Towards New Catalytic Antioxidants: A Simple and Mild Synthesis of Selenenylsulfides
by
 , ,
, ,
Damiano Tanini
* ,
,
Chiara Bonardi
, 
Caterina Viglianisi
 ,
,
Antonella Capperucci
and 
Stefano Menichetti
* Dipartimento di Chimica ‘Ugo Schiff’, Via della Lastruccia 3-13, 50019 Sesto Fiorentino (FI), Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2019, 9(4), 333; https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040333
Submission received: 14 March 2019
/
Revised: 28 March 2019
/
Accepted: 28 March 2019
/
Published: 4 April 2019
(This article belongs to the Special Issue Chalcogens in Catalysis: Synthesis and Biology)
Abstract
:A new methodology for the synthesis of small molecules containing the S-Se bond is reported. Aryl- and alkyl-selenols react smoothly with N-thiophthalimides to afford the corresponding selenenylsulfides through a clean SN2 path occurring at the sulfur atom. The reaction proceeds under very mild conditions in DMF in absence of catalysts for most of the substrates. The scope of the reaction was found to be broad, allowing a wide series of selenols and N-thiophtalimides to be efficiently employed in this procedure. Owing to the instability of the S-Se bond, selenenylsulfides exhibited a remarkable tendency to disproportionate to the corresponding symmetric diselenides and disulfides. Preliminary evaluation of the catalytic antioxidant properties of novel selenenylsulfides showed their behaviour as GPx mimics.
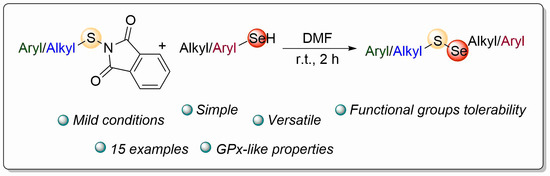
1. Introduction
Antioxidants are compounds able to prevent or inhibit the oxidation of other molecules, and can be natural or synthetic [1,2]. Natural systems include both low molecular weight derivatives, such as polyphenols, ascorbic acid, lipoic acid and vitamin A, and also several enzymes, namely glutathione peroxidase (GPx), thioredoxine reductase (TrxR), superoxide dismutase (SOD), and catalase. Elements like zinc, selenium, iron, and copper are responsible of the activity of certain enzymes. Also, synthetic antioxidants found an extensive use in medicinal and food chemistry.
In this context, selenium and its organic derivatives play an essential role in biochemistry and medicine. In fact, organoselenium compounds, together with some sulfur and tellurium derivatives, find application as antioxidants, anticancer, chemo-protectors, and enzymes modulators [3,4,5,6,7,8,9]. Several studies established the role of selenium in biological systems [10,11].
The catalytic activity of enzymes and of synthetic antioxidants has been ascribed to the heteroatom present in the active site. Selenium derivatives are well known to behave as mimics of the selenoenzyme GPx [12,13,14]. Besides this activity, the positive effect of selenium containing molecules against various diseases has been also attributed to other antioxidant mechanisms, such as ROS (reactive oxygen species) scavenging properties and metal coordination (iron and copper) to prevent DNA damage [15]. Organoselenium compounds usually behave as more efficient systems with respect to sulfur analogues in cancer prevention or as radical scavengers.
On the basis of the catalytic cycle proposed for GPx, the selenol moiety of the enzyme (EnzSeH) is oxidized by the peroxide, forming a selenenic acid (EnzSeOH), which reacts with a thiol (typically glutathione, GSH) leading to a selenenylsulfide (EnzSeSG). The mixed selenosulfide reacts with a second equivalent of GSH, to reform the selenol, together with oxidized glutathione GSSG. The selenenylsulfide, containing the Se-S bridge, represents the key intermediate for the activity of the enzyme [7,16,17,18,19]. Taking into account the importance of this process, several synthetic approaches to obtain selenated small molecules as enzyme mimics have been developed. However, to the best of our knowledge, a limited number of methods are reported for the preparation of selenenylsulfides. These methods typically exploited the reactivity of diselenides in thiol-diselenide exchange or foresee the reaction of sulfenyl derivatives with thiols [16,17,20,21,22].
It is well established that the Se-S bridge is unstable and undergoes disproportionation reaction to afford diselenides and disulfides. Some examples are reported on selenenylsulfides stabilized by sterically bulky groups or by intramolecular Se…Het (N, O) interactions [23].
Owing to the interest in these selenium containing compounds, the search for novel procedures to prepare small molecules containing the Se-S unit is of great interest.
Our long-dated interest in the chemistry of silyl chalcogenides allowed to disclose novel procedures to access a plethora of sulfur and selenium containing molecules exploiting the nucleophilic behaviour of the chalcogen atom towards various electrophiles [24,25,26,27,28,29]. Recently we reported the reaction of bis(trimethylsilyl)selenide (HMDSS) with N-thiophtalimides as a mild, general metal free procedure for the synthesis of variously substituted disulfides [30].
Indeed, despite the interest in the reactivity of selenols as nucleophilic selenium transfer reagents, their use has been critically limited by their instability. However, HMDSS was efficient in the opening of ring strained heterocycles, namely epoxides, episulfides and aziridines, leading to a wide range of β-functionalized selenols, which were stable enough to react with electrophiles under controlled catalytic conditions [31]. Differently substituted stable aryl selenols were also prepared by reduction of the parent diselenides to explore their activity as enzyme inhibitors [32].
On the basis of our findings on the behaviour of selenols we report here a new approach for the preparation of selenenylsulfides using N-thiophtalimides as suitable electrophiles.
2. Results and Discussion
We began our investigations by studying the reaction of benzeneselenol 1a with 2,4-dimethoxyphenyl N-thiophthalimide 2a (Table 1). The reaction was initially performed at 0 °C for 2 h in the presence of Et3N, using CHCl3 as the solvent (Table 1, entry 1). Interestingly, under these conditions the formation of the desired selenenylsulfide 3a was observed, albeit together with a comparable amount of diselenide 4a and disulfide 5a. Furthermore, ca. 30% of the starting N-thiophthalimide 2a remained unreacted. Similar results, both in terms of conversion and 3a:4a:5a ratio, were obtained upon performing the reaction at −78 °C for 4 h (entry 2). We evaluated whether the solvent could have an effect on the reaction outcome. We found that the use of acetonitrile in place of chloroform gave the diselenide 4a and the disulfide 5a as main reaction products, together with a larger amount of unreacted 2a (ca. 50%, entry 3). The use of toluene gave slight 3a:4a:5a ratio improvements, although poor conversion was achieved (Table 1, entry 4). Remarkably, when DMF was used as solvent in the presence of Et3N or Cs2CO3/TBAI, complete consumption of 2a was observed within 2 h. Under these conditions a comparable mixture of compounds 3a, 4a, and 5a was observed (entries 5 and 6). Finally, we were delighted to find that the desired selenenylsulfide 3a was smoothly formed as major product simply treating selenol 1a with N-thiophthalimide 2a in DMF, in absence of any catalyst (entry 7). This observation seems to suggest that the formation of selenenylsulfide 3a is the result of a clean SN2 at sulfur of 2a by the nucleophile 1a, favoured in polar aprotic solvents such as DMF.
In our hands compound 3a proved to be rather unstable, as the selenenylsulfide exhibited a remarkable tendency to form the corresponding symmetrical diselenide 4a and disulfide 5a. Indeed, selenenylsulfide 3a significantly decomposed on silica gel during flash chromatography purification (36% isolated yield). Similar behaviour was observed when using a different stationary phase such as neutral Al2O3. We also observed that both bases and acids could promote the conversion of 3a into 4a and 5a. Intriguingly, the mild acidity of chloroform proved to be sufficient to induce fast conversion of 3a, which could also be observed while acquiring the 1H NMR spectrum in CDCl3.
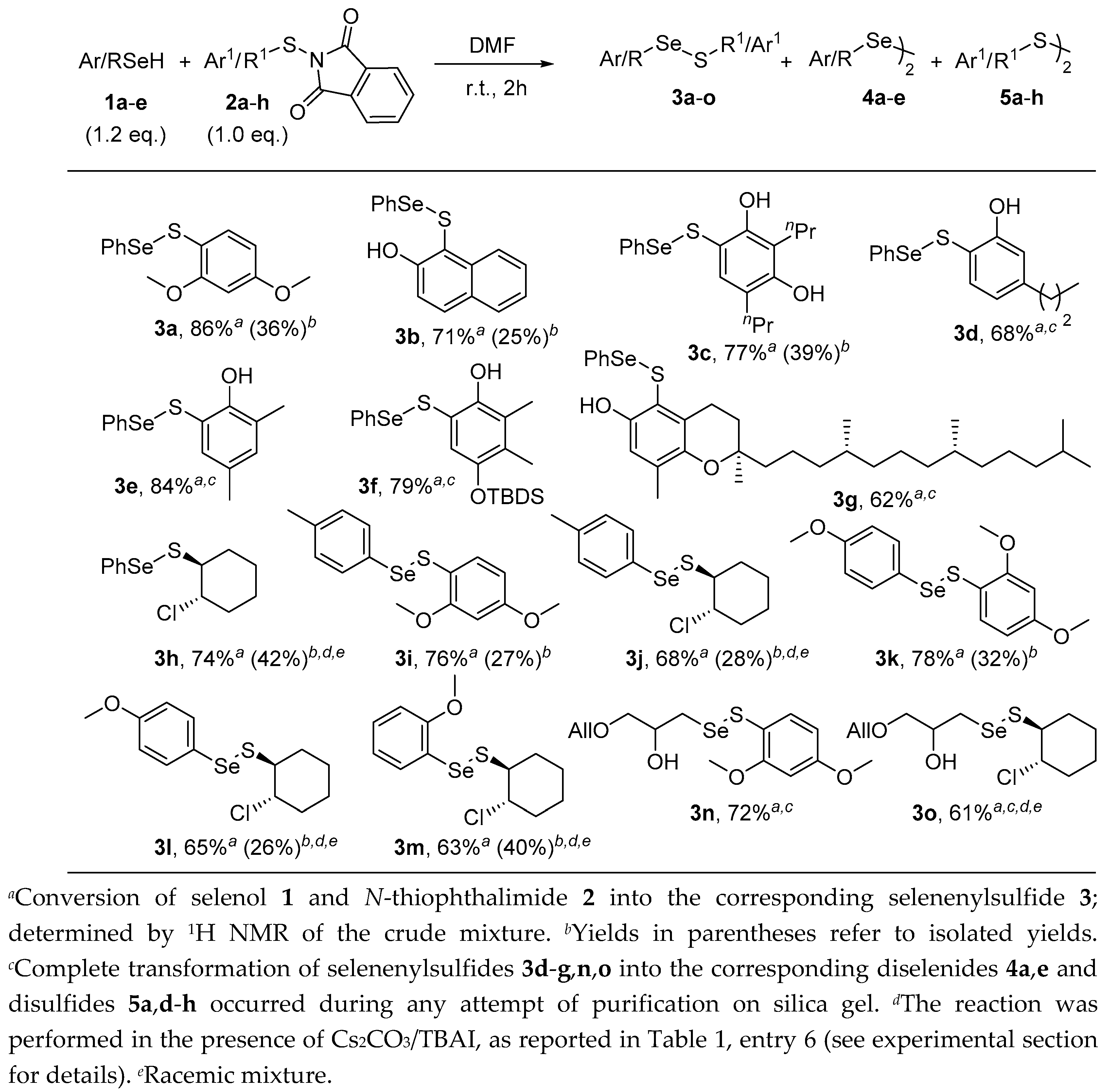
Having established optimum reaction conditions for the conversion of selenols and N-thiophthalimides into the corresponding selenenylsulfides, we explored the scope and the limitation of the reaction. We initially focused our attention on variations of the N-thiophthalimide structure. We found that, under the optimised reaction conditions, variously substituted N-arylthiophthalimides 2a–f, bearing different substituents, such as alkyl, unprotected hydroxyl, methoxy, and silyloxy moieties at different positions of the aromatic ring, reacted smoothly with benzeneselenol 1a, enabling the formation of aryl(phenylselanyl)sulfanes 3a–f (Scheme 1). Furthermore, the reaction was also applied to the N-arylthio derivative of δ-tocopherol 2g, leading to the formation of compound 3g bearing a vitamin E-like skeleton and the phenylseleno moiety. Intriguingly, the 2-chloro-1-N-cyclohexylthiophthalimide 2h proved to be scarcely reactive under the standard conditions. Nonetheless, 1a and 2h readily reacted in the presence of Cs2CO3 and TBAI to afford alkyl(phenylselanyl)sulfane 3h. However, as previously observed for compound 3a, although selenenylsulfides 3b–h were formed in good yields, a significant decomposition occurred during purification. Whereas pure 3b,c,h could be isolated in moderate yields, aryl(phenylselanyl)sulfane 3d–g gave only the corresponding diselenide 4a and disulfides 5d–g upon flash column chromatography (Scheme 1). Having demonstrated that a variety of aryl- and alkyl- N-thiophthalimides could be successfully employed in this reaction, we next explored the scope of the reaction with respect to differently substituted selenols. Pleasingly, arylselenols 1b–d, bearing methyl and methoxy groups onto different position of the aromatic ring, reacted efficiently with N-arylthiophthalimide 2a and N-alkylthiophthalimide 2h to yield the corresponding substituted selenenylsulfides 3i–m. Despite the instability exhibited by S-Se bonds, compounds 3i–m were obtained in good to moderate isolated yields (Scheme 1).
Finally, in order to evaluate whether this reactivity could also be extended to functionalized alkyl selenols, β-hydroxy selenol 1e was treated with N-thiophthalimides 2a and 2h. Unfortunately, albeit selenenylsulfides 3n and 3o were efficiently formed, complete disproportionation occurred during purification on silica gel, leading exclusively to the isolation of diselenide 4e and disulfides 5a,h (Scheme 1).
Worthy of mention is the complete chemoselectivity observed in the reactions of selenols with N-thiophthalimide 2h. In fact, despite working in the presence of a remarkably strong nucleophilic selenate ion and a potentially very good leaving group, no trace of the attack on the chloro substituted carbon was observed, being the sulfur atom of 3h the unique electrophilic partner of these reactions.
The amount of diselenides 4 and disulfides 5 could not be reduced working under strictly controlled conditions (i.e., using dry-box, in absence of light, fast manipulation). The observed behaviour is in accordance with previous reports on the low stability of this kind of structures which, amongst other factors, depends on the nature of substituents on the sulfur or selenium atoms [19,23].
As stated above, a selenenylsulfide is postulated to be the key intermediate of the GPx catalytic cycle. The selenium atom of a selenocysteine residue (Sec) of the GPx catalytic triad and the thiol group of a molecule of GSH are indeed involved in the formation of the selenenylsulfide which, upon reaction with a second molecule of GSH regenerates the catalytically active selenol moiety of Sec. This biochemical mechanism allows the reduction, in living cells, of harmful hydroperoxides to safe alcohols and water, therefore accounting for the biological relevance of S-Se bonds formation [1,3,4].
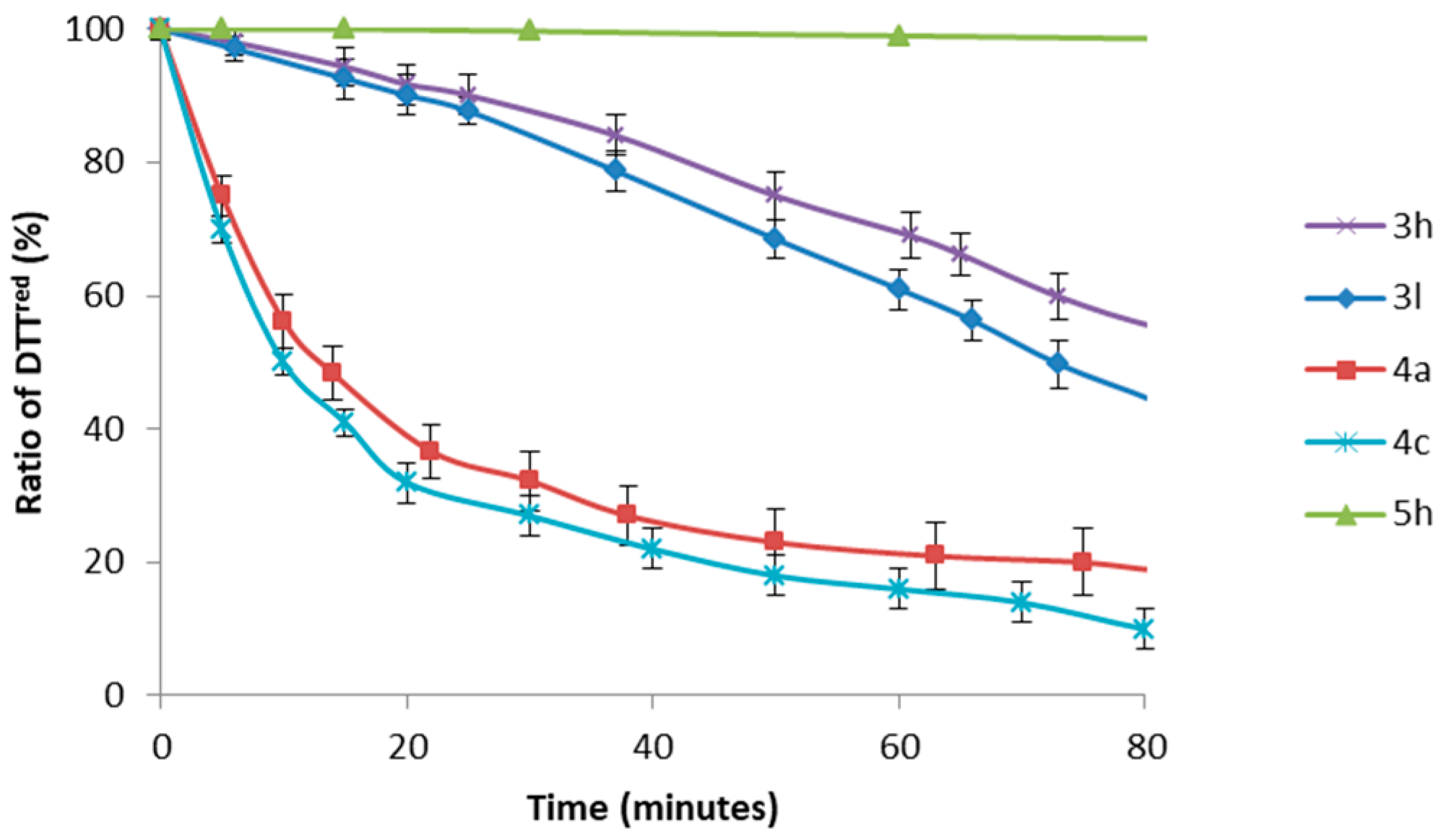
Thus, having developed a novel procedure for the synthesis of small molecules containing the S-Se moiety, we sought to preliminary test their GPx-like activity. The thiol peroxidase-like activity of compounds 3h and 3l was studied according to the DTT oxidation test (Figure 1) [33,34,35]. Indeed, selenenylsulfides 3h,l behaved as medium effective catalysts with T50 around 70 min [36] Furthermore, in line with previous findings on the antioxidant properties of different organoselenium compounds [37,38], the presence of a methoxy group onto the arylseleno moiety renders selenenylsulfide 3l more efficient than 3h (Figure 1). To get more insight on the GPx activity of these new compounds [39,40,41,42], in Figure 1 are reported data obtained for diselenides 4a and 4c and disulfide 5h measured under the same conditions. Thus, while diselenides 4a,c promoted a faster oxidation than selenenylsulfides with T50 around 20 min, disulfide 5h did not exhibit significant catalytic antioxidant properties (Figure 1).
3. Materials and Methods
3.1. Experimental Section
All the reactions were performed under a positive pressure of nitrogen and were monitored by TLC using commercially available precoated plates (silica gel 60 F 254) and compounds were visualised by fluorescence quenching or by staining the plates with acidic p-anisaldehyde solution. Silica gel 60, 230–400 mesh, was used for flash column chromatography. Dry solvents were obtained using a Pure Solv™ Micro system. Commercially available reagents were used as obtained from freshly opened containers without further purification. Aryl selenols 1a–d [32], alkyl selenol 1e [31], and N-thiophthalimides 2 [43,44,45,46,47,48] were prepared according to reported procedures. Spectroscopic data of diselenides 4a–e [26,49] and disulfides 5a–h [30] matched those previously reported in the literature.
1H and 13C NMR spectra were recorded in CDCl3 or C6D6 with a Varian Mercury Plus instrument or with a Varian INOVA instrument at 400 and 100 MHz, respectively. The corresponding residual non-deuterated solvent was used as a reference (CDCl3: 7.26 ppm for 1H and 77.0 ppm for 13C; C6D6: 7.16 ppm for 1H and 128.0 ppm for 13C). 77Se NMR spectra were recorded using Bruker 400 Ultrashield spectrometer (Bruker, Milan, Italy), operating at 76 MHz. Diphenyl diselenide (PhSe)2 was used as an external reference for 77Se NMR (δ = 461 ppm). 1H NMR data are reported as follows: chemical shift, integration, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, m = multiplet, dd = doublet of doublet, etc.), coupling constant (J) or line separation (ls), and assignment. Mass spectra (MS) were determined by ESI (Thermo Fisher Scientific, Milan, Italy). See Electronic Supplementary Material for details.
3.2. General Procedure for the Synthesis of Selenenylsulfides 3
A solution of selenol 1 (0.20 mmol, 1.0 eq.) in dry DMF (0.5 mL) was added to a solution of N-thiophthalimide 2 (0.24 mmol, 1.2 eq.) in dry DMF (1.5 mL) at room temperature under inert atmosphere (N2); the reaction mixture was stirred for 4 h (reaction progress monitored by TLC). Afterwards, the mixture was diluted with Et2O (5 mL) and then H2O (3 mL) was added. The organic phase was extracted with Et2O (5 mL), washed with water (3 × 5 mL) and brine (5 mL), and then dried over Na2SO4; filtered and concentrated in vacuo. The crude material was purified by flash chromatography to afford selenenylsulfides 3.
3.2.1. Synthesis of (2,4-Dimethoxyphenyl)(phenylselanyl)sulfane 3a
Following the general procedure, N-thiophthalimide 2a (57 mg, 0.18 mmol) and benzeneselenol 1a (24 mg, 0.15 mmol) gave, after flash chromatography (petroleum ether/Et2O 5:1, 3a: Rf = 0.47; 4a: Rf = 0.95; 5a: Rf = 0.14), selenenylsulfide 3a (18 mg, 36%) as a yellowish oil. 1H NMR (400 MHz, C6D6) δ (ppm): 7.63-7.69 (2H, m), 7.52 (1H, d, J = 8.5 Hz), 6.93-7.04 (3H, m), 6.28 (1H, d, J = 2.5 Hz), 6.11 (1H, dd, J = 2.5, 8.5 Hz), 3.22 (3H, s, CH3O), 3.17 (3H, s, CH3O). 13C NMR (100 MHz, C6D6) δ (ppm): 162.1, 160.3, 135.3, 130.9, 128.8, 128.0, 127.5, 127.1, 104.9, 99.3, 54.8, 54.6. HRMS m/z calcd for C14H15O2SSe 326.9958, found 326.9963.
3.2.2. Synthesis of 1-((Phenylselanyl)thio)naphthalen-2-ol 3b
Following the general procedure, N-thiophthalimide 2b (58 mg, 0.18 mmol) and benzeneselenol 1a (24 mg, 0.15 mmol) gave, after flash chromatography (petroleum ether/Et2O 20:1, 3b: Rf = 0.48; 4a: Rf = 0.83; 5b: Rf = 0. 38), selenenylsulfide 3b (12 mg, 25%) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.05 (1H, d, J = 8.4 Hz), 7.75–7.80 (2H, m), 7.59 (2H, ap.d., J = 8.1 Hz), 7.32–7.43 (2H, m), 7.25–7.29 (3H, m), 7.16 (1H, d, J = 8.4 Hz), 6.70 (1H, s, OH). 13C NMR (100 MHz, CDCl3) δ (ppm): 156.9, 135.0, 135.0, 132.8, 131.5, 129.5, 129.3, 129.2, 128.5, 128.5, 127.4, 125.1, 123.8, 116.7. 77Se NMR (76 MHz, CDCl3) δ (ppm): 637.9. HRMS m/z calcd for C16H12NaOSSe 354.9672, found 354.9681.
3.2.3. Synthesis of 4-((Phenylselanyl)thio)-2,6-dipropylbenzene-1,3-diol 3c
Following the general procedure, N-thiophthalimide 2c (89 mg, 0.24 mmol) and benzeneselenol 1a (31 mg, 0.20 mmol) gave, after flash chromatography (petroleum ether/Et2O 8:1, 3c: Rf = 4.2; 4a: Rf = 0.92; 5c: Rf = 0.26), selenenylsulfide 3c (30 mg, 39%) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.62 (2H, app.d, J = 8Hz ), 7.28–7.41 (3H, m), 6.89 (1H, s), 6.21 (1H, s, OH), 4.90 (1H, s, OH), 2.60 (3H, t, J = 8 Hz, CH3), 2.40 (3H, t, J = 8 Hz, CH3), 1.46–1.63 (4H, m), 0.81–1.02 (4H, m). 13C NMR (100 MHz, CDCl3) δ (ppm): 155.0, 154.7, 134.1, 134.0, 132.2, 129.2, 128.9, 120.5, 114.6, 111.1, 31.4, 26.0, 22.7, 22.0, 14.1, 13.8. 77Se NMR (76 MHz, CDCl3) δ (ppm): 641.8. HRMS m/z calcd for C18H22NaO2SSe 405.0403, found 405.0397.
3.2.4. Synthesis of (2-Chlorocyclohexyl)(phenylselanyl)sulfane 3h
Following a slightly modified general procedure, a solution of benzeneselenol 1a (31 mg, 0.20 mmol) in dry DMF (2.5 mL) was cooled at 0 °C under inert atmosphere and treated with Cs2CO3 (65 mg, 0.20 mmol, 1.0 eq.) and TBAI (74 mg, 0.20 mmol, 1.0 eq.). Afterwards, a DMF solution (0.5 mL) of N-alkylthiophthalimide 2h (70 mg, 0.24 mmol, 1.2 eq.) was added and the mixture was allowed to warm to room temperature and stirred for 4 h. Then, the mixture was diluted with Et2O (5 mL) and then saturated aq. NH4Cl (3 mL) was added. The organic phase was extracted with Et2O (5 mL), washed with brine (3 × 8 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (petroleum ether, 3h: Rf = 0.54; 4a: Rf = 0.65) to afford selenenylsulfide 3h (26 mg, 42%) as a yellowish oil. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.58–7.64 (2H, m), 7.22–7.31 (3H, m), 3.99–4.04 (1H, m, CHCl), 2.97–3.02 (1H, m, CHS), 2.27–2.29 (1H, m), 2.16–2.27 (1H, m), 1.55–1.73 (4H, m), 1.32–1.42 (2H, m). 13C NMR (100 MHz, CDCl3) δ (ppm): 131.5, 129.7, 129.1, 127.3, 62.6, 55.2, 34.5, 31.6, 24.0, 23.7. 77Se NMR (76 MHz, CDCl3) δ (ppm): 463.6. HRMS m/z calcd for C12H16ClSSe 306.9826, found 306.9833.
3.2.5. Synthesis of (2,4-Dimethoxyphenyl)(p-tolylselanyl)sulfane 3i
Following the general procedure, N-thiophthalimide 2a (57 mg, 0.18 mmol) and 4-methylbenzeneselenol 1b (26 mg, 0.15 mmol) gave, after flash chromatography (petroleum ether/Et2O 15:1, 3i: Rf = 0.43; 4b: Rf = 0.95; 5a: Rf = 0.10), selenenylsulfide 3i (14 mg, 27%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.49 (2H, ap.d., J = 8.0 Hz), 7.41 (1H, d, J = 9.2 Hz), 7.07 (2H, ap.d., J = 8.0 Hz), 6.38–6.46 (2H, m), 3.80 (3H, s), 3.79 (3H, s), 2.34 (3H, s). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.9, 155.3, 136.1, 132.9, 132.4, 130.5, 105.7, 99.6, 56.4, 56.0, 21.6. HRMS m/z calcd for C15H17O2SSe 341.0114, found 341.0102.
3.2.6. Synthesis of (2-Chlorocyclohexyl)(p-tolylselanyl)sulfane 3j
Following a slightly modified general procedure, a solution of 4-methylbenzeneselenol 1b (26 mg, 0.15 mmol) in dry DMF (2.5 mL) was cooled at 0 °C under inert atmosphere and treated with Cs2CO3 (49 mg, 0.15 mmol) and TBAI (56 mg, 0.20 mmol). Afterwards, a DMF solution (0.5 mL) of N-alkylthiophthalimide 2h (53 mg, 0.18 mmol) was added and the mixture was allowed to warm to room temperature and stirred for 4 h. Then, the mixture was diluted with Et2O (5 mL) and then saturated aq. NH4Cl (3 mL) was added. The organic phase was extracted with Et2O (5 mL), washed with brine (3 × 8 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (petroleum ether, Rf = 0.45) to afford selenenylsulfide 3j (13 mg, 28%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.53 (2H, ap.d., J = 8.1Hz), 7.12 (2H, ap.d., J = 8.1 Hz), 4.03-4.08 (1H, m, CHCl), 2.99-3.04 (1H, m, CHS), 2.34 (3H, s, CH3), 2.29–2.33 (1H, m), 2.17–2.28 (1H, m), 1.62–1.75 (4H, m), 1.36–1.43 (2H, m). 13C NMR (100MHz, CDCl3) δ (ppm): 138.2, 132.9, 131.1, 130.6, 63.2, 55.7, 34.8, 32.0, 24.5, 24.2, 21.7. HRMS m/z calcd for C13H17ClNaSSe 342.9802, found 342.9818.
3.2.7. Synthesis of (2,4-Dimethoxyphenyl)((4-methoxyphenyl)selanyl)sulfane 3k
Following the general procedure, N-thiophthalimide 2a (57 mg, 0.18 mmol) and 4-methoxybenzeneselenol 1c (28 mg, 0.15 mmol) gave, after flash chromatography (petroleum ether/Et2O 10:1, 3k: Rf = 0.25; 4c: Rf = 0.42; 5a: Rf = 0.12), selenenylsulfide 3k (17 mg, 32%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.53 (2H, ap.d., J = 8.8 Hz), 7.43 (1H, d, J = 9.1 Hz), 6.83 (2H, ap.d., J = 8.8 Hz), 6.40–6.46 (2H, m), 3.83 (3H, s), 3.82 (3H, s), 3.81 (3H, s). 13C NMR (100MHz, CDCl3) δ (ppm): 160.2, 159.9, 155.6, 136.6, 136.0, 134.0, 124.3, 115.3 105.6, 99.5, 56.4, 56.0, 55.9. 77Se NMR (76 MHz, CDCl3) δ (ppm): 504.8. HRMS m/z calcd for C15H16NaO3SSe 378.9883, found 378.9896.
3.2.8. Synthesis of (2-Chlorocyclohexyl)((4-methoxyphenyl)selanyl)sulfane 3l
Following a slightly modified general procedure, a solution of 4-methoxybenzeneselenol 1c (37 mg, 0.20 mmol) in dry DMF (2.5 mL) was cooled at 0 °C under inert atmosphere and treated with Cs2CO3 (65 mg, 0.20 mmol, 1.0 eq.) and TBAI (74 mg, 0.20 mmol, 1.0 eq.). Afterwards, a DMF solution (0.5 mL) of N-alkylthiophthalimide 2h (70 mg, 0.24 mmol, 1.2 eq.) was added and the mixture was allowed to warm to room temperature and stirred for 4 h. Then, the mixture was diluted with Et2O (5 mL) and then saturated aq. NH4Cl (3 mL) was added. The organic phase was extracted with Et2O (5 mL), washed with brine (3 × 8 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (petroleum ether, Rf = 0.26) to afford selenenylsulfide 3l (17 mg, 26%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.58 (2H, app.d., J = 8.8 Hz), 6.85 (2H, app.d., J = 8.8 Hz), 4.01–4.15 (1H, m), 3.81 (3H, s, CH3O), 2.90–3.11 (1H, m), 2.07–2.38 (2H, m), 1.52–1.81 (4H, m), 1.28–1.41 (2H, m). 13C NMR (100 MHz, CDCl3) δ (ppm): 160.4, 135.6, 134.0, 115.5, 63.2, 55.9, 55.6, 34.5, 31.8, 24.3, 24.0. 77Se NMR (76 MHz, CDCl3) δ (ppm): 503.6. HRMS m/z calcd for C13H17ClNaOSSe 358.9752, found 358.9741.
3.2.9. Synthesis of (2-Chlorocyclohexyl)((2-methoxyphenyl)selanyl)sulfane 3m
Following a slightly modified general procedure, a solution of 2-methoxybenzeneselenol 1d (28 mg, 0.15 mmol) in dry DMF (2.5 mL) was cooled at 0 °C under inert atmosphere and treated with Cs2CO3 (49 mg, 0.15 mmol) and TBAI (56 mg, 0.15 mmol). Afterwards, a DMF solution (0.5 mL) of N-alkylthiophthalimide 2h (53 mg, 0.18 mmol) was added and the mixture was allowed to warm to room temperature and stirred for 4 h. Then, the mixture was diluted with Et2O (5 mL) and then saturated aq. NH4Cl (3 mL) was added. The organic phase was extracted with Et2O (5 mL), washed with brine (3 × 8 mL), dried over Na2SO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (petroleum ether/Et2O 100:1, Rf = 0.30) to afford selenenylsulfide 3m (20 mg, 40%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.82 (1H, dd, J = 1.6, 7.7 Hz), 7.23–7.28 (1H, m), 7.00–7.04 (1H, m), 6.85 (1H, dd, J = 1.1, 8.1 Hz), 4.05–4.09 (1H, m, CHCl), 3.91 (3H, s, CH3O), 2.96–3.01 (1H, m, CHS), 2.34–2.36 (1H, m), 2.23–2.33 (1H, m), 1.62–1.78 (4H, m), 1.36-1.41 (2H, m). 13C NMR (100 MHz, CDCl3) δ (ppm): 157.0, 129.1. 128.5, 126.1, 122.4, 110.9, 63.5, 56.5, 55.5, 35.2, 32.3, 24.6, 24.4. HRMS m/z calcd for C13H17ClNaOSSe 358.9752, found 358.9745.
3.3. GPx-Like Catalytic Activity Measurments
GPx-like activity was determined according to a reported NMR assay [28,29,30]. DTTred (0.14 mmol) and catalyst (0.014 mmol) were dissolved in CD3OD (0.6 mL), and the solution was added to 35% H2O2 (14 μL, 0.14 mmol) to start the reaction. 1H NMR spectra were measured at a variable reaction time at 25 °C. The relative populations of DTTred and DTTox were determined by integration of the 1H NMR signals.
4. Conclusions
In summary, we have disclosed a novel procedure to convert selenols and N-thiophthalimides into the corresponding selenenylsulfides under very mild procedure. These small molecules, containing the S-Se bond, resemble the key intermediate involved in the GPx catalytic cycle and, therefore, could be employed as useful and simple models to gain insight into the GPx mechanism. Furthermore, preliminary evaluation of the catalytic antioxidant properties of selenenylsulfides showed that they possess GPx-like activity.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/9/4/333/s1.
Author Contributions
Conceptualization, D.T. and S.M.; methodology, C.B.; data curation, C.V.; writing—original draft preparation, D.T. and A.C.; writing—review and editing, D.T., A.C. and S.M.
Funding
This research was founded by Ente Cassa Risparmio Firenze.
Conflicts of Interest
The authors declare no conflict of interest.
Notes and References
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Thiol cofactors for selenoenzymes and their synthetic mimics. Org. Biomol. Chem. 2008, 6, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; du Mont, W.W.; Sies, H. Chemistry of biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Koketsu, M. Biologically significant selenium-containing heterocycles. Coord. Chem. Rev. 2011, 255, 2968–2990. [Google Scholar]
- Mónica Álvarez-Pérez, M.; Wesam, A.; Małgorzata, A.M.; Handzlik, J.; Domínguez-Álvarez, E. Selenides and diselenides: A review of their anticancer and chemopreventive activity. Molecules 2018, 23, 628. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Schneider, A. Synthesis of selenocysteine and its derivatives with an emphasis on selenenylsulfide (-Se-S-) formation. Chem. Biodivers. 2008, 5, 375–388. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Capperucci, A.; Malevolti, G.; Turco, F.; Ferraroni, M.; Supuran, C.T. Synthesis of different thio-scaffolds bearing sulfonamide with subnanomolar carbonic anhydrase II and IX inhibitory properties and X-ray investigations for their inhibitory mechanism. Bioorg. Chem. 2018, 81, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Tanini, D.; Capperucci, A.; Supuran, C.T. First evaluation of organotellurium derivatives as carbonic anhydrase I, II, IV, VII and IX inhibitors. Bioorg. Chem. 2018, 76, 268–272. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Bhuyan, B.J.; Mugesh, G. Biological and biochemical aspects of selenium compounds. In Organoselenium Chemistry: Synthesis and Reactions; Wirth, T., Ed.; Wiley-VCH Verlag & Co.: Weinheim, Germany, 2012. [Google Scholar]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef] [PubMed]
- Orian, L.; Toppo, S. Organochalcogen peroxidase mimetics as potential drugs: A long story of a promise still unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G.; Singh, H.B. Synthetic organoselenium compounds as antioxidants: Glutathione peroxidase activity. Chem. Soc. Rev. 2000, 29, 347–357. [Google Scholar] [CrossRef]
- Zimmerman, M.T.; Bayse, C.A.; Ramoutar, R.R.; Brumaghim, J.L. Sulfur and selenium antioxidants: Challenging radical scavenging mechanisms and developing structure-activity relationships based on metal binding. J. Inorg. Biochem. 2015, 145, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Haratake, M.; Tachibana, Y.; Emaya, Y.; Yoshida, S.; Fuchigami, T.; Nakayama, M. Synthesis of nanovesicular Glutathione Peroxidase mimics with a selenenylsulfide-bearing lipid. ACS Omega 2016, 1, 58–65. [Google Scholar] [CrossRef]
- Boutureira, O.; Bernardes, G.J.L.; Fernàndez-Gonzàlez, M.; Anthony, D.C.; Davis, B.G. Selenenylsulfide-linked homogeneous glycopeptides and glycoproteins: Synthesis of human “Hepatic Se Metabolite A”. Angew. Chem. Int. Ed. 2012, 57, 1432–1436. [Google Scholar] [CrossRef]
- Cheng, Q.; Sandalova, T.; Lindqvist, Y.; Arnér, E.S.J. Crystal structure and catalysis of the selenoprotein Thioredoxin Reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; Andersson, C.; Morgenstern, R.; Cotgreave, I.A.; Andersson, C.-M.; Hallberg, A. Evidence for a common selenolate intermediate in the glutathione peroxidase-like catalysis of α-(phenylselenenyl) ketones and diphenyl diselenide. Tetrahedron 1994, 50, 2929–2938. [Google Scholar] [CrossRef]
- Venkateswarlu, C.; Gautam, V.; Chandrasekaran, S. Synthesis of mixed glycosyl disulfides/selenosulfides using benzyltriethylammonium tetrathiomolybdate as a sulfur transfer reagent. Carbohydr. Res. 2015, 402, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Ahrika, A.; Auger, J.; Paris, J. Stabilisation of 2-nitrophenyl-selenosulfide, -diselenide and -thioselenide ions in N,N-dimethylacetamide. N. J. Chem. 1999, 23, 679–681. [Google Scholar] [CrossRef]
- Potapov, V.A.; Amosova, S.V.; Petrov, P.A.; Romanenko, L.S.; Keiko, V.V. Exchange reactions of dialkyl dichalcogenides. Sulfur Lett. 1992, 15, 121–126. [Google Scholar]
- Kumar, K.; Kandasamy, K.; Singh, H.B.; Wolmershäuser, G.; Butcher, R.J. Chelate ring size effect on the reactivity of [2-(2-phenyl-5,6-dihydro-4h-1,3-oxazinyl)]lithium and Se···N interactions in low-valent organoselenium compounds: facile isolation of diorganotriselenide. Organometallics 2004, 23, 4199–4208. [Google Scholar] [CrossRef]
- Tanini, D.; Grechi, A.; Dei, S.; Teodori, E.; Capperucci, A. An easy one-step procedure for the synthesis of novel β-functionalised tellurides. Tetrahedron 2017, 73, 5646–5653. [Google Scholar] [CrossRef]
- Capperucci, A.; Salles, C.; Scarpelli, S.; Tanini, D. Selective access to sulfurated and selenated heterocycles by intramolecular cyclization of β-substituted sulfides and selenides. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 172–174. [Google Scholar] [CrossRef]
- Tanini, D.; Capperucci, A.; Degl’Innocenti, A. Bis-(trimethylsilyl)selenide in the Selective Synthesis of β-Hydroxy, β-Mercapto, and β-Amino Diorganyl Diselenides and Selenides Through Ring Opening of Strained Heterocycles. Eur. J. Org. Chem. 2015, 357–369. [Google Scholar] [CrossRef]
- Capperucci, A.; Tanini, D. Silicon-assisted synthesis and functionalization of sulfurated and selenated compounds. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1320–1338. [Google Scholar] [CrossRef]
- Tanini, D.; Barchielli, G.; Benelli, F.; Degl’Innocenti, A.; Capperucci, A. Aziridines ring opening by silyl chalcogenides: A stereoselective access to polyfunctionalized molecules as precursor of sulfurated and selenated heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1265–1270. [Google Scholar] [CrossRef]
- Capperucci, A.; Tanini, D.; Borgogni, C.; Degl’Innocenti, A. Thiosilane- and organoselenosilane-mediated novel access to 3,7- disubstituted-1,2,5- trithiepanes and −1,2,5-dithiaselenepanes. Heteroat. Chem. 2014, 25, 678–683. [Google Scholar] [CrossRef]
- Viglianisi, C.; Bonardi, C.; Ermini, E.; Capperucci, A.; Menichetti, S.; Tanini, D. Selenosilane-Promoted Selective Mild Transformation of N-Thiophthalimides into Symmetric Disulfides. Synthesis 2019, 51, 1819–1824. [Google Scholar] [CrossRef]
- Tanini, D.; Tiberi, C.; Gellini, C.; Salvi, P.R.; Capperucci, A. A Straightforward access to stable β-functionalized alkyl selenols. Adv. Synth. Catal. 2018, 360, 3367–3375. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Nocentini, A.; Capperucci, A.; Ferraroni, M.; Gratteri, P.; Supuran, C.T. Selenols: A new class of carbonic anhydrase inhibitors. Chem. Commun. 2019, 55, 648–651. [Google Scholar] [CrossRef]
- Tanini, D.; Grechi, A.; Ricci, L.; Dei, S.; Teodori, E.; Capperucci, A. Novel functionalized organotellurides with enhanced thiol peroxidase catalytic activity. New J. Chem. 2018, 42, 6077–6083. [Google Scholar] [CrossRef] [Green Version]
- Tanini, D.; D’Esopo, V.; Chen, D.; Barchielli, G.; Capperucci, A. Novel sulfur and selenium-containing antioxidants: Synthesis and evaluation of their GPx-like activity. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 166–168. [Google Scholar] [CrossRef]
- Kumakura, F.; Mishra, B.; Priyadarsini, K.I.; Iwaoka, M. A watersoluble cyclic selenide with enhanced glutathione peroxidase-like catalytic activities. Eur. J. Org. Chem. 2010, 3, 440–445. [Google Scholar] [CrossRef]
- T50 is the time required, in seconds, to halve the initial thiol concentration after the addition of H2O2.
- Bhabak, K.P.; Mugesh, G. A simple and efficient strategy to enhance the antioxidant activities of amino-substituted glutathione peroxidase mimics. Chem. Eur. J. 2008, 14, 8640–8651. [Google Scholar] [CrossRef]
- Bailly, F.; Azaroual, N.; Bernier, J.L. Design, synthesis and glutathione peroxidase-like properties of ovothiol-derived diselenides. Bioorg. Med. Chem. 2003, 11, 4623–4630. [Google Scholar] [CrossRef]
- In our hands, S-Se bond exhibited a certain degree of instability. However, selenenylsulfides 3h and 3l proved to be stable for longer times (few days) with respect to those required to perform the GPx-like activity measurements (1 or 2 h). A possible mechanism for the catalytic action of selenylsulfides involves: (i) nucleophilic attack of one of the thiol moieties of DTT onto the Se atom of 3, with the formation of a selenenylsulfide bearing the DTT skeleton; (ii) intramolecular nucleophilic attack of the residual SH moiety onto the S atom of the selenenylsulfide with formation of DTTox and an arylselenol; (iii) H2O2 oxidation of the selenol to the corresponding selenenic acid (ArSeOH); (iv) eventually, ArSeOH undergoes redox reactions with DTT and H2O2, as reported for the catalytic activity of diselenides (see reference [42]). Notably, in this first step (i), likely the rate-determining one, the attack at selenium is both kinetically and thermodynamically favoured over attack at sulfur (see references [40,41,42,43]). On the other hand, the attack at sulfur is favoured in the second step (ii), due to the formation of a six membered cyclic disulfide (DTTox). Further investigations in order to better elucidate the reaction mechanism are currently ongoing.
- Bortoli, M.; Wolters, L.P.; Orian, L.; Bickelhaupt, F.M. Addition-Elimination or Nucleophilic Substitution? Understanding the Energy Profiles for the Reaction of Chalcogenolates with Dichalcogenides. J. Chem. Theory Comput. 2016, 12, 2752–2761. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Mugesh, G. Functional Mimics of Glutathione Peroxidase: Bioinspired Synthetic Antioxidants. Acc. Chem. Res. 2010, 43, 1408–1419. [Google Scholar] [CrossRef]
- Bachrach, S.M.; Demoin, D.W.; Luk, M.; Miller, J.V., Jr. Nucleophilic Attack at Selenium in Diselenides and Selenosulfides. A Computational Study. J. Phys. Chem. A 2004, 108, 4040–4046. [Google Scholar] [CrossRef]
- Capozzi, G.; Gori, L.; Menichetti, S.; Nativi, C. Phthalimidesulfenyl Chloride. Part 4. Addition to Acetylenes and Synthetic Utilization of their Adducts. J. Chem. Soc. Perkin Trans. 1 1992, 1923–1928. [Google Scholar] [CrossRef]
- Capozzi, G.; Nativi, C.; Menichetti, S.; Rosi, A.; Franck, R.W.G. Phthalimidesulfenyl Chloride Part 6. The First Example of an alpha-Oxothione Acting as Heterodiene: Synthesis of 2,3-Dihydro-1,4-Oxathiines. Tetrahedron Lett. 1993, 34, 4253–4257. [Google Scholar] [CrossRef]
- Capozzi, G.; Menichetti, S.; Neri, S.; Skowronska, A. Fluoride Ion Promoted Synthesis of Thiiranes. Synlett 1994, 267–268. [Google Scholar] [CrossRef]
- Menichetti, S.; Aversa, M.C.; Cimino, F.; Contini, A.; Tomaino, A.; Viglianisi, C. Synthesis and ‘double-faced’ antioxidant activity of polyhydroxylated 4-thiaflavans. Org. Biomol. Chem. 2005, 3, 3066–3072. [Google Scholar] [CrossRef]
- Lodovici, M.; Menichetti, S.; Viglianisi, C.; Caldini, S.; Giuliani, E. Polyhydroxylated 4-Thiaflavans as Multipotent Antioxidants: Protective Effect on Oxidative DNA Damage in vitro. Bioorg. Med. Chem. Lett. 2006, 16, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Viglianisi, C.; Marcantoni, E.; Carapacchi, V.; Menichetti, S.; Marsili, L. A base-mediated mild sulfenylation of indoles and pyrrole with α-acylthiones. Eur. J. Org. Chem. 2014, 6405–6410. [Google Scholar] [CrossRef]
- Singh, D.; Deobald, A.M.; Camargo, L.R.S.; Tabarelli, G.; Rodrigues, O.E.D.; Braga, A.L. An Efficient One-Pot Synthesis of Symmetrical Diselenides or Ditellurides from Halides with CuO Nanopowder/Se0 or Te0/Base. Org. Lett. 2010, 12, 3288–3291. [Google Scholar] [CrossRef]
Scheme 1.
Scope of the synthesis of selenenylsulfides from selenols and N-thiophthalimides.

Figure 1.
Oxidation of DTTred with H2O2 in the presence of Se- or S-containing catalysts (10 mol%). Reaction conditions: [DTTred]0 = 0.14 M, [H2O2]0 = 0.14 M, [catalyst] = 0.014 M, CD3OD (0.6 mL). Reaction progress was monitored by means of 1H NMR. The mean ± SD values of three separate experiments are reported.
Figure 1.
Oxidation of DTTred with H2O2 in the presence of Se- or S-containing catalysts (10 mol%). Reaction conditions: [DTTred]0 = 0.14 M, [H2O2]0 = 0.14 M, [catalyst] = 0.014 M, CD3OD (0.6 mL). Reaction progress was monitored by means of 1H NMR. The mean ± SD values of three separate experiments are reported.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions for the synthesis of 3a from 1a and 2a.

| Entry | Solvent | Base | Temperature | Time | 3a:4a:5a a | Conversion (%) a |
|---|---|---|---|---|---|---|
| 1 | CHCl3 | Et3N | 0 °C to r.t. | 2 h b | 36:32:32 | 71 c |
| 2 | CHCl3 | Et3N | −78 °C | 4 h | 34:33:33 | 88 c |
| 3 | CH3CN | Et3N | 0 °C to r.t. | 4 h | 18:41:41 | 52 c |
| 4 | PhMe | Et3N | 0 °C to r.t. | 24 h | 42:29:29 | 67 c |
| 5 | DMF | Et3N | 0 °C to r.t. | 2 h | 32:34:34 | >95 |
| 6 | DMF | Cs2CO3/TBAl | 0 °C to r.t. | 2 h | 44:28:28 | >95 |
| 7 | DMF | - | r.t. | 4 h | 72:14:14 | >95 |
a Determined by 1H NMR of the crude reaction mixture. b Longer reaction time led to improved conversion but lower 3a:4a:5a ratio. c 29%, 12%, 48%, and 33% of starting N-thiophthalimide 2a remained unreacted, respectively for entries 1, 2, 3, and 4.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tanini, D.; Bonardi, C.; Viglianisi, C.; Capperucci, A.; Menichetti, S. Towards New Catalytic Antioxidants: A Simple and Mild Synthesis of Selenenylsulfides. Catalysts 2019, 9, 333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040333
AMA Style
Tanini D, Bonardi C, Viglianisi C, Capperucci A, Menichetti S. Towards New Catalytic Antioxidants: A Simple and Mild Synthesis of Selenenylsulfides. Catalysts. 2019; 9(4):333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040333
Chicago/Turabian StyleTanini, Damiano, Chiara Bonardi, Caterina Viglianisi, Antonella Capperucci, and Stefano Menichetti. 2019. "Towards New Catalytic Antioxidants: A Simple and Mild Synthesis of Selenenylsulfides" Catalysts 9, no. 4: 333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040333
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.