Stereoselective Enzymatic Reduction of 1,4-Diaryl-1,4-Diones to the Corresponding Diols Employing Alcohol Dehydrogenases

 , and
, and
Abstract
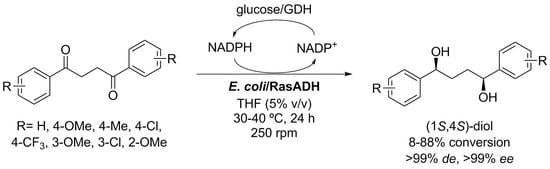
:
1. Introduction
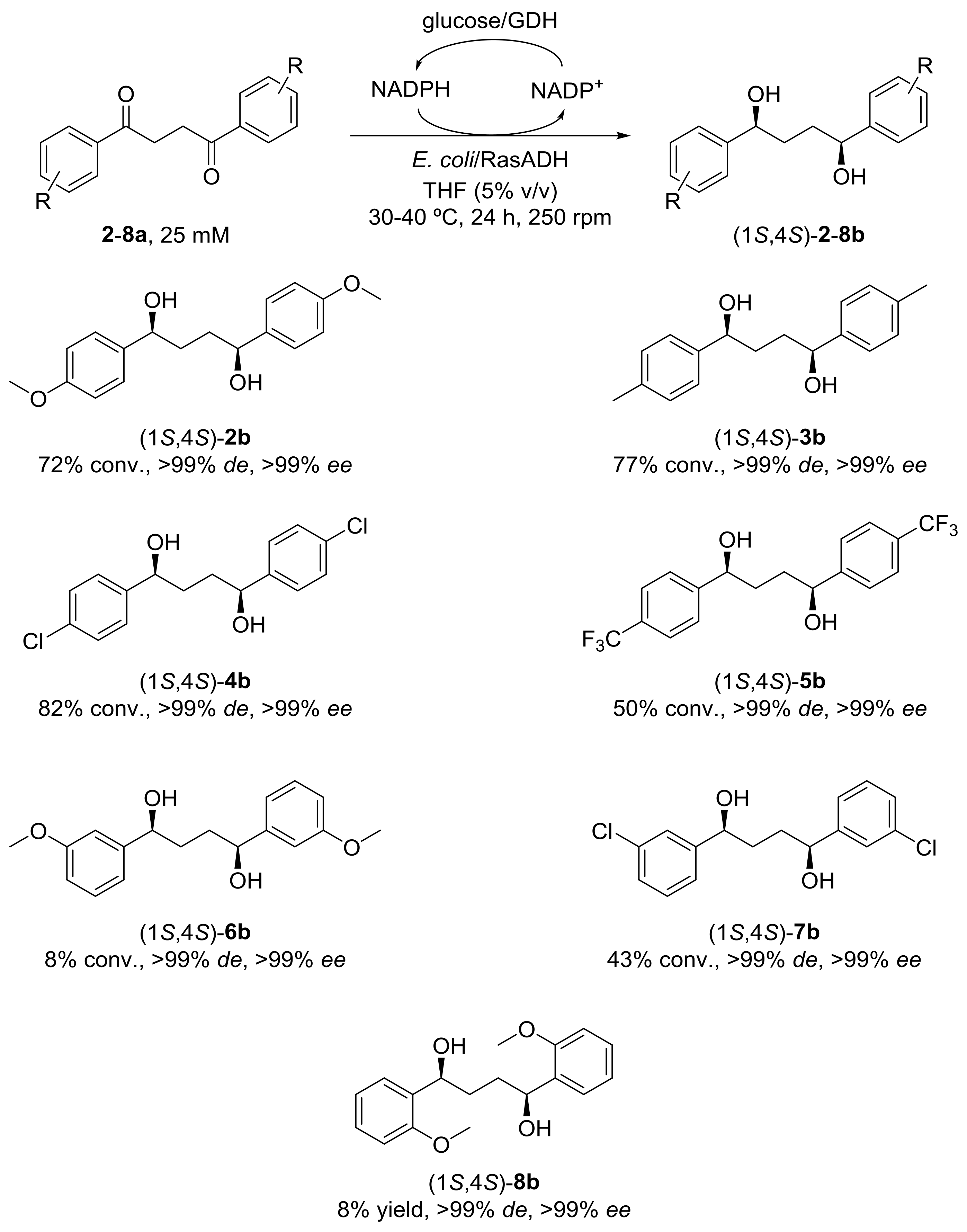
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Materials and Methods
4.2. General Procedure for the Synthesis of 1,4-Diaryl-1,4-Diketones 1–8a
4.3. General Procedure for the Synthesis of Racemic 1,4-Diaryl-1,4-Diols 1–8b
4.4. General Procedure for the Synthesis of Racemic 4-Hydroxy-1,4-Bis(2-methoxyphenyl)butan-1-one 8c
4.5. General Procedure for the Enzymatic Conversion of 1,4-Diaryl-1,4-Diols 1–8b Using Overexpressed E. coli/RasADH
4.6. General Procedure for the Bioreduction of 1,4-Diphenylbutane-1,4-Dione 1a Using Commercial Alcohol Dehydrogenases
4.7. Preparative Bioreductions of 1,4-Diarylbutane-1,4-Diones 1–5a, 7a, and 8a Using Overexpressed E. coli/RasADH
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Robinson, A.; Aggarwal, V. Asymmetric total synthesis of solandelactone E: Stereocontrolled synthesis of the 2-ene-1,4-diol core through a lithiation–borylation–allylation sequence. Angew. Chem. 2010, 122, 6823–6825. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Heckel, A. TADDOLs, Their Derivatives, and TADDOL Analogues: Versatile Chiral Auxiliaries. Angew. Chem. Int. Ed. 2001, 40, 92–138. [Google Scholar] [CrossRef]
- Denmark, S.E.; Chang, W.-T.T.; Houk, K.N.; Liu, P. Development of chiral bis-hydrazone ligands for the enantioselective cross-coupling reactions of aryldimethylsilanolates. J. Org. Chem. 2015, 80, 313–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sweet, J.A.; Lam, K.-C.; Rheingold, A.; McGrath, D.V. Chiral amine–imine ligands based on trans-2,5-disubstituted pyrrolidines and their application in the palladium-catalyzed allylic alkylation. Tetrahedron Asymmetry 2009, 20, 1672–1682. [Google Scholar] [CrossRef]
- Melchiorre, P.; Jorgensen, K.A. Direct enantioselective Michael addition of aldehydes to vinyl ketones catalyzed by chiral amines. J. Org. Chem. 2003, 68, 4151–4157. [Google Scholar] [CrossRef] [PubMed]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef]
- Hynes, J.T.; Klinman, J.P.; Limbach, H.-H.; Schowen, R.L. (Eds.) Hydrogen Transfer Reactions; Wiley-VCH: Weinheim, Germany, 2007; Volumes 1–4. [Google Scholar]
- Zheng, L.-S.; Llopis, Q.; Echeverria, P.-G.; Férand, C.; Guillamot, G.; Phansavath, P.; Ratovelomanana-Vidal, V. Asymmetric transfer hydrogenation of (hetero)arylketones with tethered Rh(III)−N-(p-tolylsulfonyl)-1,2-diphenylethylene-1,2-diamine complexes: Scope and limitations. J. Org. Chem. 2017, 82, 5607–5615. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Helal, C.J. Reduction of carbonyl compounds with chiral oxazaborolidine catalyst: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Ed. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Mathre, D.J.; Thomson, A.S.; Douglas, A.W.; Hoogsteen, K.; Carroll, J.D.; Corley, E.G.; Grabowski, E.J.J. A practical process for the preparation of tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c][1,3,2]oxazaborole-borane. A highly enantioselective stoichiometric and catalytic reducing agent. J. Org. Chem. 1993, 58, 2880–2888. [Google Scholar] [CrossRef]
- Li, X.; Zhao, G.; Cao, W.-G. An efficient method for catalytic asymmetric reduction of diketones and application of synthesis to chiral 2,5-diphenylpirrolidine and 2,5-diphenylthiolane. Chin. J. Chem. 2006, 24, 1402–1405. [Google Scholar] [CrossRef]
- Aldous, D.J.; Dutton, W.M.; Steel, P.G. A simple enantioselective preparation of (2S,5S)-2,5-diphenylpyrrolidine and related diaryl amines. Tetrahedron Asymmetry 2000, 11, 2455–2462. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, C.M.; Pelletier, J.M. Expanding the organic toolbox: A guide to integrating biocatalysis in synthesis. Chem. Soc. Rev. 2012, 41, 1585–1605. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.E.; Maguire, A.R. Recent trends in whole cell and isolated enzymes in enantioselective synthesis. Arkivoc 2012, 2012, 321–382. [Google Scholar]
- Torrelo, G.; Hanefeld, U.; Hollmann, F. Biocatalysis. Catal. Lett. 2015, 145, 309–345. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; González-Martínez, D.; Gotor-Fernández, V. Stereoselective Biocatalysis. A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransform. 2018, 36, 102–130. [Google Scholar] [CrossRef]
- Mattson, A.; Öhrner, N.; Hult, K.; Norin, T. Resolution of diols with C2-symmetry by lipase catalysed transesterification. Tetrahedron Asymmetry 1993, 4, 925–930. [Google Scholar] [CrossRef]
- Nagai, H.; Morimoto, T.; Achiwa, K. Facile enzymatic synthesis of optically active 2,5-hexanediol derivatives and its application to the preparation of optically pure cyclic sulfate for chiral ligands. Synlett 1994, 1994, 289–290. [Google Scholar] [CrossRef]
- Caron, G.; Kazlauskas, R.J. Isolation of racemic 2,4-pentanediol and 2,5-hexanediol from commercial mixtures of racemic and meso isomers by way of cyclic sulfites. Tetrahedron Asymmetry 1994, 6, 657–664. [Google Scholar] [CrossRef]
- Persson, B.A.; Larsson, A.L.E.; Le Ray, M.; Bäckvall, J.-E. Ruthenium- and enzyme-catalyzed dynamic kinetic resolution of secondary alcohols. J. Am. Chem. Soc. 1999, 121, 1645–1650. [Google Scholar] [CrossRef]
- Persson, B.A.; Huerta, F.; Bäckvall, J.-E. Dynamic kinetic resolution of secondary diols via coupled ruthenium and enzyme catalysis. J. Org. Chem. 1999, 64, 5237–5240. [Google Scholar] [CrossRef]
- Edin, M.; Bäckvall, J.-E. On the mechanism of the unexpected facile formation of meso-diacetate products in enzymatic acetylation of alkanediols. J. Org. Chem. 2003, 68, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Martín-Matute, B.; Edin, M.; Bäckvall, J.-E. Highly efficient synthesis of enantiopure diacetylated C2-symmetric diols by ruthenium- and enzyme-catalyzed dynamic kinetic asymmetric transformation (DYKAT). Chem. Eur. J. 2006, 12, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Borén, L.; Leijondahl, K.; Bäckvall, J.-E. Dynamic kinetic asymmetric transformation of 1,4-diols and the preparation of trans-2,5-disubstituted pyrrolidines. Tetrahedron Lett. 2009, 50, 3237–3240. [Google Scholar] [CrossRef]
- Martín-Matute, B.; Bäckvall, J.-E. Ruthenium- and enzyme-catalyzed dynamic kinetic asymmetric transformation of 1,4-diols: Synthesis of γ-hydroxy ketones. J. Org. Chem. 2004, 69, 9191–9195. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lihammar, R.; Bäckvall, J.-E. Investigation of the impact of water on the enantioselectivity displayed by CALB in the kinetic resolution of δ-functionalized alkan-2-ol derivatives. Chem. Eur. J. 2014, 20, 13517–13521. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Arends, I.W.C.E.; Holtmann, D. Enzymatic reductions for the chemist. Green Chem. 2011, 13, 2285–2313. [Google Scholar] [CrossRef]
- Magano, J.; Dunetz, J.R. Large-scale carbonyl reductions in the pharmaceutical industry. Org. Process Res. Dev. 2012, 16, 1156–1184. [Google Scholar] [CrossRef]
- Nealon, C.M.; Musa, M.M.; Patel, J.M.; Phillips, R.S. Controlling substrate specificity and stereospecificity of alcohol dehydrogenases. ACS Catal. 2015, 5, 2100–2114. [Google Scholar] [CrossRef]
- Kratzer, R.; Woodley, J.M.; Nidetzky, B. Rules for biocatalyst and reaction engineering to implement effective, NAD(P)H-dependent, whole cell bioreductions. Biotechnol. Adv. 2015, 33, 1641–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurina-Sanz, M.; Bisogno, F.R.; Lavandera, I.; Orden, A.A.; Gotor, V. Promiscuous substrate-binding explains the enzymatic stereo- and regiocontrolled synthesis of enantiopure hydroxy ketones and diols. Adv. Synth. Catal. 2009, 351, 1842–1848. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Wu, X. Dicarbonyl reduction by single enzyme for the preparation of chiral diols. Chem. Soc. Rev. 2012, 41, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, K.; Edegger, K.; Kroutil, W.; Liese, A. Overcoming the thermodynamic limitation in asymmetric hydrogen transfer reactions catalyzed by whole cells. Biotechnol. Bioeng. 2006, 95, 192–198. [Google Scholar] [CrossRef] [PubMed]
- De Gonzalo, G.; Lavandera, I.; Faber, K.; Kroutil, W. Enzymatic reduction of ketones in “micro-aqueous” media catalyzed by ADH-A from Rhodococcus ruber. Org. Lett. 2007, 9, 2163–2166. [Google Scholar] [CrossRef] [PubMed]
- Machielsen, R.; Leferink, N.G.H.; Hendriks, A.; Brouns, S.J.J.; Hennemann, H.-G.; Daussmann, T.; Oost, J. Laboratory evolution of Pyrococcus furiosus alcohol dehydrogenase to improve the production of (2S,5S)-hexanediol at moderate temperatures. Extremophiles 2008, 12, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Katzberg, M.; Bertau, M.; Hummel, W. Highly efficient and stereoselective biosynthesis of (2S,5S)-hexanediol with a dehydrogenase from Saccharomyces cerevisiae. Org. Biomol. Chem. 2010, 8, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Mizar, P.; Wirth, T. Flexible stereoselective functionalizations of ketones through umpolung with hypervalent iodine reagents. Angew. Chem. Int. Ed. 2014, 53, 5993–5997. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Henry, J.B.; Li, Y.; Mount, A.R.; Slawin, A.M.Z.; Woollins, J.D. Synthesis of novel 2,5-diarylselenophenes from selenation of 1,4-diarylbutane-1,4-diones or methanol/arylacetylenes. Org. Biomol. Chem. 2010, 8, 1655–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevar, N.M.; Kel’in, A.V.; Kulinkovich, O.G. One step preparation of 1,4-diketones from methyl ketones and α-bromomethyl ketones in the presence of ZnCl2 • t-BuOH·• Et2NR as a condensation agent. Synthesis 2000, 9, 1259–1262. [Google Scholar] [CrossRef]
- Lavandera, I.; Kern, A.; Ferreira-Silva, B.; Glieder, A.; de Wildeman, S.; Kroutil, W. Stereoselective bioreduction of bulky-bulky ketones by a novel ADH from Ralstonia sp. J. Org. Chem. 2008, 73, 6003–6005. [Google Scholar] [CrossRef] [PubMed]
- Leuchs, S.; Greiner, L. Alcohol dehydrogenase from Lactobacillus brevis: A versatile robust catalyst for enantioselective transformations. Chem. Biochem. Eng. Q. 2011, 25, 267–281. [Google Scholar]
- Lavandera, I.; Kern, A.; Resch, V.; Ferreira-Silva, B.; Glieder, A.; Fabian, W.M.F.; de Wildeman, S.; Kroutil, W. One-way biohydrogen transfer for oxidation of sec-alcohols. Org. Lett. 2008, 10, 2155–2158. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Laivenieks, M.; Zeikus, J.G.; Phillips, R.S. Mutation of cysteine-295 to alanine in secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus affects the enantioselectivity and substrate specificity of ketone reductions. Bioorg. Med. Chem. 2001, 9, 1659–1666. [Google Scholar] [CrossRef]
- Stampfer, W.; Kosjek, B.; Moitzi, C.; Kroutil, W.; Faber, K. Biocatalytic asymmetric hydrogen transfer. Angew. Chem. Int. Ed. 2002, 41, 1014–1017. [Google Scholar] [CrossRef]
- Abu, R.; Woodley, J. Application of enzyme coupling reactions to shift thermodynamically limited biocatalytic reactions. ChemCatChem 2015, 7, 3094–3105. [Google Scholar] [CrossRef]
- Man, H.; Kędziora, K.; Kulig, J.; Frank, A.; Lavandera, I.; Gotor-Fernández, V.; Rother, D.; Hart, S.; Turkenburg, J.P.; Grogan, G. Structures of alcohol dehydrogenases from Ralstonia and Sphingobium spp. reveal the molecular basis for their recognition of ‘bulky–bulky’ ketones. Top. Catal. 2014, 57, 356–365. [Google Scholar] [CrossRef]
- García-Cerrada, S.; Redondo-Gallego, L.; Martínez-Olid, F.; Rincón, J.A.; García-Losada, P. Practical manufacture of 4-alkyl-4-aminocyclohexylalcohols using ketoreductases. Org. Process Res. Dev. 2017, 21, 779–784. [Google Scholar] [CrossRef]
- Ceylan, M.; Gürdere, M.B.; Budak, Y.; Kazaz, C.; Seçen, H. One-step preparation of symmetrical 1,4-diketones from α-halo ketones in the presence of Zn-I2 as a condensation agent. Synthesis 2004, 35, 1750–1754. [Google Scholar] [CrossRef]
- Fujita, K.; Shinokubo, H.; Oshima, K. Highly diastereoselective tandem reduction–allylation reactions of 1,4-diketones with zirconocene–olefin complexes. Angew. Chem. Int. Ed. 2003, 42, 2550–2552. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Feng, Z.-J.; Chen, J.-R.; Lu, L.-Q.; Xiao, W.-J. Visible-light-induced C‒S bond activation: Facile access to 1,4-diketones from β-ketosulfones. Chem: Eur. J. 2014, 20, 3045–3049. [Google Scholar] [CrossRef] [PubMed]
- Domin, D.; Benito-Garagorri, D.; Mereiter, K.; Hametner, C.; Fröhlich, J.; Kirchner, K. Synthesis and characterization of new chiral palladium β-diimine complexes. J. Organomet. Chem. 2007, 692, 1048–1057. [Google Scholar] [CrossRef]




{kind=link}
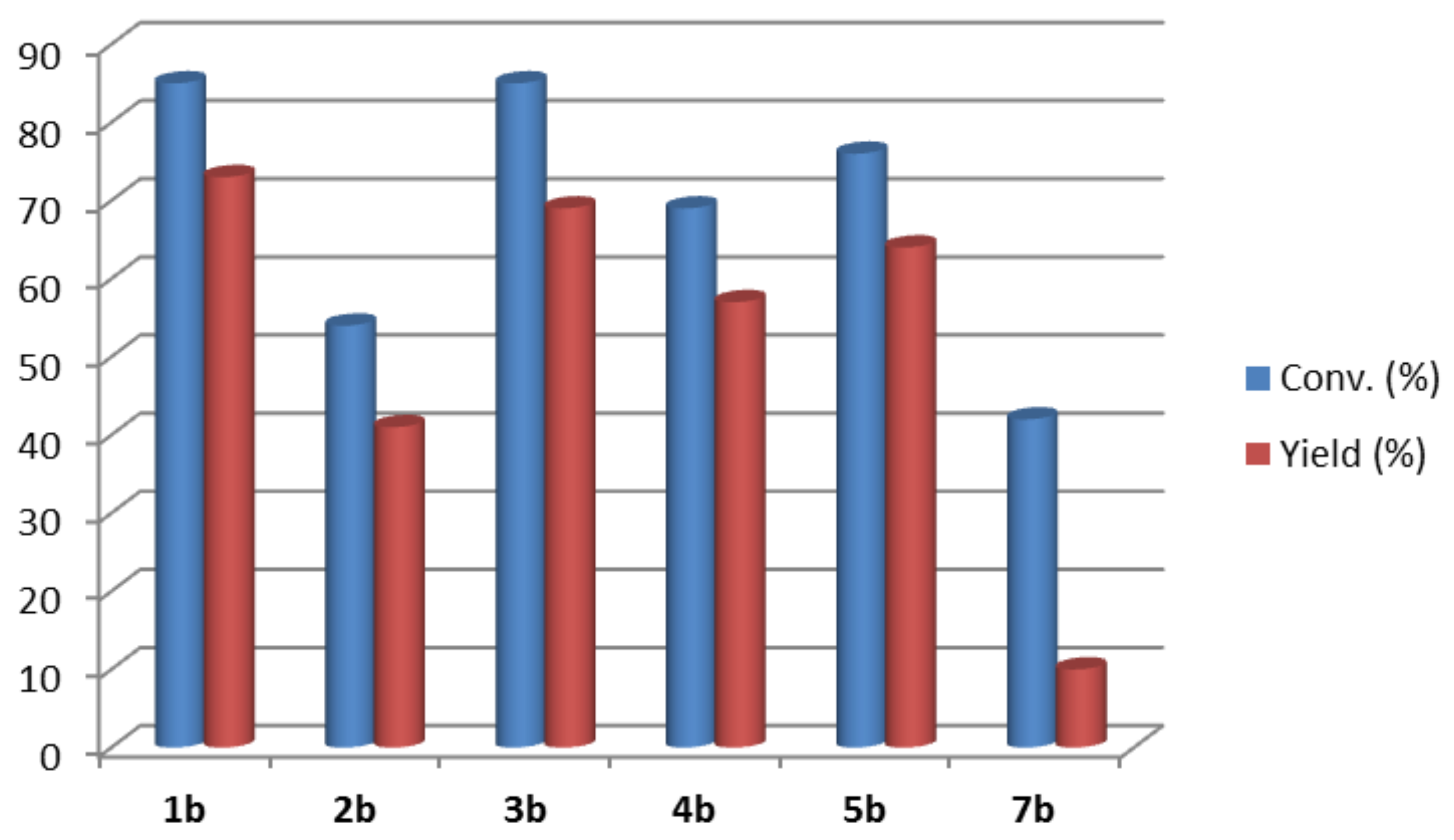{kind=link}
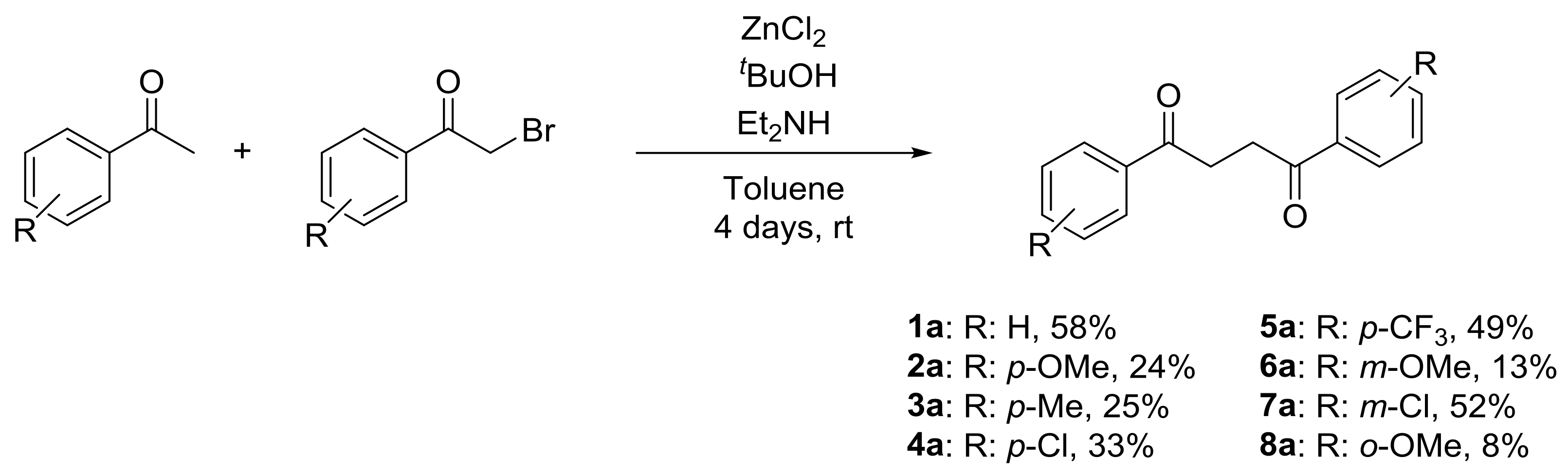{kind=link}
{kind=link}

| Entry | Regeneration System | Cosolvent | % (v/v) | t (h) | Conversion (%) b | de b,c | ee (%) b,d |
|---|---|---|---|---|---|---|---|
| 1 | Glucose/GDH | DMSO | 2.5 | 24 | 56 | 99:1 | >99 |
| 2 | Glucose/GDH | DMSO | 2.5 | 48 | 82 | 99:1 | >99 |
| 3 | Glucose/GDH | - | - | 48 | 72 | 99:1 | >99 |
| 4 | IPA | - | - | 48 | 72 | 89:11 | >99 |
| 5 | Glucose/GDH | EtOH | 2.5 | 24 | 57 | 99:1 | >99 |
| 6 | Glucose/GDH | 1,4-dioxane | 2.5 | 24 | 72 | 99:1 | >99 |
| 7 | Glucose/GDH | 1,4-dioxane | 5 | 24 | 78 | >99 | >99 |
| 8 | Glucose/GDH | 1,4-dioxane | 10 | 24 | 79 | 99:1 | >99 |
| 9 | Glucose/GDH | 1,4-dioxane | 10 | 48 | 80 | 99:1 | >99 |
| 10 | Glucose/GDH | MTBE | 2.5 | 24 | 70 | >99 | >99 |
| 11 | Glucose/GDH | MTBE | 5 | 24 | 83 | >99 | >99 |
| 12 | Glucose/GDH | MTBE | 10 | 24 | 77 | >99 | >99 |
| 13 | Glucose/GDH | MTBE | 10 | 48 | 77 | >99 | >99 |
| 14 | Glucose/GDH | THF | 2.5 | 24 | 72 | 99:1 | >99 |
| 15 | Glucose/GDH | THF | 5 | 24 | 88 | >99 | >99 |
| 16 | Glucose/GDH | THF | 10 | 24 | 80 | 99:1 | >99 |
| 17 | Glucose/GDH | THF | 10 | 48 | 82 | 99:1 | >99 |

| Entry | ADH | Conversion (%) b | de b,c | ee (%) b,d |
|---|---|---|---|---|
| 1 | P1-B02 | 85 | >99 | >99 |
| 2 | P1-B10 | 79 | >99 | >99 |
| 3 | P1-B12 | 75 | 99:1 | >99 |
| 4 | P2-D03 | 89 | 87:13 | >99 e |
| 5 | P2-D11 | 69 | 98:2 | >99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourelle-Insua, Á.; De Gonzalo, G.; Lavandera, I.; Gotor-Fernández, V. Stereoselective Enzymatic Reduction of 1,4-Diaryl-1,4-Diones to the Corresponding Diols Employing Alcohol Dehydrogenases. Catalysts 2018, 8, 150. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8040150
Mourelle-Insua Á, De Gonzalo G, Lavandera I, Gotor-Fernández V. Stereoselective Enzymatic Reduction of 1,4-Diaryl-1,4-Diones to the Corresponding Diols Employing Alcohol Dehydrogenases. Catalysts. 2018; 8(4):150. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8040150
Chicago/Turabian StyleMourelle-Insua, Ángela, Gonzalo De Gonzalo, Iván Lavandera, and Vicente Gotor-Fernández. 2018. "Stereoselective Enzymatic Reduction of 1,4-Diaryl-1,4-Diones to the Corresponding Diols Employing Alcohol Dehydrogenases" Catalysts 8, no. 4: 150. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8040150