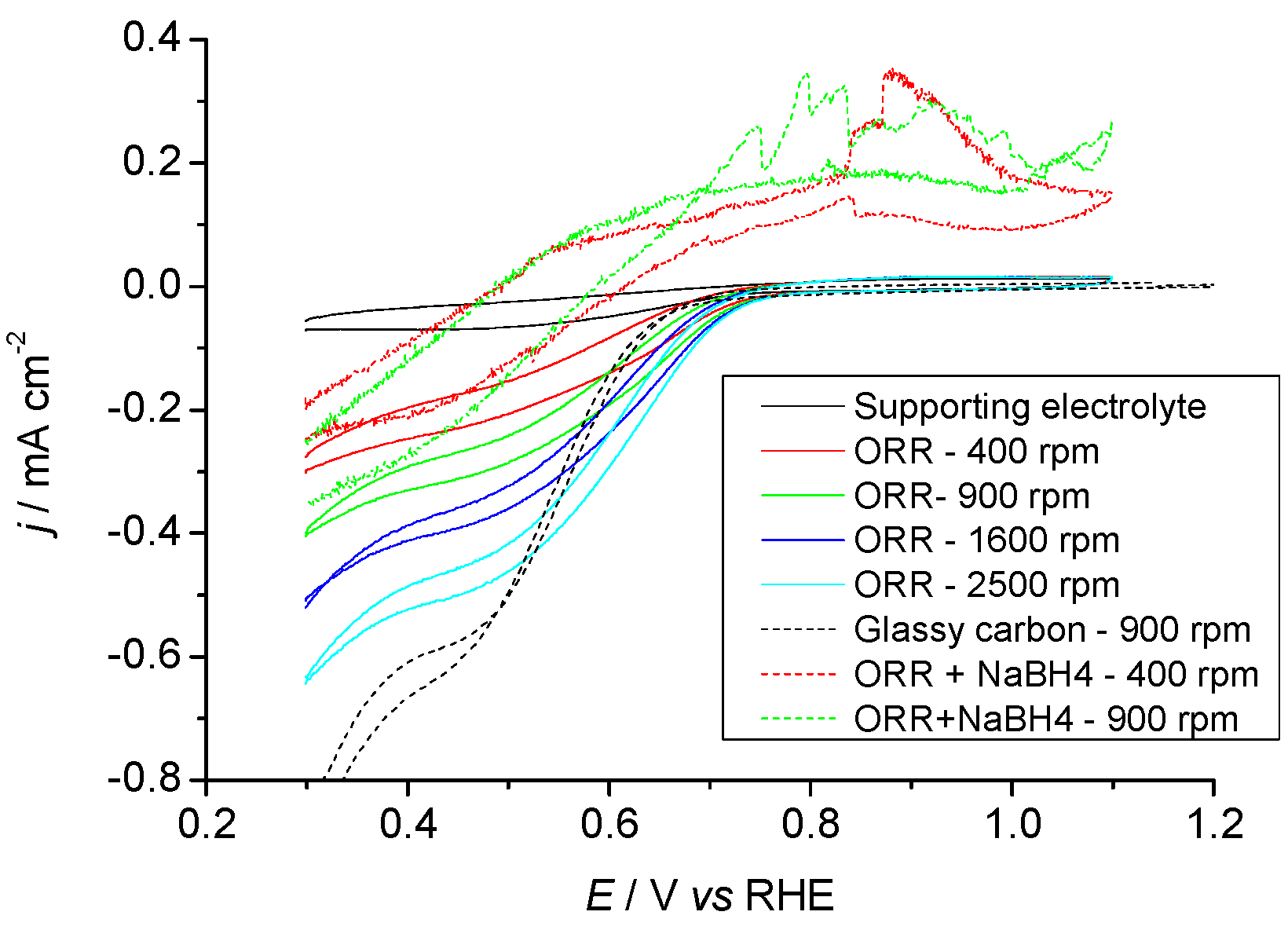
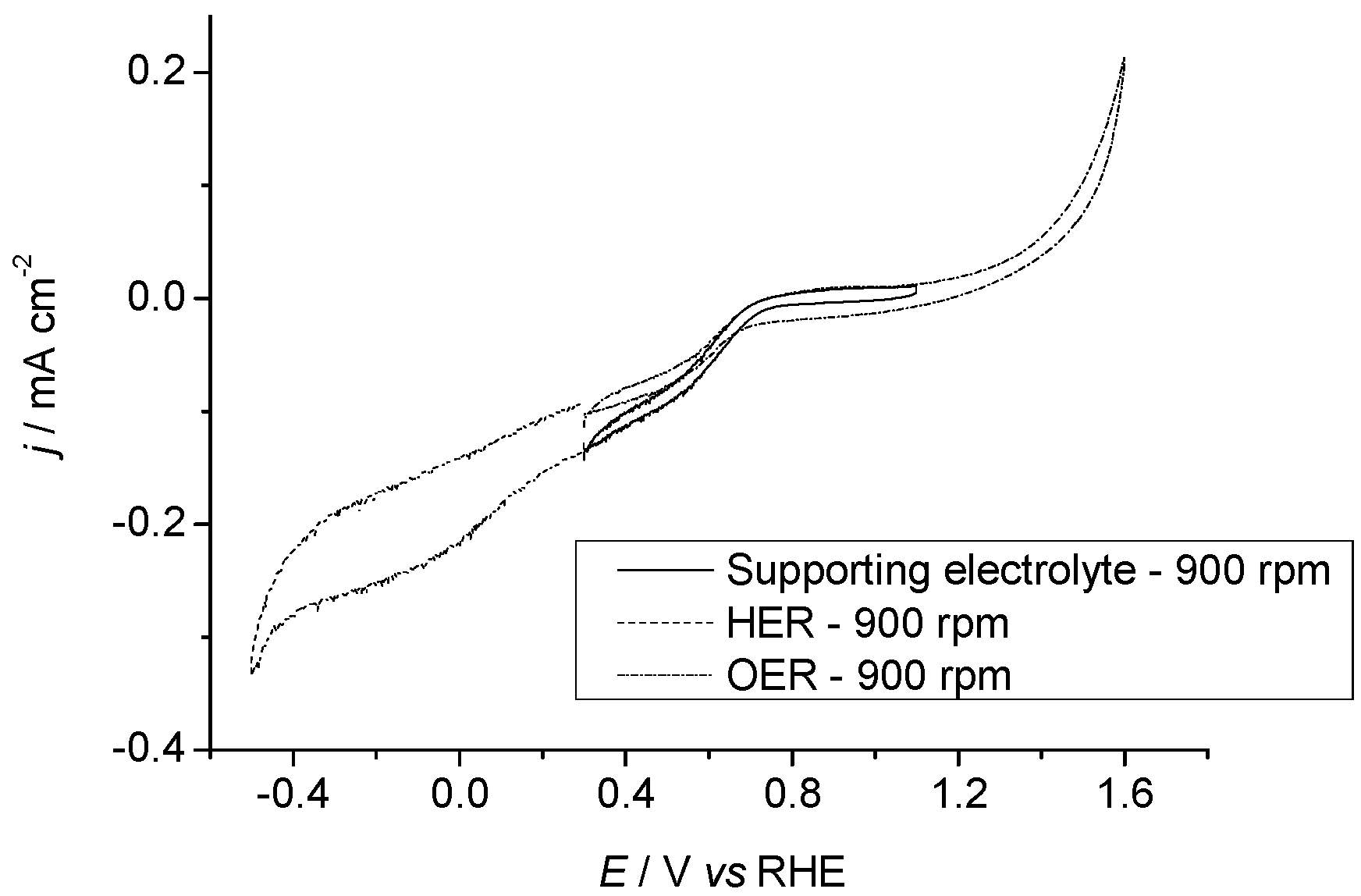
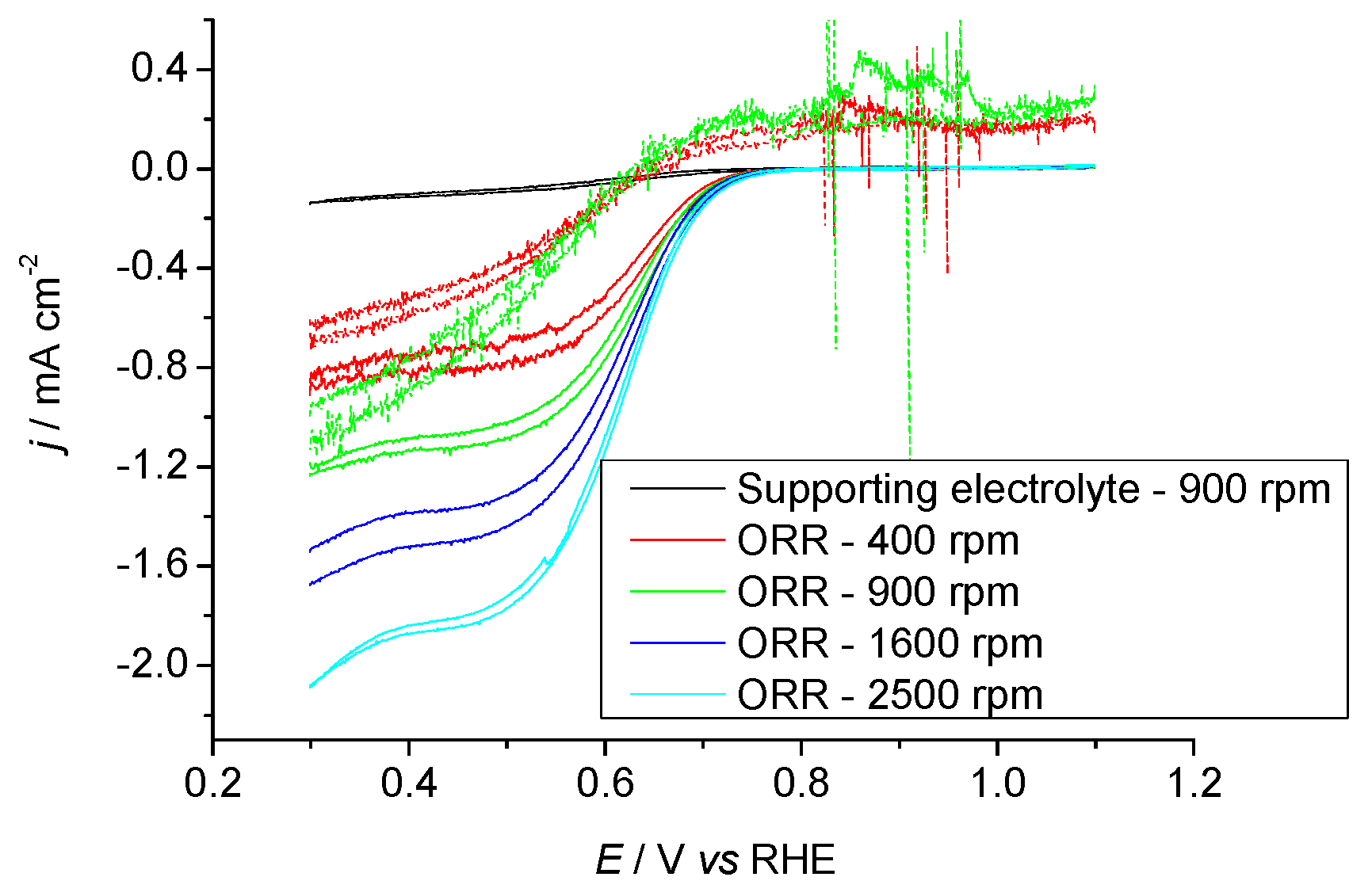
2.3.1. Pr6O11 As-Prepared (Amorphous)
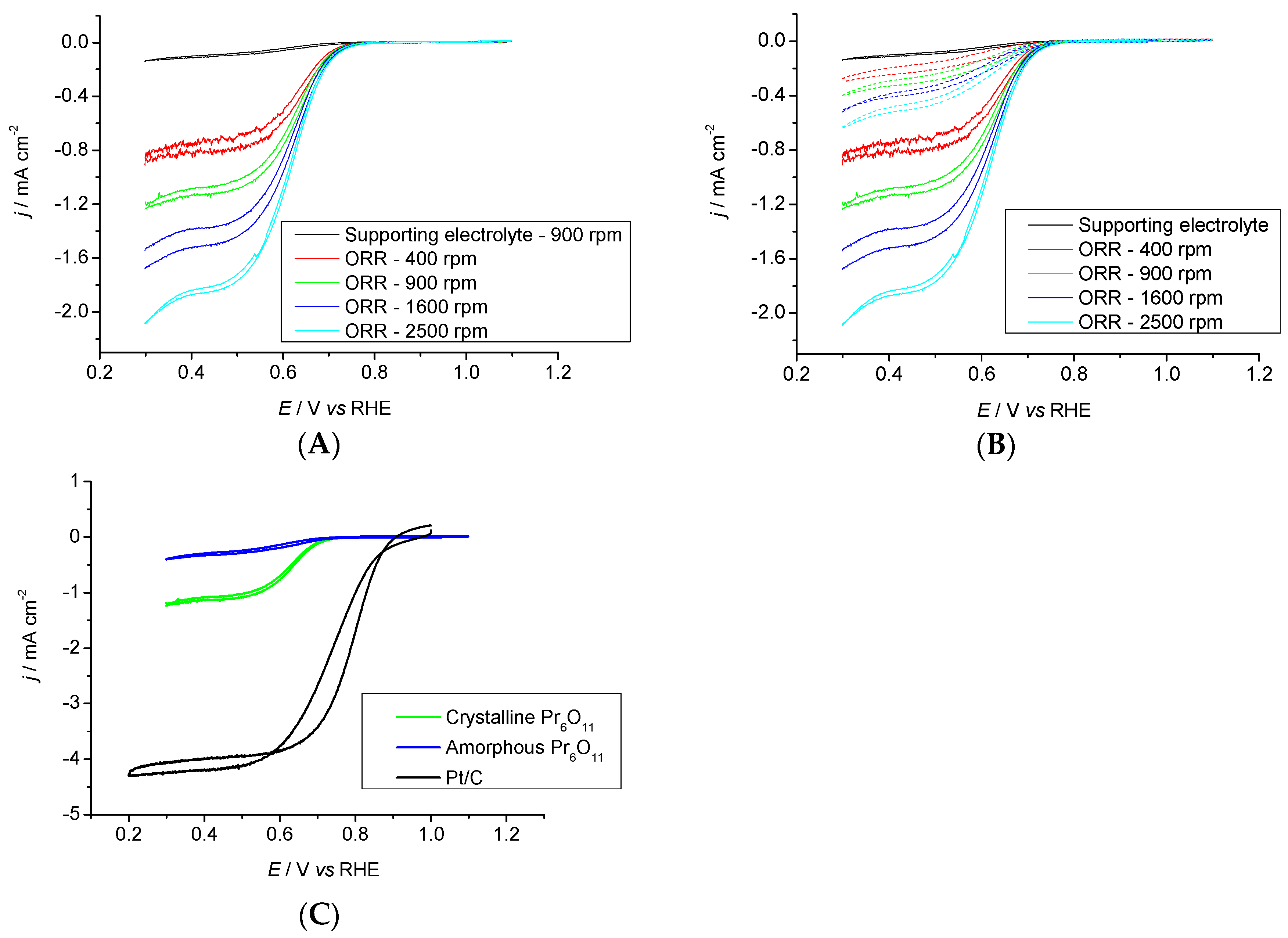
2.3.2. Pr6O11 Sintered (Crystalline)
3. Materials and Methods
3.1. Pr6O11 Film Preparation
3.2. Microstructural and Electrochemical Characterization
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghoniem, A.F. Needs, resources and climate change: Clean and efficient conversion technologies. Prog. Energ. Comb. Sci. 2011, 37, 15–51. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Marini, S.; Salvi, P.; Nelli, P.; Pesenti, R.; Villa, M.; Berrettoni, M.; Zangari, G.; Kiros, Y. Advanced alkaline water electrolysis. Electrochim. Acta 2012, 82, 384–391. [Google Scholar] [CrossRef]
- Niether, C.; Faure, S.; Bordet, A.; Deseure, J.; Chatenet, M.; Carrey, J.; Chaudret, B.; Rouet, A. Improved water electrolysis using magnetic heating of FeC–Ni core–shell nanoparticles. Nat. Energy 2018, 3, 476–483. [Google Scholar] [CrossRef]
- Chen, G.; Bare, S.R.; Mallouk, T.E. Development of Supported Bifunctional Electrocatalysts for Unitized Regenerative Fuel Cells. J. Electrochem. Soc. 2002, 149, A1092–A1099. [Google Scholar] [CrossRef]
- Narayan, S.R.; Manohar, A.K.; Mukerjee, S. Bi-functional oxygen electrodes challenges and prospects. Electrochem. Soc. Interface 2015, 24, 65–69. [Google Scholar] [CrossRef]
- Liao, P.; Carter, E.A. New concepts and modeling strategies to design and evaluate photo-electro-catalysts based on transition metal oxides. Chem. Soc. Rev. 2013, 42, 2401–2422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bieberle-Hütter, A. Modeling and Simulations in Photoelectrochemical Water Oxidation: From Single Level to Multiscale Modeling. ChemSusChem 2016, 9, 1223–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, H.A.; Vielstich, W.; Yokokawa, H. Handbook of Fuel Cells; John Wiley & Sons Ltd.: Chichester, UK, 2009. [Google Scholar]
- Vielstich, W.; Lamm, A.; Gasteiger, H.A. Handbook of Fuel Cells; Wiley: Chichester, UK, 2003. [Google Scholar]
- Jörissen, L. Bifunctional oxygen/air electrodes. J. Power Sour. 2006, 155, 23–32. [Google Scholar] [CrossRef]
- Vidakovic, T.R.; Avramov-Ivic, M.L.; Ivanovic, V.Z.; Zecevic, S.K.; Nikolic, B.Z. Electrochemical study of metal-tetrasulfonated phthalocyanines adsorbed on gold, platinum and iron electrodes and their role in electrochemical oxygen reduction. J. Serbian Chem. Soc. 1998, 63, 41–51. [Google Scholar]
- Chatenet, M.; Génies-Bultel, L.; Aurousseau, M.; Durand, R.; Andolfatto, F. Oxygen reduction on silver catalysts in solutions containing various concentrations of sodium hydroxide—Comparison with platinum. J. Appl. Electrochem. 2002, 32, 1131–1140. [Google Scholar] [CrossRef]
- Roche, I.; Chainet, E.; Chatenet, M.; Vondrak, J. Carbon-supported manganese oxide nanoparticles as electrocatalysts for the Oxygen Reduction Reaction (ORR) in alkaline medium: Physical characterizations and ORR mechanism. J. Phys. Chem. C 2007, 111, 1434–1443. [Google Scholar] [CrossRef]
- Piana, M.; Catanorchi, S.; Gasteiger, H.A. Kinetics of Non-Platinum Group Metal Catalysts for the Oxygen Reduction Reaction in Alkaline Medium. ECS Trans. 2008, 16, 2045–2055. [Google Scholar]
- Garcia, A.C.; Lima, F.H.B.; Ticianelli, E.A.; Chatenet, M. Carbon-supported nickel-doped manganese oxides as electrocatalysts for the oxygen reduction reaction in the presence of sodium borohydride. J. Power Sources 2013, 222, 305–312. [Google Scholar] [CrossRef]
- Benhangi, P.H.; Alfantazi, A.; Gyenge, E. Manganese Dioxide-based bifunctional oxygen reduction/evolution electrocatalysts: Effect of perovskite doping and potassium ion insertion. Electrochim. Acta 2014, 123, 42–50. [Google Scholar] [CrossRef]
- Tarasevich, M.R.; Davydova, E.S. Nonplatinum cathodic catalysts for fuel cells with alkaline electrolyte (Review). Rus. J. Electrochem. 2016, 52, 193–219. [Google Scholar] [CrossRef]
- Singh, R.N.; Madhu; Awasthi, R.; Tiwari, S.K. Iron molybdates as electrocatalysts for O2 evolution reaction in alkaline solutions. Int. J. Hydrogen Energy 2009, 34, 4693–4700. [Google Scholar] [CrossRef]
- Moureaux, F.; Stevens, P.; Toussaint, G.; Chatenet, M. Development of an oxygen-evolution electrode from 316L stainless steel: Application to the oxygen evolution reaction in aqueous lithium–air batteries. J. Power Sources 2013, 229, 123–132. [Google Scholar] [CrossRef]
- Suntivich, J.; Perry, E.; Gasteiger, H.; Shao-Horn, Y. The Influence of the Cation on the Oxygen Reduction and Evolution Activities of Oxide Surfaces in Alkaline Electrolyte. Electrocatalysis 2013, 4, 49–55. [Google Scholar] [CrossRef]
- Gerken, J.B.; Shaner, S.E.; Massé, R.C.; Porubsky, N.J.; Stahl, S.S. A survey of diverse earth abundant oxygen evolution electrocatalysts showing enhanced activity from Ni-Fe oxides containing a third metal. Energy Environ. Sci. 2014, 7, 2376–2382. [Google Scholar] [CrossRef]
- Bates, M.K.; Jia, Q.; Doan, H.; Liang, W.; Mukerjee, S. Charge-Transfer Effects in Ni-Fe & Ni-Fe-Co Mixed-Metal-Oxides for the Alkaline Oxygen Evolution Reaction. ACS Catal. 2015, 6, 155–161. [Google Scholar]
- Li, N.; Xia, W.Y.; Wang, J.; Liu, Z.L.; Li, Q.Y.; Chen, S.Z.; Xu, C.W.; Lu, X.H. Manganese oxides supported on hydrogenated TiO2 nanowire array catalysts for the electrochemical oxygen evolution reaction in water electrolysis. J. Mater. Chem. A 2015, 3, 21308–21313. [Google Scholar] [CrossRef]
- Schäfer, H.; Chatenet, M. Steel: The Resurrection of a Forgotten Water-Splitting Catalyst. ACS Energy Lett. 2018, 3, 574–591. [Google Scholar] [CrossRef]
- Davodi, F.; Mühlhausen, E.; Tavakkoli, M.; Sainio, J.; Jiang, H.; Gökce, B.; Marzun, G.; Kallio, T. Catalyst Support Effect on the Activity and Durability of Magnetic Nanoparticles: Toward Design of Advanced Electrocatalyst for Full Water Splitting. ACS Appl. Mater. Interfaces 2018, 10, 31300–31311. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Martí-Sànchez, S.; Nafria, R.; Joshua, G.; de la Mata, M.; Guardia, P.; Flox, C.; Martínez-Boubeta, C.; Simeonidis, K.; Llorca, J.; et al. Fe3O4@NiFexOy Nanoparticles with Enhanced Electrocatalytic Properties for Oxygen Evolution in Carbonate Electrolyte. ACS Appl. Mater. Interfaces 2016, 8, 29461–29469. [Google Scholar] [CrossRef] [PubMed]
- Davodi, F.; Tavakkoli, M.; Lahtinen, J.; Kallio, T. Straightforward synthesis of nitrogen-doped carbon nanotubes as highly active bifunctional electrocatalysts for full water splitting. J. Catal. 2017, 353, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Neburchilov, V.; Wang, H.; Martin, J.J.; Qu, W. A review on air cathodes for zinc-air fuel cells. J. Power Sources 2010, 195, 1271–1291. [Google Scholar] [CrossRef]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; Li, B.; Goh, F.W.T.; Hor, T.S.A.; Zong, Y.; Liu, Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- Risch, M. Perovskite electrocatalysts for the Oxygen reduction reaction in alkaline media. Catalysts 2017, 7, 154. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Shao, Z. Perovskite/Carbon Composites: Applications in Oxygen Electrocatalysis. Small 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Poux, T.; Napolskiy, F.S.; Dintzer, T.; Kéranguéven, G.; Istomin, S.Y.; Tsirlina, G.A.; Antipov, E.V.; Savinova, E.R. Dual role of carbon in the catalytic layers of perovskite/carbon composites for the electrocatalytic oxygen reduction reaction. Catal. Today 2012, 189, 83–92. [Google Scholar] [CrossRef]
- Sunarso, J.; Torriero, A.A.J.; Zhou, W.; Howlett, P.C.; Forsyth, M. Oxygen reduction reaction activity of La-based perovskite oxides in alkaline medium: A thin-film rotating ring-disk electrode study. J. Phys. Chem. C 2012, 116, 5827–5834. [Google Scholar] [CrossRef]
- Fabbri, E.; Mohamed, R.; Levecque, P.; Conrad, O.; Kötz, R.; Schmidt, T.J. Composite electrode boosts the activity of Ba0.5Sr0.5Co0.8Fe0.2O3−δ perovskite and carbon toward oxygen reduction in alkaline media. ACS Catal. 2014, 4, 1061–1070. [Google Scholar] [CrossRef]
- Poux, T.; Bonnefont, A.; Kéranguéven, G.; Tsirlina, G.A.; Savinova, E.R. Electrocatalytic oxygen reduction reaction on perovskite oxides: Series versus direct pathway. ChemPhysChem 2014, 15, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Catalytic roles of perovskite oxides in electrochemical oxygen reactions in alkaline media. J. Electrochem. Soc. 2014, 161, F694–F697. [Google Scholar] [CrossRef]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Influence of Surface Orientation on the Catalytic Activities of La0.8Sr0.2CoO3 Crystal Electrodes for Oxygen Reduction and Evolution Reactions. ChemElectroChem 2016, 3, 214–217. [Google Scholar] [CrossRef]
- Celorrio, V.; Dann, E.; Calvillo, L.; Morgan, D.J.; Hall, S.R.; Fermin, D.J. Oxygen Reduction at Carbon-Supported Lanthanides: TheRole of the B-Site. ChemElectroChem 2016, 3, 283–291. [Google Scholar] [CrossRef]
- Celorrio, V.; Calvillo, L.; Granozzi, G.; Russell, A.E.; Fermin, D.J. AMnO3 (A = Sr, La, Ca, Y) Perovskite Oxides as Oxygen Reduction Electrocatalysts. Top. Catal. 2018, 61, 154–161. [Google Scholar] [CrossRef]
- Gobaille-Shaw, G.P.A.; Celorrio, V.; Calvillo, L.; Morris, L.J.; Granozzi, G.; Fermín, D.J. Effect of Ba Content on the Activity of La1−xBaxMnO3 Towards the Oxygen Reduction Reaction. ChemElectroChem 2018, 5, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.I.; Risch, M.; Park, S.; Kim, M.G.; Nam, G.; Jeong, H.Y.; Shao-Horn, Y.; Cho, J. Optimizing nanoparticle perovskite for bifunctional oxygen electrocatalysis. Energy Environ. Sci. 2016, 9, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Stoerzinger, K.A.; Choi, W.S.; Jeen, H.; Lee, H.N.; Shao-Horn, Y. Role of strain and conductivity in oxygen electrocatalysis on LaCoO3 thin films. J. Phys. Chem. Lett. 2015, 6, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Gwon, O.; Jo, H.; Ok, K.M.; Kim, G. Major Role of Surface Area in Perovskite Electrocatalysts for Alkaline Systems. ChemElectroChem 2017, 4, 468–471. [Google Scholar] [CrossRef]
- Huang, K.; Yuan, L.; Jiang, Y.; Zhang, J.; Geng, Z.; Luo, L.; Feng, S. Hydrothermal shape controllable synthesis of La0.5Sr0.5MnO3 crystals and facet effect on electron transfer of oxygen reduction. Inorg. Chem. Front. 2018, 5, 732–738. [Google Scholar] [CrossRef]
- Yu, J.; Chen, D.; Saccoccio, M.; Lam, K.; Ciucci, F. Promotion of Oxygen Reduction with Both Amorphous and Crystalline MnOx through the Surface Engineering of La0.8Sr0.2MnO3−δ Perovskite. ChemElectroChem 2018, 5, 1105–1112. [Google Scholar] [CrossRef]
- Hua, B.; Zhang, Y.Q.; Yan, N.; Li, M.; Sun, Y.F.; Chen, J.; Li, J.; Luo, J.L. The Excellence of Both Worlds: Developing Effective Double Perovskite Oxide Catalyst of Oxygen Reduction Reaction for Room and Elevated Temperature Applications. Adv. Funct. Mater. 2016, 26, 4106–4112. [Google Scholar] [CrossRef]
- Chiba, R.; Aono, H.; Kato, K. An SOFC cathode infiltrated with Pr6O11. ECS Trans. 2013, 57, 1831–1840. [Google Scholar] [CrossRef]
- Ding, X.; Zhu, W.; Hua, G.; Li, J.; Wu, Z. Enhanced oxygen reduction activity on surface-decorated perovskite La0.6Ni0.4FeO3 cathode for solid oxide fuel cells. Electrochim. Acta 2015, 163, 204–212. [Google Scholar] [CrossRef]
- Sharma, R.K.; Djurado, E. An efficient hierarchical nanostructured Pr6O11 electrode for solid oxide fuel cells. J. Mater. Chem. A 2018, 6, 10787–10802. [Google Scholar] [CrossRef]
- Chen, C.H.; Emond, M.H.J.; Kelder, E.M.; Meester, B.; Schoonman, J. Electrostatic sol-spray deposition of nanostructured ceramic thin films. J. Aerosol Sci. 1999, 30, 959–967. [Google Scholar] [CrossRef]
- Jaworek, A. Electrospray droplet sources for thin film deposition. J. Mater. Sci. 2007, 42, 266–297. [Google Scholar] [CrossRef]
- Marinha, D.; Rossignol, C.; Djurado, E. Influence of electrospraying parameters on the microstructure of La0.6Sr0.4Co0.2F0.8O3−δ films for SOFCs. J. Solid State Chem. 2009, 182, 1742–1748. [Google Scholar] [CrossRef]
- Marinha, D.; Hayd, J.; Dessemond, L.; Ivers-Tiffée, E.; Djurado, E. Performance of (La,Sr)(Co,Fe)O3−x double-layer cathode films for intermediate temperature solid oxide fuel cell. J. Power Sources 2011, 196, 5084–5090. [Google Scholar] [CrossRef]
- Sharma, R.K.; Burriel, M.; Djurado, E. La4Ni3O10−δ as an efficient solid oxide fuel cell cathode: Electrochemical properties versus microstructure. J. Mater. Chem. A 2015, 3, 23833–23843. [Google Scholar] [CrossRef]
- Sharma, R.K.; Burriel, M.; Dessemond, L.; Martin, V.; Bassat, J.M.; Djurado, E. An innovative architectural design to enhance the electrochemical performance of La2NiO4+δ cathodes for solid oxide fuel cell applications. J. Power Sources 2016, 316, 17–28. [Google Scholar] [CrossRef]
- Marinha, D.; Dessemond, L.; Djurado, E. Comprehensive Review of Current Developments in IT-SOFCs. Curr. Inorg. Chem. 2013, 3, 2–22. [Google Scholar] [CrossRef]
- Gañán-Calvo, A.M.; Dávila, J.; Barrero, A. Current and droplet size in the electrospraying of liquids. Scaling laws. J. Aerosol Sci. 1997, 28, 249–275. [Google Scholar] [CrossRef]
- Jung, K.N.; Jung, J.H.; Im, W.B.; Yoon, S.; Shin, K.H.; Lee, J.W. Doped lanthanum nickelates with a layered perovskite structure as bifunctional cathode catalysts for rechargeable metal-air batteries. ACS Appl. Mater. Interfaces 2013, 5, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; McCrory, C.C.L.; Ferrer, I.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking nanoparticulate metal oxide electrocatalysts for the alkaline water oxidation reaction. J. Mater. Chem. A 2016, 4, 3068–3076. [Google Scholar] [CrossRef] [Green Version]
- Menezes, P.W.; Indra, A.; Bergmann, A.; Chernev, P.; Walter, C.; Dau, H.; Strasser, P.; Driess, M. Uncovering the prominent role of metal ions in octahedral: Versus tetrahedral sites of cobalt-zinc oxide catalysts for efficient oxidation of water. J. Mater. Chem. A 2016, 4, 10014–10022. [Google Scholar] [CrossRef]
- Menezes, P.W.; Indra, A.; González-Flores, D.; Sahraie, N.R.; Zaharieva, I.; Schwarze, M.; Strasser, P.; Dau, H.; Driess, M. High-Performance Oxygen Redox Catalysis with Multifunctional Cobalt Oxide Nanochains: Morphology-Dependent Activity. ACS Catal. 2015, 5, 2017–2027. [Google Scholar] [CrossRef]
- Menezes, P.W.; Indra, A.; Sahraie, N.R.; Bergmann, A.; Strasser, P.; Driess, M. Cobalt–Manganese-Based Spinels as Multifunctional Materials that Unify Catalytic Water Oxidation and Oxygen Reduction Reactions. ChemSusChem 2015, 8, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-Y.; Gorlin, Y.; Man, I.C.; Calle-Vallejo, F.; Norskov, J.K.; Jaramillo, T.F.; Rossmeisl, J. Identifying active surface phases for metal oxide electrocatalysts: A study of manganese oxide bi-functional catalysts for oxygen reduction and water oxidation catalysis. Phys. Chem. Chem. Phys. 2012, 14, 14010–14022. [Google Scholar] [CrossRef] [PubMed]
- Poux, T.; Bonnefont, A.; Ryabova, A.; Kéranguéven, G.; Tsirlina, G.A.; Savinova, E.R. Electrocatalysis of hydrogen peroxide reactions on perovskite oxides: Experiment versus kinetic modeling. Phys. Chem. Chem. Phys. 2014, 16, 13595–13600. [Google Scholar] [CrossRef] [PubMed]
- Case, B. The diffusivity of oxygen in dilute alkaline solution from 0° to 65 °C. Electrochim. Acta 1973, 18, 293–299. [Google Scholar] [CrossRef]
- Yang, X.; Li, S.; Liiu, Y.; Wei, X.; Liu, Y. LaNi0.8Co0.2O3 as a cathode catalyst for a direct borohydride fuel cell. J. Power Sources 2011, 196, 4992–4995. [Google Scholar] [CrossRef]
- Serov, A.; Aziznia, A.; Benhangi, P.H.; Artyushkova, K.; Atanassov, P.; Gyenge, E. Borohydride-tolerant oxygen electroreduction catalyst for mixed-reactant Swiss-roll direct borohydride fuel cells. J. Mater. Chem. A 2013, 1, 14384–14391. [Google Scholar] [CrossRef]
- Liu, B.H.; Li, Z.P. Current status and progress of direct borohydride fuel cell technology development. J. Power Sources 2009, 187, 291–297. [Google Scholar] [CrossRef]
- Cheng, H.; Scott, K. Investigation of non-platinum cathode catalysts for direct borohydride fuel cells. J. Electroanal. Chem. 2006, 596, 117–123. [Google Scholar] [CrossRef]
- Baranton, S.; Coutanceau, C.; Roux, C.; Hahn, F.; Léger, J.M. Oxygen reduction reaction in acid medium at iron phthalocyanine dispersed on high surface area carbon substrate: Tolerance to methanol, stability and kinetics. J. Electroanal. Chem. 2005, 577, 223–234. [Google Scholar] [CrossRef]
- Chatenet, M.; Micoud, F.; Roche, I.; Chainet, E.; Vondrak, J. Kinetics of sodium borohydride direct oxidation and oxygen reduction in sodium hydroxide electrolyte—Part II. O2 reduction. Electrochim. Acta 2006, 51, 5452–5458. [Google Scholar] [CrossRef]
- Garcia, A.; Linares, J.; Chatenet, M.; Ticianelli, E. NiMnOx/C: A Non-noble Ethanol-Tolerant Catalyst for Oxygen Reduction in Alkaline Exchange Membrane DEFC. Electrocatalysis 2014, 5, 41–49. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.A.; Goya, M.C.; Arévalo, M.C.; Rodríguez, J.L.; Pastor, E. Carbon supported Ag and Ag–Co catalysts tolerant to methanol and ethanol for the oxygen reduction reaction in alkaline media. Int. J. Hydrogen Energy 2016, 41, 19789–19798. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Yu, J.; Chen, Y.; Liu, M.; Shao, Z. Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution Reactions. Chem. Mater. 2016, 28, 1691–1697. [Google Scholar] [CrossRef]
- Li, Z.; Lv, L.; Wang, J.; Ao, X.; Ruan, Y.; Zha, D.; Hong, G.; Wu, Q.; Lan, Y.; Wang, C.; et al. Engineering phosphorus-doped LaFeO3−δ perovskite oxide as robust bifunctional oxygen electrocatalysts in alkaline solutions. Nano Energy 2018, 47, 199–209. [Google Scholar] [CrossRef]
- Liu, X.; Gong, H.; Wang, T.; Guo, H.; Song, L.; Xia, W.; Gao, B.; Jiang, Z.; Feng, L.; He, J. Cobalt-Doped Perovskite-Type Oxide LaMnO3 as Bifunctional Oxygen Catalysts for Hybrid Lithium–Oxygen Batteries. Chem. Asian J. 2018, 13, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Afzal, R.A.; Park, K.Y.; Cho, S.H.; Kim, N.I.; Choi, S.R.; Kim, J.H.; Lim, H.T.; Park, J.Y. Oxygen electrode reactions of doped BiFeO3 materials for low and elevated temperature fuel cell applications. RSC Adv. 2017, 7, 47643–47653. [Google Scholar] [CrossRef]
- Dzara, M.J.; Christ, J.M.; Joghee, P.; Ngo, C.; Cadigan, C.A.; Bender, G.; Richards, R.M.; O’Hayre, R.; Pylypenko, S. La and Al co-doped CaMnO3 perovskite oxides: From interplay of surface properties to anion exchange membrane fuel cell performance. J. Power Sources 2018, 375, 265–276. [Google Scholar] [CrossRef]
- Hua, B.; Sun, Y.F.; Li, M.; Yan, N.; Chen, J.; Zhang, Y.Q.; Zeng, Y.; Shalchi Amirkhiz, B.; Luo, J.L. Stabilizing Double Perovskite for Effective Bifunctional Oxygen Electrocatalysis in Alkaline Conditions. Chem. Mater. 2017, 29, 6228–6237. [Google Scholar] [CrossRef]
- Chen, C.F.; King, G.; Dickerson, R.M.; Papin, P.A.; Gupta, S.; Kellogg, W.R.; Wu, G. Oxygen-deficient BaTiO3−x perovskite as an efficient bifunctional oxygen electrocatalyst. Nano Energy 2015, 13, 423–432. [Google Scholar] [CrossRef]
- Sharma, R.K.; Burriel, M.; Dessemond, L.; Bassat, J.M.; Djurado, E. Lan+1NinO3n+1 (n = 2 and 3) phases and composites for solid oxide fuel cell cathodes: Facile synthesis and electrochemical properties. J. Power Sources 2016, 325, 337–345. [Google Scholar] [CrossRef]
- Sharma, R.K.; Burriel, M.; Dessemond, L.; Bassat, J.M.; Djurado, E. Design of interfaces in efficient Ln2NiO4+δ (Ln = La, Pr) cathodes for SOFC applications. J. Mater. Chem. A 2016, 4, 12451–12462. [Google Scholar] [CrossRef]
- Djurado, E.; Salaün, A.; Mignardi, G.; Rolle, A.; Burriel, M.; Daviero-Minaud, S.; Vannier, R.N. Electrostatic spray deposition of Ca3Co4O9 + δ layers to be used as cathode materials for IT-SOFC. Solid State Ionics 2016, 286, 102–110. [Google Scholar] [CrossRef]
- Çelikbilek, Ӧ.; Jauffrès, D.; Siebert, E.; Dessemond, L.; Burriel, M.; Martin, C.L.; Djurado, E. Rational design of hierarchically nanostructured electrodes for solid oxide fuel cells. J. Power Sources 2016, 333, 72–82. [Google Scholar] [CrossRef]
- Constantin, G.; Rossignol, C.; Barnes, J.P.; Djurado, E. Interface stability of thin, dense CGO film coating on YSZ for solid oxide fuel cells. Solid State Ionics 2013, 235, 36–41. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Phys. Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Chatenet, M.; Concha, B.M.; Parrour, G.; Diard, J.-P.; Lima, F.H.B.; Ticianelli, E.A. Potential and limitation of the Direct Borohydride Fuel Cell. Special emphasis on the Borohydride Oxidation Reaction (BOR) mechanism and kinetics on gold electrocatalysts. In Boron Hydrides, High Potential Hydrogen Storage Materials; Demirci, U.B., Miele, P., Eds.; Nova Science: New York, NY, USA, 2011; pp. 103–135. [Google Scholar]
- Olu, P.-Y.; Deschamps, F.; Caldarella, G.; Chatenet, M.; Job, N. Investigation of platinum and palladium as potential anodic catalysts for direct borohydride and ammonia borane fuel cells. J. Power Sources 2015, 297, 492–503. [Google Scholar] [CrossRef]
- Olu, P.-Y.; Job, N.; Chatenet, M. Evaluation of anode (electro)catalytic materials for the direct borohydride fuel cell: Methods and benchmarks. J. Power Sources 2016, 327, 235–257. [Google Scholar] [CrossRef]
- Chen, J.G.; Jones, C.W.; Linic, S.; Stamenkovic, V.R. Best Practices in Pursuit of Topics in Heterogeneous Electrocatalysis. ACS Catal. 2017, 7, 6392–6393. [Google Scholar] [CrossRef]
- Zadick, A.; Dubau, L.; Sergent, N.; Berthomé, G.; Chatenet, M. Huge Instability of Pt/C Catalysts in Alkaline Medium. ACS Catal. 2015, 5, 4819–4824. [Google Scholar] [CrossRef]
- Lafforgue, C.; Zadick, A.; Dubau, L.; Maillard, F.; Chatenet, M. Selected review of the degradation of Pt and Pd-based carbonsupported electrocatalysts for alkaline fuel cells: Towards mechanisms of degradation. Fuel Cells 2018, 18, 229–238. [Google Scholar] [CrossRef]
- Cherevko, S.; Zeradjanin, A.R.; Keeley, G.P.; Mayrhofer, K.J.J. A comparative study on gold and platinum dissolution in acidic and alkaline media. J. Electrochem. Soc. 2014, 161, H822–H830. [Google Scholar] [CrossRef]
- Schalenbach, M.; Kasian, O.; Ledendecker, M.; Speck, F.D.; Mingers, A.M.; Mayrhofer, K.J.J.; Cherevko, S. The Electrochemical Dissolution of Noble Metals in Alkaline Media. Electrocatalysis 2018, 9, 153–161. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Wiberg, G.K.H.; Arenz, M. Impact of Glass Corrosion on the Electrocatalysis on Pt Electrodes in Alkaline Electrolyte. J. Electrochem. Soc. 2008, 155, P1–P5. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.K.; Müller, V.; Chatenet, M.; Djurado, E. Oxygen Reduction Reaction Electrocatalysis in Alkaline Electrolyte on Glassy-Carbon-Supported Nanostructured Pr6O11 Thin-Films. Catalysts 2018, 8, 461. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100461
Sharma RK, Müller V, Chatenet M, Djurado E. Oxygen Reduction Reaction Electrocatalysis in Alkaline Electrolyte on Glassy-Carbon-Supported Nanostructured Pr6O11 Thin-Films. Catalysts. 2018; 8(10):461. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100461
Chicago/Turabian StyleSharma, Rakesh K., Verónica Müller, Marian Chatenet, and Elisabeth Djurado. 2018. "Oxygen Reduction Reaction Electrocatalysis in Alkaline Electrolyte on Glassy-Carbon-Supported Nanostructured Pr6O11 Thin-Films" Catalysts 8, no. 10: 461. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100461