In Silico Acetylene [2+2+2] Cycloadditions Catalyzed by Rh/Cr Indenyl Fragments



Dipartimento di Scienze Chimiche Universita’ degli Studi di Padova, Via Marzolo 1, 35131 Padova, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(8), 679; https://0-doi-org.brum.beds.ac.uk/10.3390/catal9080679
Submission received: 20 July 2019
/
Revised: 7 August 2019
/
Accepted: 8 August 2019
/
Published: 9 August 2019
(This article belongs to the Special Issue Advanced Strategies for Catalyst Design)
Abstract
:Metal-catalyzed alkyne [2+2+2] cycloadditions provide a variety of substantial aromatic compounds of interest in the chemical and pharmaceutical industries. Herein, the mechanistic aspects of the acetylene [2+2+2] cycloaddition mediated by bimetallic half-sandwich catalysts [Cr(CO)3IndRh] (Ind = (C9H7)−, indenyl anion) are investigated. A detailed exploration of the potential energy surfaces (PESs) was carried out to identify the intermediates and transition states, using a relativistic density functional theory (DFT) approach. For comparison, monometallic parent systems, i.e., CpRh (Cp = (C5H5)−, cyclopentadienyl anion) and IndRh, were included in the analysis. The active center is the rhodium nucleus, where the [2+2+2] cycloaddition occurs. The coordination of the Cr(CO)3 group, which may be in syn or anti conformation, affects the energetics of the catalytic cycle as well as the mechanism. The reaction and activation energies and the turnover frequency (TOF) of the catalytic cycles are rationalized, and, in agreement with the experimental findings, our computational analysis reveals that the presence of the second metal favors the catalysis.
1. Introduction
The [2+2+2] cycloadditions of small unsaturated molecules, such as alkynes and nitriles, afford a variety of aromatic, heterocyclic and polycyclic compounds of paramount importance in the chemical and pharmaceutical industries [1]. In 1867, the synthesis of benzene by thermal cyclotrimerization of acetylene was reported for the first time [2]. Despite the reaction being highly exothermic, it is strongly disfavored by entropic factors, and this limits its synthetic utility. Reppe and Schweckendieck demonstrated that low valent metal nuclei catalyze these [2+2+2] cycloadditions [1]. Further studies assessed that a wide variety of metals such as Ti, Zr, Ru, Co, Rh, Ir, Ni, and Pd may play an important role as catalysts in the synthesis of benzene and its derivatives and, in the past decades, experimental results and theoretical insight have been reported to establish the correct mechanism and tune the efficiency and regioselectivity [3,4,5,6,7].
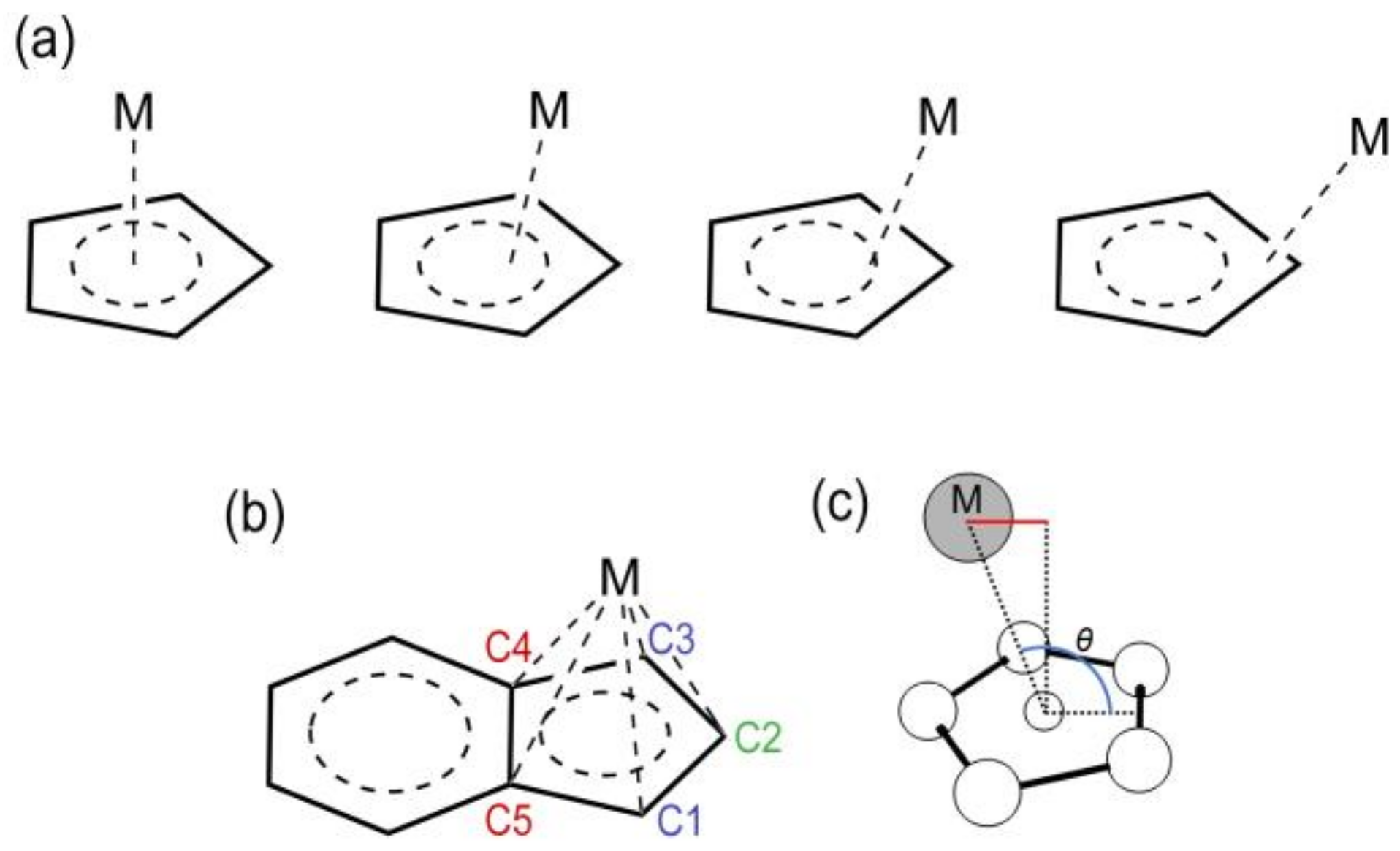
One important class of catalysts for alkyne [2+2+2] cycloadditions are the half-sandwich complexes, i.e., metal-cyclopentadienyl (CpM) or metal-ligand complexes in which the metal is coordinated to the Cp moiety of larger polycyclic ligands. These compounds possess peculiar structural features and reactivity properties; for this reason, and for their synthetic versatility, they are largely used [8,9,10,11,12,13]. They are denoted here with the general formula Cp’MLn, where Cp’ = Cp, Ind, and Ln are the ancillary ligands coordinated to the metal center (M), so that the 18-electrons rule is satisfied. Group 9 metals, i.e., cobalt, rhodium, and iridium, have been largely employed and, particularly, cobalt and rhodium have revealed significant catalytic efficiency [14,15,16,17]. The bonding mode of the metal to the Cp moiety (hapticity) is not perfectly symmetric (η5), but typically exhibits a distortion toward allylic (η3) coordination [18,19], and eventually to an extreme structure in which a σ metal-carbon bond forms (η1). This phenomenon is called metal slippage, and the different bonding modes are shown in Scheme 1a.
To quantify the slippage, Basolo and coworkers [20,21] introduced the geometrical parameter Δ (Equation (1)).
As shown in Scheme 1b, M-C4 and M-C5 are the longest distances between M and two adjacent C atoms of the Cp ring, and M-C3 and M-C1 are the distances between M and the C atoms adjacent to C4 and C5, respectively.
The slippage variations occurring during the catalytic cycle can be quantified referring to the value of Δ, which changes from 0 Å (η5) to nearly 0.3 Å (η3) till 0.6 Å or even higher values (η1). Some of us have recently pointed out that this definition suitably applies to symmetric systems [22], and thus have introduced another descriptor, i.e., the label independent slippage parameter (LISP) (Equation (2)).
is actually the sum of the five average minimum distances from a normal vector passing through the centroid and the metal; is the distance between the metal and the centroid of the ring of atoms (Scheme 1c). Importantly, is also suitable for describing non-symmetric displacements. In the same paper [22], a relationship between the catalytic activity of several half-sandwich group 9 metal complexes for alkyne [2+2+2] cycloadditions and the slippage span expressed in terms of (computed as the difference between the maximum and the minimum value of LISP along the catalytic cycle) was established. The slippage span was then related to the turn-over frequency () values, calculated with the energy span model [23], and it emerged that the lower the is, the higher the is. Finally, in order to improve the sensitivity of , was introduced:
In this descriptor (Equation (3)), the first term indicates how far/close each intermediate/transition state of the catalytic cycle is from/to the starting point. The second term accounts for the slippage difference between two consecutive states along the whole catalytic cycle. The last term includes the slippage variation between the last intermediate and the initial state. The availability of a flexible parameter to quantify the metal slippage, which intuitively influences the catalytic activity, and of a relationship between the metal slippage and the turn-over frequency is a valuable tool. So far, the slippage span model has been applied to monometallic Co and Rh half-sandwich catalysts for alkyne [2+2+2] cycloadditions [22]; in this work, we apply it to bimetallic Rh/Cr indenyl catalysts to assess its general validity.
Different strategies have been developed to tune the regioselectivity and the efficiency of the half-sandwich catalysts. Ingrosso et al. [24,25] experimentally studied the influence of organic moieties, i.e., cyclopentadienyl (Cp), indenyl (Ind) and fluorenyl (FN), in Rh(I) half-sandwich catalysts on alkyne [2+2+2] cycloadditions. Booth et al. [26] reported that IndRh initiates the reaction ten times faster as compared to CpRh, and this was related to the so-called indenyl effect (a phenomenon firstly reported by Adam J. Hart-Davis and Roger J. Mawby in 1969 [27], it was thoroughly explored and named by Fred Basolo [28]; it consists in an enhancement of the rate of the substitution reactions at the metal when indenyl is used instead of cyclopentadienyl aromatic ligand). It was also observed that the presence of an electron withdrawing group in the Cp ligand reduces the catalytic activity at low temperature [29,30]. Based on these studies, it emerged that structural and electronic modifications to the aromatic moiety of the half-sandwich catalyst influence its efficiency.
Changing the metal also plays a role. For example, considering group 9 metals, Co is highly preferred when compared to Rh and Ir [31,32].
Finally, another modification to a half-sandwich catalyst is the coordination of a second metal to form a bimetallic complex [1] when the aromatic ligand is polycyclic; the second metal can be in syn or anti position. A very nice example, reported by Ceccon et al. [33,34,35], is [Cr(CO)3IndRh]L2 (Scheme 2). The idea behind the design of these compounds was that the presence of two metal centers within the same molecule may profoundly affect both the physical properties and the reactivity of the catalyst. In fact, it was found that the presence of a second metal in the anti position strongly enhances the reactivity as compared to monometallic complexes (extra-indenyl effect) [36].
Nowadays, DFT computational methodologies make it possible to investigate the mechanistic details of catalytic reactions, defining with accuracy their thermodynamic (reaction energies) as well as their kinetic (activation energies) features. After the pioneering work by Albright and co-workers on CpCo catalyzed acetylene [2+2+2] cycloaddition to benzene [37], several important computational studies were carried out by Calhorda and Kirchner on CpRuCl [38,39,40], Orian and Bickelhaupt on CpRh and IndRh [41,42,43] and analogous heteroaromatic Rh(I) catalysts [44], Koga and co-workers on CpCo [31,45,46], and Hapke et al. on CpIr [45]. To the best of our knowledge, no theoretical mechanistic investigation on the use of bimetallic half-sandwich catalysts has been reported so far. The use of bimetallic complexes as catalysts in organic synthesis is interesting, because the reaction rate and selectivity can be tuned via possible inter-metal cooperative effects. Ceccon et al. have provided rather complete information on anti-and syn-[Cr(CO)3IndRhL2]. Particularly, they studied the cyclotrimerization of methylpropriolate (MP) and dimethyl-acetylenecarboxylate (DMAD) with mono and bimetallic catalysts, i.e., IndRh(COD), p-NO2-IndRh(COD) and bimetallic anti-[Cr(CO)3IndRh(COD)]. They found that the Rh/Cr catalyst leads to a greatly enhanced catalytic efficiency compared to the monometallic one. This increase of catalytic activity was ascribed to a synergic or “cooperative” interaction between the two metals in activating the substrate of interest [33,34,35].
The main goal of this study is the detailed investigation of the mechanism of acetylene [2+2+2] cycloaddition catalyzed by Rh/Cr indenyl fragments, particularly focusing on (i) the presence of the second metal, i.e., Cr, on the mechanism and energetics; (ii) the relationship between rhodium slippage and catalytic activity, and (iii) the outline of general guidelines for the design of Rh(I) half-sandwich catalysts based on the slippage span model. Points (i) and (ii) will be presented in the Results section, while point (iii) will be treated in the Discussion.
2. Results
The computational mechanistic investigation of acetylene [2+2+2] cycloaddition to benzene catalyzed by the monometallic catalysts CpRh and IndRh was first reported in 2006 [41]. In Scheme 3a (Path I), the well-known and widely accepted mechanism, proposed by Albright for CpCo catalysis [47,48] is shown. The catalytic cycle begins with the replacement of the ancillary ligands L of the catalyst precursor, i.e., Cp-or Ind-RhL2 (L = CO, PPh3 or COD (1,5-cyclooctadiene)) by two molecules of acetylene, leading to the bis-acetylene complex Z-1. The coordinated acetylene molecules undergo oxidative coupling and the unsaturated 16-electrons rhodacycle Z-2 forms. This elementary step typically has the highest activation energy and was recently discussed in detail for group 9 metal-Cp fragments [49]. The subsequent coordination of a third acetylene molecule occurs without an appreciable activation energy and leading to Z-3, and, after its addition to the π-electron system of the rhodacycle, the intermediate Z-4 is obtained, which is characterized by an unsaturated bent six-membered ring. By further stepwise addition of two acetylene molecules, the intermediate Z-5 first forms and then the initial catalyst is regenerated with the cleavage of benzene.
An alternative mechanism was postulated by Booth and co-workers [26] on the basis of their experimental findings, which implies that a ligand of the catalyst precursor remains bonded to the metal center throughout the whole catalytic cycle; this is shown in Scheme 3b (Path II). This mechanism was rationalized by Orian et al. [43] in a recent systematic study on the indenyl effect and its connection to metal slippage. The presence of an ancillary ligand imposes strong hapticity variations in both CpRh and IndRh catalysis. In addition, the bicyclic CO-Z-b, and the heptacyclic CO-Z-h were located along this catalytic path, which resembles the one described for CpRuCl catalysis [38].
Inspired by the experimental work by Ceccon et al. [33,34,35], we chose the bimetallic catalyst [Cr(CO)3 IndRhL2], where the second metal group Cr(CO)3 can be coordinated both in anti and syn conformations and examined both paths I and II of Scheme 3. The optimized molecular structures (ZORA-BLYP/TZ2P) of anti- and syn-[Cr(CO)3IndRh(CO)2] (Figure S1) are in good agreement with the X-ray crystallographic structures (labelled as HEXPOP [50] and HAPPOD [34] in the Cambridge database (CSD) [51]). Significant geometry parameters are compared in Table S1.
For simplicity, in the ongoing discussion, anti-and syn-[Cr(CO)3IndRh] fragments are abbreviated as anti-or syn-IndRh (Scheme 3).
2.1. Acetylene [2+2+2] Cycloaddition Catalyzed by Anti-[Cr(CO)3IndRh] Fragment: Reaction Mechanism and PES (Path I)
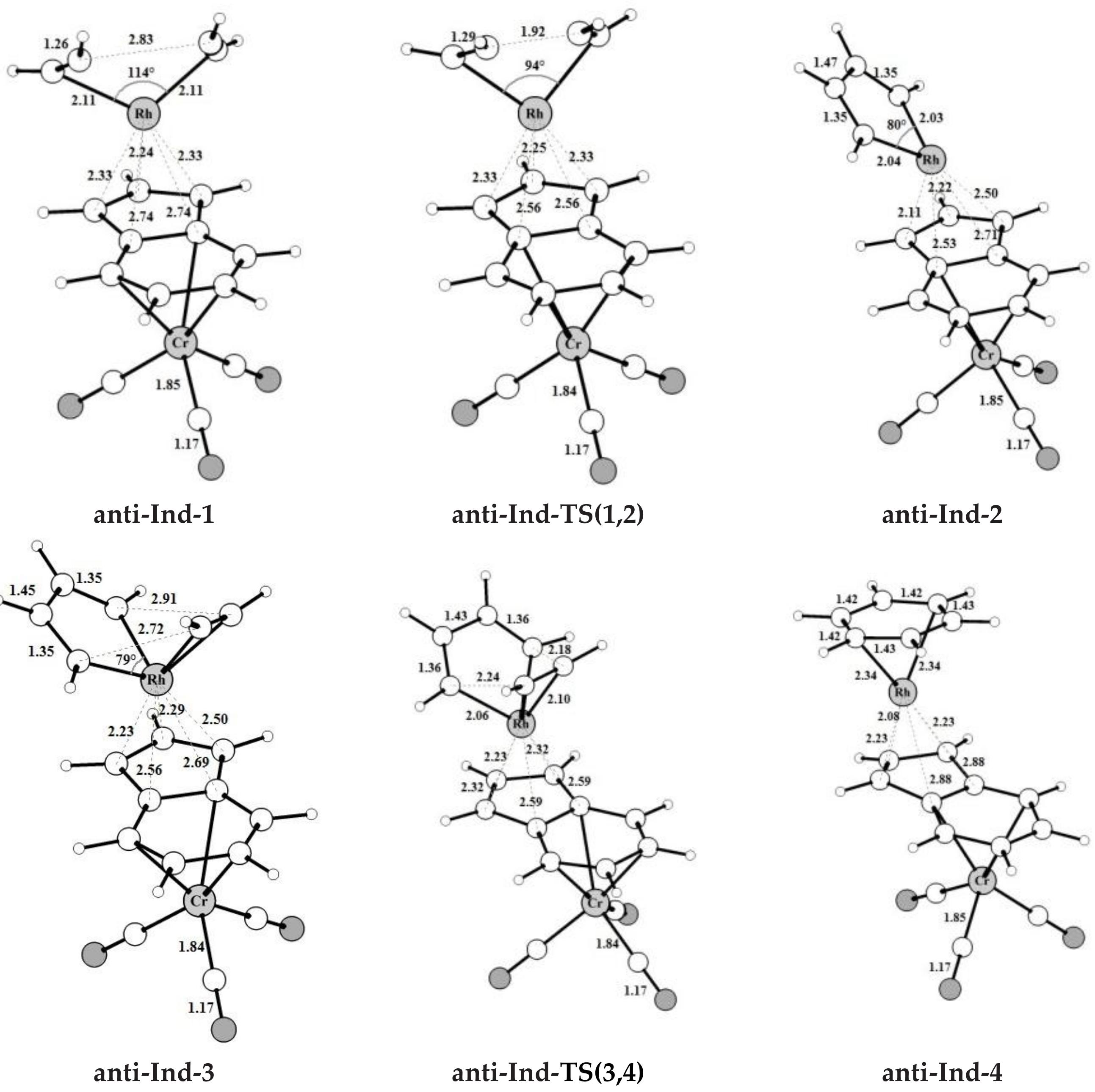
The intermediates and transition states found for acetylene [2+2+2] cycloaddition catalyzed by the bimetallic anti-IndRh along Path I (Scheme 3a), are shown in Figure 1. Those found on the PESs of the parent monometallic catalysts, i.e., CpRh and IndRh, are in Figures S2 and S3, respectively. The computed energy profile for anti-RhInd is shown in Figure 2a.
The catalytic cycle mediated by anti-IndRh is very similar to the cycle described for the monometallic parent catalysts CpRh and IndRh [41] since the Cr(CO)3 group is coordinated in anti, and thus there is no steric effect. It begins with replacement of the ancillary ligands L by two acetylene molecules to form a bis-acetylene complex labelled anti-Ind-1 (Scheme 3a). This process usually occurs experimentally by thermal or photochemical activation and might be dissociative or associative depending on the nature of the metal, on the electrophilicity of the ligands and on the substituents on the Cp ring [21,28,52]. In anti-Ind-1, the acetylene molecules are slanted with respect to the plane of the indenyl ring and the C-C bond length is 1.26 Å. The Rh-Cα and Rh-Cβ bond lengths are 2.11 Å and 2.13 Å, respectively; they are shorter as compared to those of Ind-1 (2.13 Å and 2.16 Å). This suggests that acetylene is more tightly bonded, likely due to the electron withdrawing effect of the second metal group Cr(CO)3. Additionally, the Rh-Cp coordination is more distorted in anti-Ind-1 than in the parent Cp-1 and Ind-1, as also quantified by the metal slippage parameters Δ and , which were calculated for the intermediates and the transition states along the whole catalytic cycle (Figure 2b).
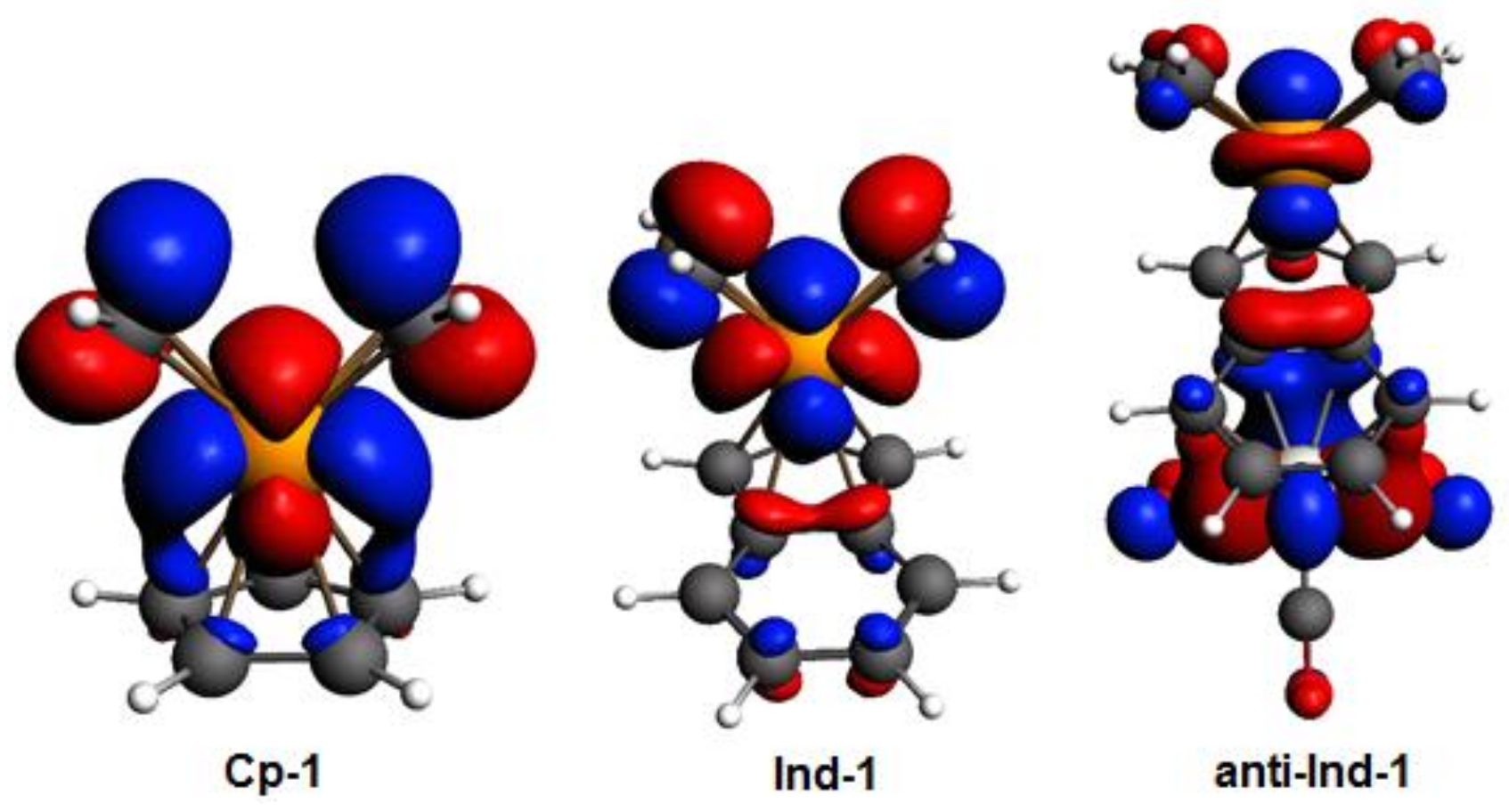
By inspecting the frontier molecular orbitals of Cp-1, Ind-1 and anti-Ind-1 shown in Figure 3, the π-antibonding character between Cp’-π system and valence d orbitals of Rh is found to increase in the order Cp-1 < Ind-1 < anti-Ind-1, leading to a corresponding increase of metal slippage. Calhorda et al. have reported the same observation in their pioneering work on the nature of indenyl effect, which was related to the nodal characteristics of Cp’-π orbitals of CpRh and IndRh [19,44]. Herein, it is found that the metal-π anti-bonding nature with Ind-π system is further enhanced in the presence of Cr(CO)3 in anti-Ind-1 compared to the parent Ind-1, leading to extra slippage of rhodium in the former.
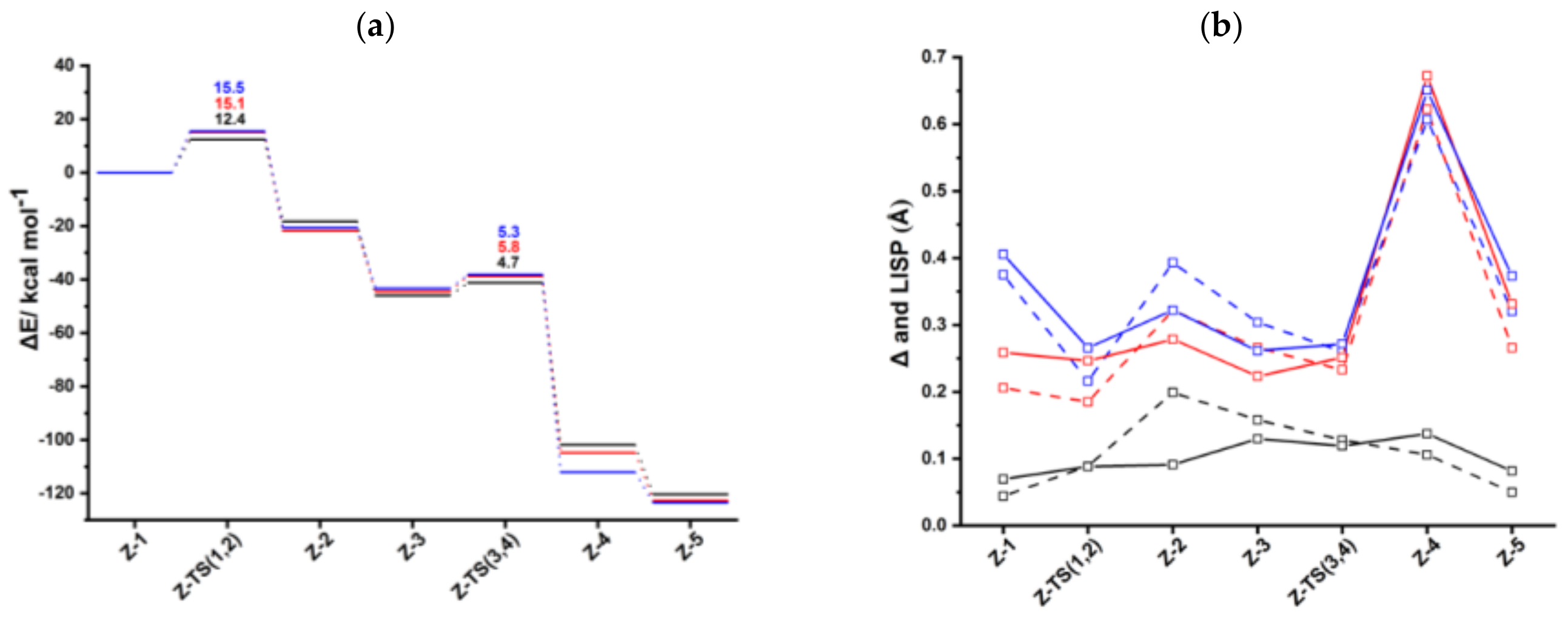
The oxidative coupling of the two coordinated acetylene molecules in anti-Ind-1 leads to the 16-electrons unsaturated rhodacyclic intermediate anti-Ind-2 (Figure 2), crossing an activation barrier of 15.5 kcal mol−1, which is higher than those computed for the formation of Ind-2 (15.1 kcal mol−1) and Cp-2 (12.4 kcal mol−1). This reaction step is exothermic by 20.6 kcal mol−1. These metallacycles are generally described by two resonating structures, i.e., a metallacyclopentadiene, as found for CpCo-(C4H4) [37,49] and CpRh-(C4H4) [41,49], and a metallacyclopentatriene, as for CpRuCl-(C4H4) [38]. In anti-Ind-2, the Cα-Cβ and Cβ-Cβ’ distances are 1.35 Å and 1.47 Å, respectively, which are rather well matched to the length of the ethylene double bond and to the length of the σ-bond between two carbon atoms, respectively. This suggests the character of rhodacyclopentadiene of anti-Ind-2.
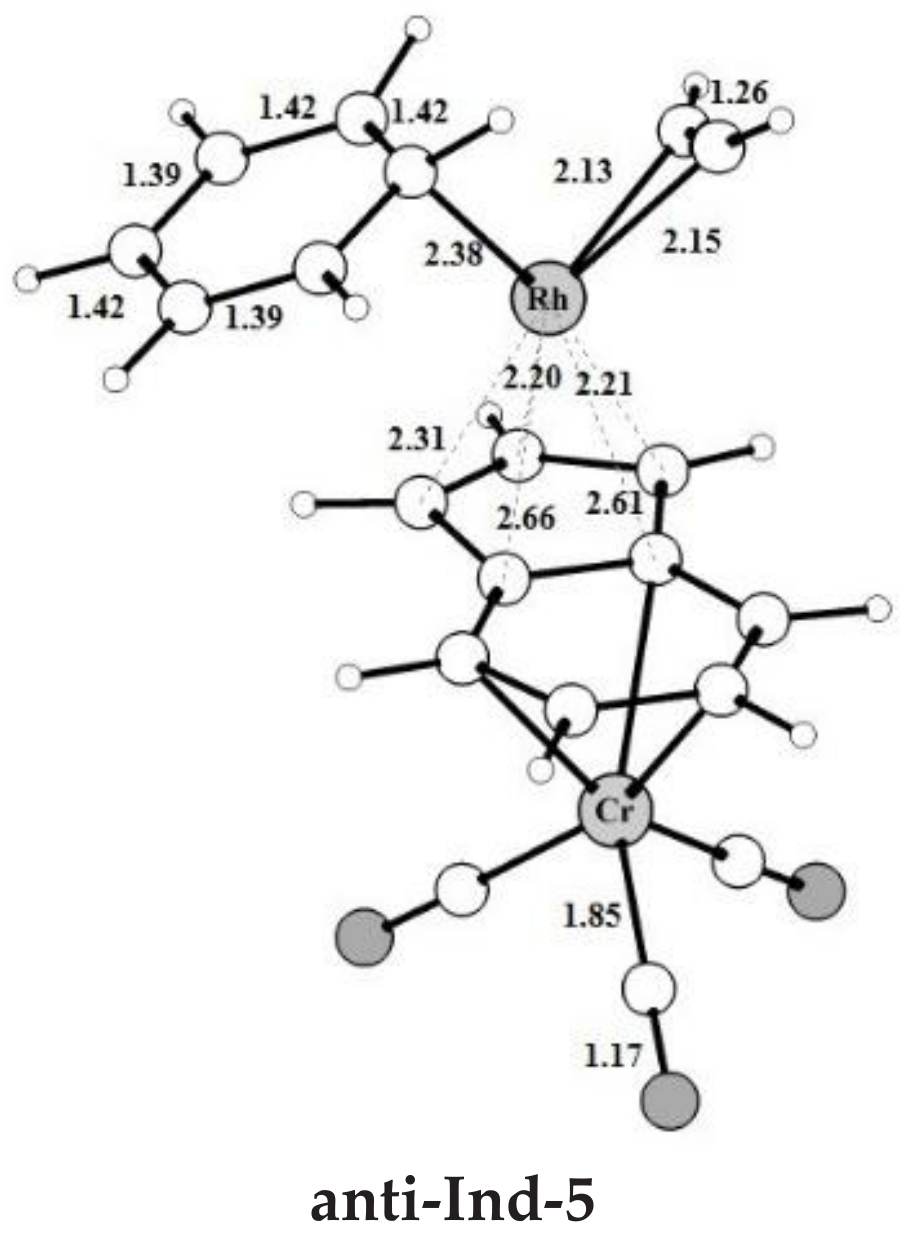
A third acetylene molecule easily coordinates to anti-Ind-2, which is converted into anti-Ind-3. This step is barrierless and exothermic by 22.8 kcal mol−1, about 5 kcal mol−1 less exothermic than the formation of CpRh-3; this value is almost identical to the reaction energy computed for the formation of IndRh-3. Subsequently, by Diels-Alder-like [4+2] addition of the coordinated acetylene to the rhodacycle, anti-Ind-4 forms, with an activation energy of 5.3 kcal mol−1 (Figure 2a). This step is strongly exothermic by 68.6 kcal mol−1, 8.4 kcal mol−1 more exothermic than the same step in the IndRh catalytic cycle; conversely, the energy barriers are very similar. Structurally, anti-Ind-4 (Figure 1) is characterized by the presence of a six-carbon arene ring coordinated to rhodium in η6 fashion, while η3 coordination is found in the Cp-Rh moiety. A similar bonding mode of rhodium is found in Ind-4; conversely, in Cp-4, η4 hapticity is observed with the arene ring and η3 +η2 coordination to Cp moiety. This pronounced slippage in Ind-4 and anti-Ind-4 explains the spikes in the profiles of Δ and (Figure 2b). The coordination of another acetylene leads to the formation of anti-Ind-5 (Figure 2a), accompanied by a variation of hapticity from η3 to η3 + η2 in the anti-IndRh fragment and by the release of 11.5 kcal mol−1. The cleavage of benzene by the incoming second acetylene completes the cycle with the regeneration of the catalyst. The released energy is 16.4 kcal mol−1. Summarizing, the η3 + η2 coordination is found along the whole cycle catalyzed by anti-IndRh except in intermediate anti-Ind-4, where η3 coordination is predicted.
Consistently with CpRh and IndRh catalysis, the first step Z-1 → Z-2, that is the oxidative coupling of the acetylene molecules, has the highest energy barrier along the cycle (Figure 2a). To gain insight on the origin of this barrier, an activation strain analysis (ASA) has been carried out and compared to those already reported for CpRh and IndRh [49]. For this purpose, the complexes were divided into two fragments, i.e., Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C4H4 moiety. Being an intramolecular reaction, the activation energy is conveneintly given as the change, upon going from the reactant to the TS, in strain within the two fragments plus the change, upon going from the reactant to the TS, in the interaction between these two fragments [53].
The results are shown in Table 1. The contributions increase from CpRh to IndRh and to anti-IndRh and are very similar for the Cp’Rh fragments (ranging from 2.4 to 2.7 kcal mol−1), but increase significantly for the C4H4 fragment going from 33.2 to 37.0 and 37.3 kcal mol−1, respectively. Since are very similar, varying from -23.2 kcal mol−1 (CpRh) to −24.6 kcal mol−1 (IndRh) and −24.5 kcal mol−1 (anti-IndRh), the increase of the barrier in the indenyl catalysts is mainly due to the strain effects localized on the bis-acetylene moiety. Based on the identical for IndRh and anti-IndRh, no influence of the second metal of the latter is found in the barrier of this oxidative coupling.
We also examined Z-4, which was found to be more stable in the case of anti-IndRh than in the cases of IndRh and CpRh. ASA was carried out on Z-4, by considering these two fragments: Cp’Rh, where (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C6H6 moiety; the results are listed in Table 2. A very high total is found in Cp-4 compared to Ind-4 and anti-Ind-4, which comes out from the benzene fragment and reflects the structural differences. In fact, benzene is bent in Cp-4 with η4 coordination (Figure S2), while in Ind-4 and anti-Ind-4, the ring is almost planar and coordinated to rhodium in η6 fashion and η3 coordination is found for the Rh coordination to the Cp ring. On the other hand, the large of Cp-4 is well balanced by a large negative , which leads to larger by about 12.2 kcal/mol than Ind-4 and anti-Ind-4.
Although the catalytic center is rhodium, we also investigated the coordination of Cr(CO)3 in all the intermediates and transition states of the anti-IndRh catalyzed cycle. ASA was carried out using as fragments Cr(CO)3 and IndRhXi (Table 3). Significantly larger values are found for anti-Ind-TS (1,2) and anti-Ind-TS (3,4); the biggest contribution comes from the IndRhLn fragment, reflecting the changes occurring at the Rh center. The of Cr(CO)3 fragment remains almost equal along the path, reflecting the fact that no important structural changes occur within the Cr(CO)3 moiety. values fluctuate in the range~47–54 kcal mol−1. In anti-Ind-4, a sudden increase of is computed. In fact, on the opposite side of the indenyl ligand, Rh-Cp coordination is highly slipped (Figure 2). Therefore, in order to compensate for the weakening of Rh-Cp coordination, Cr(CO)3 binds more tightly the benzene moiety of the aromatic ligand. In anti-Ind-4, the increased is due to a larger electrostatic contribution which is not sufficiently counterbalanced by an increase of .
2.2. Acetylene [2+2+2] Cycloaddition Catalyzed by Syn-[Cr(CO)3IndRh] Fragment: Reaction Mechanism and PES (Path I)
As stated above, the coordination of the Cr(CO)3 group to the benzene moiety of IndRh catalyst may occur in anti or syn conformation [54]. Ceccon and co-workers have illuminated the stereochemistry of the syn catalyst, i.e., syn-[Cr(CO)3IndRh], and found that it is kinetically more stable because of a Rh-Cr interaction.
First, we investigated the acetylene [2+2+2] cycloaddition to benzene catalyzed by syn-IndRh catalyst along Path I (Scheme 3a). The structures of the intermediates and transition states found on the PES with their relevant parameters are shown in Figure S4, while the computed energy profile is in Figure 4a. Like for anti-IndRh catalysis, at the beginning of the cycle, the ancillary ligands L are replaced by two molecules of acetylene, leading to the formation of the bis-acetylene complex syn-Ind-1 (Figure S4). In syn-Ind-1, the Rh-Cα bond length is 2.15 Å, the distance Cβ-Cβ’ of two acetylenes coordinated to rhodium is 2.91 Å, and the angle Cα-Rh-Cα’ is 91°. To quantify the Rh-Cp slippage along the catalytic cycle, Δ and parameters were computed for all the intermediates and transition states and are shown in Figure 4b. In syn-Ind-1, their values are 0.40 Å and 0.37 Å, respectively, with no appreciable difference with respect to anti-Ind-1; this might be ascribed to Rh-Cr interaction, despite high steric effects are present.
The oxidative coupling in syn-Ind-1 leads to the formation of the five-membered ring rhodacycle syn-Ind-2 with an activation energy of 15.2 kcal mol−1, nearly the same as found for anti-IndRh. Notably, in syn-Ind-2 complex, one CO of Cr(CO)3 interacts with Rh. In fact, the distance between the CO ligand and rhodium is 2.30 Å, and the distance between Cr and Rh center is smaller by 0.34 Å than in syn-Ind-1, implying stabilization of syn-Ind-2. In this situation, the rhodacycle syn-Ind-2 is a nearly 18-electron saturated complex, in contrast to anti-Ind-2 and the parent Ind-2 and Cp-2. The conversion of syn-Ind-1 into syn-Ind-2 is exothermic by 26.4 kcal mol−1, about 6 kcal mol−1 more than in the anti-catalyst and in the parent IndRh (Figure 3a). Finally, in this step, an hapticity shift from η3+η2 to η3 occurs (Figure 4b).
The coordination of the third acetylene to syn-Ind-2 occurs from the upper side with a low barrier of 0.7 kcal mol−1, via syn-Ind-TS (2,3) (Figure S4) and a large slippage variation ( changes by approximately 0.6 Å) leading to almost η1 coordination. The conversion syn-Ind-2 → syn-Ind-3 occurs only with a slight modification in the carbon-carbon bonds of the rhodacycle. The reaction is exothermic by 15.6 kcal mol−1. In the next step, syn-Ind-4 forms, crossing a barrier of 4.6 kcal mol−1. The value drops from 1.09 Å to 0.42 Å (Figure 4b). syn-Ind-4 has a six-carbon arene ring coordinated to rhodium in η4 fashion, while in Rh-Cp the coordination is η3 +η2. Thus, the bonding mode of rhodium is different from anti-Ind-4 (Figure 2), where η6 coordination and η3 coordination are found between the six-carbon arene ring and rhodium and the Cp ring and rhodium, respectively. However, syn-Ind-4 resembles the case of the parent Cp-4 [41]. This step is exothermic by 62.3 kcal mol−1.
Finally, the coordination of another acetylene leads to the formation of syn-Ind-5 with no appreciable activation energy and with the release of 16.8 kcal mol−1 (Figure 4a). It is less exothermic than the formation of Ind-5 but more exothermic by 5 kcal/mol than the formation of anti-Ind-5. The cleavage of benzene promoted by another acetylene completes the cycle and leads to the regeneration of the catalyst. The energy released in this last step is 18.8 kcal mol−1. During the catalysis, the Rh-Cr distance varies in the range 3.05–3.51 Å, but remains close to the crystallographic value of 3.1 Å [34].
The hapticity variations along the catalytic cycle are definitively more pronounced in syn-IndRh catalysis than in the anti-IndRh one.
By inspecting the energy profile (Figure 4a), in the syn-IndRh catalyzed process the oxidative coupling Z-1 → Z-2 has the highest energy barrier. From ASA (Table 1), a very high ΔΔEstrain contribution to the barrier has been found compared to anti-IndRh and also to the parent catalysts IndRh and CpRh. However, is a pretty strongly stabilizing term and compensates for the strain, resulting in a lowering of . In contrast to anti-IndRh, the strain arising from the Cp’Rh fragment is very high (Table 1) because of the steric effects and because Cr(CO)3 undergoes deformation to interact with the Rh center.
To further assess the interaction and the role of Cr(CO)3 in the syn-IndRh catalyzed process, ASA was carried out for the intermediates and transition states of the whole catalytic cycle, considering Cr(CO)3 and IndRhXi fragments; the results are reported in Table 4. are in the range ~49–58 kcal mol−1, larger than those computed for the anti-IndRh molecular species (Table 3), suggesting Rh-Cr stabilizing interaction in the former case. On the other hand, is also larger. This is mainly ascribed to steric factors. In particular, the contribution of Cr(CO)3 to in syn-Ind-2, syn-Ind-TS (2,3) and syn-Ind-3 is higher, suggesting some structural differences from the anti-analogous species. In fact, in these structures one carbonyl ligand is coordinated to Rh (Figure S4). This feature also leads to larger in syn-Ind-2.
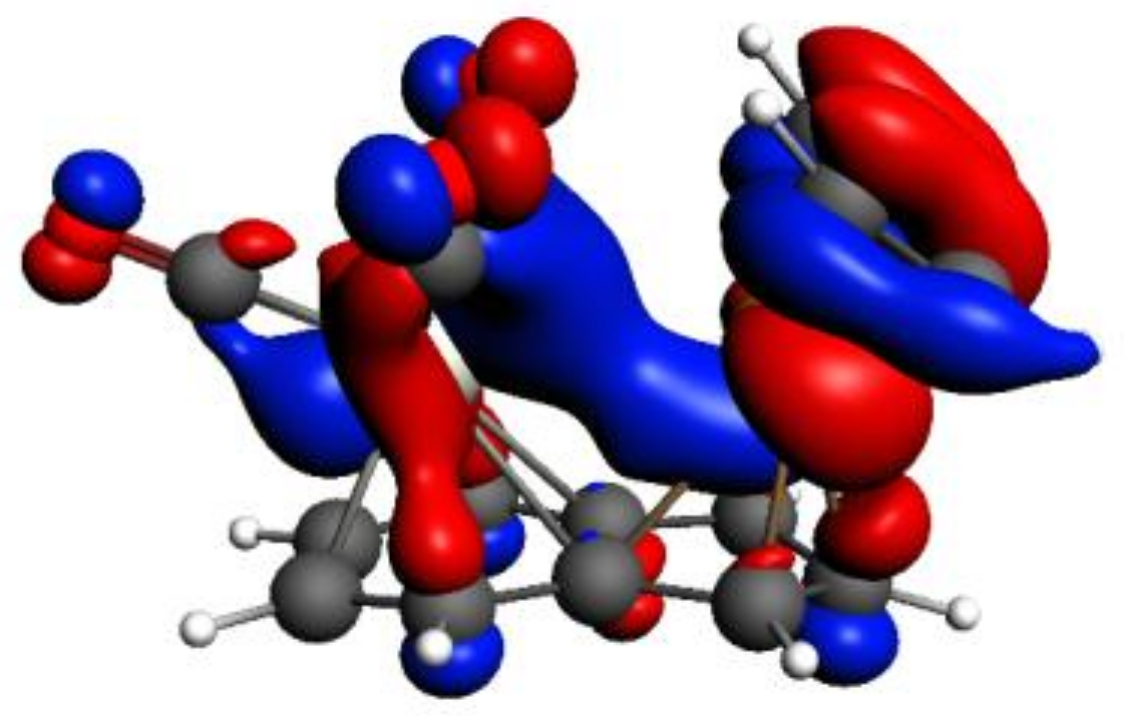
As an example of an existing favorable inter-metal interaction, in Figure 5, we show HOMO-3 of syn-Ind-1, which is formed by the contributions of the HOMOs of Cr(CO)3 and IndRh(C2H2)2 fragments, whose metal d-based molecular orbitals (MOs) indicate stabilizing d-d bonding between Rh-Cr.
2.3. Acetylene [2+2+2] Cycloaddition Catalyzed by Anti-[Cr(CO)3IndRh] Fragment: Reaction Mechanism and PES (Path II)
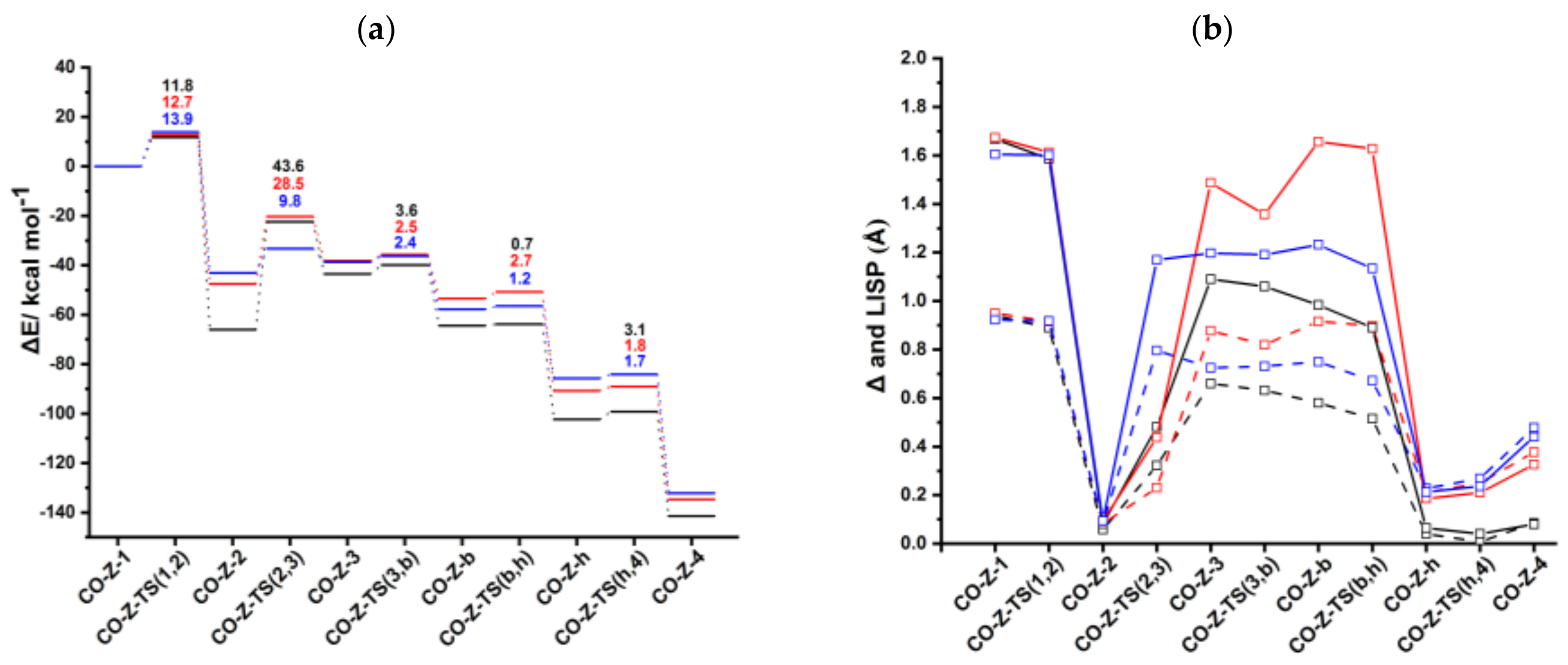
According to the alternative mechanistic path by Booth et al. [26], an ancillary ligand of the catalyst precursor remains bonded to rhodium during the catalytic cycle; this mechanism is denoted Path II (Scheme 3b). For CpRh(CO) and IndRh(CO), this path has been thoroughly explored, making it possible to give an interpretation of the higher efficiency of the indenyl catalyst observed in the experiment [35]. Thus, here we considered the same hypothesis for anti-IndRh(CO) catalyzed acetylene [2+2+2] cycloaddition. The structures of the intermediates and transition states found on the PES with their relevant parameters are in Figures S5–S7. The energy profile of the process is shown in Figure 6a.
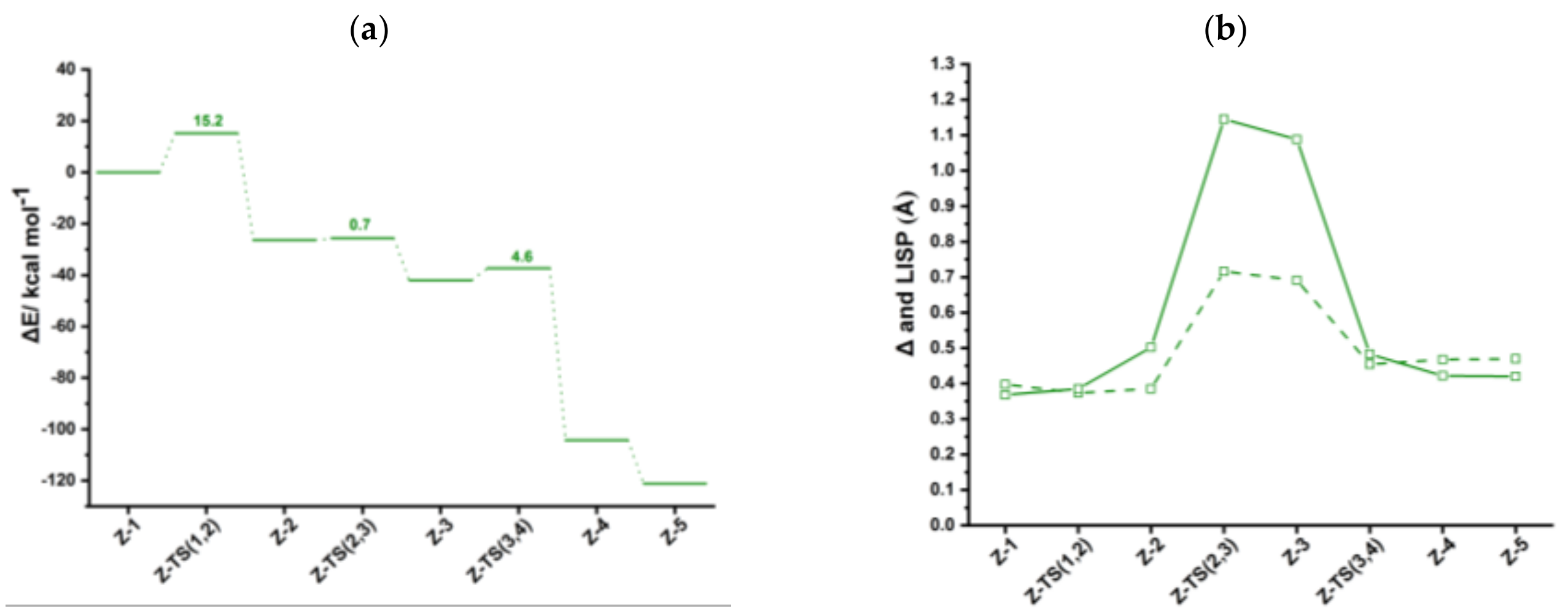
CO-anti-Ind-1 is characterized by η1 coordination which is due to the presence of two acetylenes and the CO ligand, as observed for CpRh(CO) and IndRh(CO) catalysis [43]. Δ and values are 0.92 Å and 1.60 Å, respectively (Figure 6b). The initial oxidative coupling leads to the formation of CO-anti-Ind-2, in which rhodium is coordinatively saturated due to the presence of CO. The energy barrier required to cross CO-anti-Ind-TS (1,2) is 13.9 kcal mol−1 slightly higher if compared to those computed for CpRh(CO) and IndRh(CO) catalysis (by about 1–2 kcal mol−1). This is the same energy trend observed along Path I. The conversion of CO-anti-Ind-1 into CO-anti-Ind-2 is accompanied by the hapticity change from η1 to distorted η5 and is exothermic by 43.1 kcal mol−1 (Figure 6a), 4.5 kcal mol−1 and 23.0 kcal mol−1 less than the analogous step in IndRh(CO) and CpRh(CO) catalysis, respectively. The addition of the third acetylene leads to the formation of the η1 CO-anti-Ind-3 with an activation energy of 9.8 kcal mol−1. The barriers for this step are much higher in the cases of IndRh(CO) and CpRh(CO) catalysis, i.e., 28.5 and 43.6 kcal mol−1, respectively. Thus, this step is kinetically favored with the bimetallic anti-IndRh(CO). In addition, the formation of CO-anti-Ind-3 is endothermic by 4.4 kcal mol−1, that is overall less endothermic if compared to the same step in IndRh(CO) and CpRh(CO) catalysis, for which 9.3 kcal mol−1 and 22.5 kcal mol−1 are computed, respectively. Thus, the bimetallic catalyst has also a thermodynamic advantage. In the next step, the activation energy of 2.4 kcal mol−1 is necessary to cross CO-anti-Ind-TS (3,b) and generate the bicyclic intermediate CO-anti-Ind-b (Figure S7), with negligible difference from the IndRh(CO) catalyzed step and lower by 1.2 kcal mol−1 than in CpRh(CO) catalyzed step. The formation of CO-anti-Ind-b is accompanied by the release of 19.1 kcal mol−1, being 3.8 kcal mol−1 more exothermic than the formation of the parent CO-Ind-b but 1.8 kcal mol−1 less exothermic than CO-Cp-b. One can notice that the bicyclic intermediate in CpRh(CO) catalysis has an higher energy while in bimetallic anti-IndRh(CO) and IndRh(CO) catalysis it lies at a lower energy (Figure 6a), suggesting a better catalytic efficiency in these latter cases. By crossing a modest barrier of 1.2 kcal mol−1, the bicyclic CO-anti-Ind-b readily transforms into the heptacyclic intermediate CO-Ind-h; this step is accompanied by the haptotropic shift η1 → distorted η5 and by the release of 28.0 kcal mol−1, a value lower than those computed for the parent catalysts, i.e., −37.4 kcal mol−1 in the case of IndRh(CO) and −37.8 kcal mol−1 in the case of CpRh(CO), respectively. CO-anti-Ind-h undergoes reductive elimination with an activation energy of 1.7 kcal mol−1. CO-anti-Ind-4 forms and 46.4 kcal mol−1 are released. Finally, benzene is cleaved from CO-anti-Ind-4 by stepwise addition of two acetylene molecules and the catalyst is regenerated.
With the formation of the 18-electron intermediate CO-Z-2, a rather flat portion of the PES begins (Figure 6a). ASA was carried out on CO-Z-2 considering CO-Cp’Rh (Cp’ = Cp, Ind and anti-[Cr(CO)3Ind]) and the C4H4 moiety as fragments; the results are listed in Table 5. The similar strain in fragment C4H4 computed for CO-Cp-2, CO-Ind-2, and CO-anti-Ind-2 (Table 5) revealed that the structure of this moiety is very similar in the three catalysts (see also Figures S5–S7). The strain contribution of the fragment CO-Cp’Rh is much higher in CO-anti-Ind-2 and CO-Ind-2 than in CO-Cp-2. This is related to the least slippage predicted for this last species. The stronger interaction ΔEint in CO-Ind-2 and CO-anti-Ind-2 does not counterbalance their strain term and thus CO-Cp-2 results the most stabilized among the three.
In the subsequent step, i.e., the addition of third acetylene to the 18-electrons rhodacycle CO-Z-2, an energy barrier is found along the catalytic Path II (Figure 6a). CO-Z-TS (2,3) was divided into two fragments, i.e., CO-Cp’Rh(C4H4) (Cp’ = Cp, Ind and anti-[Cr(CO)3Ind] and acetylene and ASA was performed. The results are shown in Table 6. The acetylene in all cases is only slightly deformed, whereas CO-Cp’Rh(C4H4) is highly strained.
To summarize, we found that most of the steps are kinetically as well as thermodynamically more favored in the bimetallic anti-IndRh(CO) catalysis. At a glance, this can be seen also from the energy profile (Figure 6a), which is flatter in the case of anti-IndRh(CO) than in the parent IndRh(CO) and CpRh(CO) catalyzed processes. Alternatively, the slippage variations quantified with Δ and (Figure 6b) are less pronounced in anti-IndRh(CO) than those computed for the parent monometallic catalysts. Thus, the smaller slippage variations and the flatter potential energy profile of the bimetallic anti-IndRh(CO) suggest an higher catalytic efficiency than the monometallic IndRh(CO) and CpRh(CO), which is consistent with the experimental findings [26]. The phenomena of indenyl effect for IndRh and extra-indenyl effect for the bimetallic anti-IndRh can be fully explained when considering the mechanism of path II.
Notably, path II has been excluded for syn-IndRh(CO) because in this particular case the rhodium center would be too much crowded.
3. Discussion
To quantify the catalytic efficiency, we calculated the turnover frequencies () for the studied catalytic cycles by using the energy span model [23,55]. Calculations were run at standard room temperature (298.15 K), as well as at the reflux temperature of toluene, i.e., 383.65 K, which is occasionally used in alkyne [2+2+2] cycloadditions as solvent [25,26,29]; the values are listed in Table 7.
Following the catalytic Path I (Scheme 3a), the are in the order CpRh > IndRh > anti-IndRh, suggesting that the bimetallic catalyst anti-IndRh is worse than the monometallic parent catalysts IndRh and CpRh. This is in contrast with the experimental results [36]. Also, no appreciable difference in terms of TOF is found between IndRh and anti-IndRh, suggesting that the influence of the second metal group, i.e., Cr(CO)3, on the efficiency of the catalyst is negligible. In all these cases, the TOF determining intermediate (TDI) is the bis-acetylene intermediate Z-1 and the TOF determining transition state (TDTS) is the subsequent transition state Z-TS (1,2) (Figure 2a).
Conversely, along the catalytic Path II (Scheme 3b), in terms of , a significant enhancement of the catalytic activity of the bimetallic anti-IndRh(CO) is found compared to those of the parent catalysts IndRh(CO) and CpRh(CO). On the basis of the values reported in Table 7, the following trend can be established anti-IndRh(CO) > IndRh(CO) > CpRh(CO), which is consistent with the experimental observation [26]. In this case, the intermediate CO-Z-2 is the TDI and the subsequent transition state CO-Z-TS (2,3) is the TDTS (Figure 6a). We can thus confirm that the coordination of Cr(CO)3 favors the catalytic activity.
As recently reported [22], the slippage variations in intermediates and transition states along the catalytic cycle may be related to the chemical activity of the half-metallocene catalysts in this class of reactions. For this purpose, the slippage span was calculated. Along Path I (Scheme 3a), values follow the trend anti-IndRh > IndRh > CpRh. Since ’ trend is reverse, i.e., anti-IndRh < IndRh < CpRh, this indicates that the higher the slippage span along the catalytic cycle is, the lower the performance of the catalyst is [22]. When following Path II, values follow the trend anti-IndRh(CO) < IndRh(CO) < CpRh(CO), while TOFs follow the reverse trend anti-IndRh(CO) > IndRh(CO) > CpRh(CO), in agreement with the presence of extra indenyl and indenyl effect in the bimetallic and monometallic Ind catalysts, respectively.
The mechanism for syn-IndRh catalyzed acetylene [2+2+2] cycloaddition is different, and so we cannot directly compare the values. However, the higher energy barriers and higher slippage variations certainly do not favor the syn catalyst.
4. Materials and Methods
All the equilibrium and transition state geometries were fully optimized (i.e., without any constraint) in gas-phase using density functional theory (DFT) approach as implemented in the Amsterdam Density Functional (ADF 2016, SCM, Vrije Universiteit: Amsterdam, The Netherlands, 2016) program [56,57]. The BLYP [58,59,60,61] function in combination with the TZ2P basis set was applied for all elements. The scalar relativistic effects were accounted for within the zeroth-order regular approximation (ZORA), which is an excellent approximation of the relativistic Dirac equation [62,63,64]. The TZ2P basis set [65] is a large uncontracted set of Slater-type orbitals (STOs) of triple-ζ quality and has been augmented with two sets of polarization functions on each atom: 2p and 3d for H, 3d, and 4f for C and O, 4p and 4f for Cr, and 5p and 4f for Rh. The frozen-core approximation was adopted for core electrons: up to 1s for C and O, up to 2p for Cr and up to 3d for Rh. This level of theory has been applied with success in previous studies [22,41,42,43,44,49].
Frequency calculations were computed to confirm that all the intermediates have positive frequencies, whereas the transition states have one imaginary frequency. The character of the normal mode associated with this imaginary frequency was carefully examined to verify that the correct transition state was found.
Activation strain analyses (ASA) were performed on selected geometries [66,67]. ASA is an approach based on molecule fragmentation, useful to understand the properties of the chemical bonding. For this purpose, the total energy of a complex can be decomposed as the sum of a strain contribution and an interaction contribution (Equation (5)):
is the energy required for the geometrical deformation of the fragments when they are brought from infinite distance to the geometry they acquire in the complex, while is the actual energy change when the deformed fragments are combined to form the overall complex. can be further decomposed into electrostatic interaction , Pauli repulsion and orbital interactions in the framework of Kohn-Sham molecular orbital (MO) theory (Equation (6)):
According to Fernandez et al. [53], who proposed an extension to the activation strain model for unimolecular reaction steps, the activation barrier can be given as the change, upon going from the reactant to the TS, in strain within the two defined fragments plus the change, upon going from the reactant to the TS, in the interaction between the same two fragments:
This situation is encountered in the present study, when the five-membered rhodacycle is formed from the bis-acetylene precursor.
The turnover frequency () was calculated by using the energy span model proposed by Kozuch and Shaik [23,55]. The expression is:
where is reaction Gibbs free energy, Ti and Ij are Gibbs free energies of the ith transition state and intermediate, respectively. , called the energy span, is equal to if or to 0 if . Instead of Gibbs free energies, electronic energies were used, as it was demonstrated that there are no significant differences in the energy profiles and corresponding ratios for analogous catalytic cycles [44].
5. Conclusions
We performed a theoretical investigation of the bimetallic system [Cr(CO)3IndRh]-mediated [2+2+2] cycloaddition of acetylene to benzene. Through a detailed exploration of the potential energy surfaces (PESs), the intermediates and transition states were located using density functional theory (DFT) methods following two mechanistic paths, i.e., Path I and II (Scheme 3a,b). The bimetallic catalysts anti- and syn-[Cr(CO)3IndRh] were tested in silico and compared to the monometallic parent catalysts CpRh and IndRh. The anti or syn coordination of Cr(CO)3 affects the energetics of the cycle and also to the mechanism. The reaction energies and barriers, the turn over frequency () and the change of the slippage parameters along the catalytic cycles are discussed.
Considering Path I, the established trend for the slippage span is anti-IndRh > IndRh > CpRh while TOF values follow the opposite order, i.e., CpRh > IndRh > anti-IndRh. This leads to the conclusion that the lower slippage span along the catalytic cycle, the higher the catalytic performance. In any case, this does not explain the highest catalytic efficiency of the bimetallic Rh/Cr compound (extra-indenyl effect).
Conversely, if we follow the catalytic cycle along Path II, a dramatic TOF enhancement of the bimetallic system anti-IndRh(CO) relative to the parent CpRh(CO) and IndRh(CO) is found. On the basis of the , the following trend of catalytic efficiency can be established anti-IndRh(CO) > IndRh(CO) > CpRh(CO), in agreement with experimental findings. In this case, the slippage span is inverted, too, i.e., CpRh(CO) > IndRh(CO) > anti-IndRh(CO), which again leads us to conclude that the lower the slippage span is, the higher the catalytic efficiency is. We can thus conclude that the coordination of Cr(CO)3 in the bimetallic indenyl catalyst improves the efficiency along Path II.
The hapticity variations of intermediates and transition states along the catalytic cycle are highly pronounced in the syn bimetallic conformer, implying low values and leading to the conclusion that the syn-[Cr(CO)3IndRh] catalyzed process is not favored.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/9/8/679/s1: Figure S1: Optimized geometries of anti- and syn-[Cr(CO)3IndRh(CO)2]. Level of theory: ZORA-BLYP/TZ2P; Figure S2: Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (I) for acetylene [2+2+2] cycloaddition catalyzed by CpRh fragment; Figure S3. Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (I) for acetylene [2+2+2] cycloaddition catalyzed by IndRh fragment; Figure S4: Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (I) for acetylene [2+2+2] cycloaddition catalyzed by syn-IndRh fragment; Figure S5: Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (II) for acetylene [2+2+2] cycloaddition catalyzed by CpRh(CO) fragment; Figure S6: Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (II) for acetylene [2+2+2] cycloaddition catalyzed by IndRh(CO) fragment; Figure S7: Optimized structures with relevant geometric parameters, i.e., bond lengths (Å) and angles (deg) of the intermediates and transition states along Path (II) for acetylene [2+2+2] cycloaddition catalyzed by anti-IndRh(CO) fragment, Table S1: Selected geometric parameters of the anti- and syn-[Cr(CO)3IndRh(CO)2], computed at ZORA-BLYP/TZ2P level of theory, Table S2: Cartesian coordinates (in Å) and ADF total energies (in kcal mol−1) of all the intermediates and the transition states along Path I catalyzed by anti-[Cr(CO)3IndRh] fragment, computed at ZORA-BLYP/TZ2P level of theory, Table S3: Cartesian coordinates (in Å) and ADF total energies (in kcal mol−1) of all the intermediates and the transition states along Path I catalyzed by syn-[Cr(CO)3IndRh] fragment, computed at ZORA-BLYP/TZ2P level of theory, Table S4: Cartesian coordinates (in Å) and ADF total energies (in kcal mol−1) of all the intermediates and the transition states along Path I catalyzed by anti-[Cr(CO)3IndRh] fragment, computed at ZORA-BLYP/TZ2P level of theory.
Author Contributions
Conceptualization, L.O.; investigation, S.M.A. and M.D.T.; data curation, S.M.A. and M.D.T.; writing—original draft preparation, S.M.A. and L.O.; writing—review and editing, L.O.; visualization, M.D.T.; supervision, L.O.; funding acquisition, L.O.
Funding
This research was funded by CINECA (Casalecchio di Reno, Italy) ISCRA Grant STREGA (Filling the STructure-REactivity GAp: in silico multiscale approaches to rationalize the design of molecular catalysts), who provided generous allocation of computational time on Galileo; P. I.: L. O. The APC was waived by the journal.
Acknowledgments
M.D.T. is grateful to Fondazione CARIPARO for financial support (PhD grant). This work has been inspired by the thorough synthetic and reactivity studies of heterobimetallic half-sandwich compounds, carried out in the past three decades by Alberto Ceccon and his co-workers, whom are all gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Jones, G. Pyridines and their Benzo Derivatives: (V) Synthesis. Compr. Heterocycl. Chem. 1984, 2, 395–510. [Google Scholar]
- Berthelot, M. Ueber die Polymeren des Acetylens. Ann. Chem. Pharm. 1867, 141, 173–184. [Google Scholar]
- Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. Cyclisierende Polymerisation von Acetylen I Über Cyclooctatetraen. Justus Liebigs Ann. Chem. 1948, 560, 1–92. [Google Scholar] [CrossRef]
- Wakatsuki, Y.; Yamazaki, H. Novel synthesis of heterocyclic compounds from acetylenes. J. Chem. Soc. Chem. Commun. 1973, 280a. [Google Scholar] [CrossRef]
- Wakatsuki, Y.; Yamazaki, H. Improved Catalytic Activity of Cyclopentadienylcobalt in the Preparation of Pyridines from Acetylenes and Nitrites. Bull. Chem. Soc. Jpn. 1985, 58, 2715–2716. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, D.; Urabe, H.; Sato, F. Metalative Reppe Reaction. Organized Assembly of Acetylene Molecules on Titanium Template Leading to a New Style of Acetylene Cyclotrimerization. J. Am. Chem. Soc. 2001, 123, 7925–7926. [Google Scholar] [CrossRef]
- Takahashi, T.; Li, Y.; Stepnicka, P.; Kitamura, M.; Liu, Y.; Nakajima, K.; Kotora, M. Coupling Reaction of Zirconacyclopentadienes with Dihalonaphthalenes and Dihalopyridines: A New Procedure for the Preparation of Substituted Anthracenes, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2002, 124, 576–582. [Google Scholar] [CrossRef]
- Werner, H. Electron-Rich Half-Sandwich Complexes?Metal Basespar excellence. Angew. Chem. Int. Ed. Engl. 1983, 22, 927–949. [Google Scholar] [CrossRef]
- Consiglio, G.; Morandini, F. Half-sandwich chiral ruthenium complexes. Chem. Rev. 1987, 87, 761–778. [Google Scholar] [CrossRef]
- Bauer, E.B. Chiral-at-metal complexes and their catalytic applications in organic synthesis. Chem. Soc. Rev. 2012, 41, 3153–3167. [Google Scholar] [CrossRef]
- Saito, S.; Yamamoto, Y. Recent Advances in the Transition-Metal-Catalyzed Regioselective Approaches to Polysubstituted Benzene Derivatives. Chem. Rev. 2000, 100, 2901–2916. [Google Scholar] [CrossRef]
- Collman, J.P. Priciples and Applications of Organotransition Metal Chemistry. Univ. Sci. Books 1987, 324. [Google Scholar]
- Varela, J.A.; Saá, C. Construction of Pyridine Rings by Metal-Mediated [2 + 2 + 2] Cycloaddition. Chem. Rev. 2003, 103, 3787–3802. [Google Scholar] [CrossRef]
- Bönnemann, H. Cobalt-Catalyzed Pyridine Syntheses from Alkynes and Nitriles. Angew. Chem. Int. Ed. Engl. 1978, 17, 505–515. [Google Scholar] [CrossRef]
- Bönnemann, H. Organocobalt Compounds in the Synthesis of Pyridines-An Example of Structure-Effectivity Relationships in Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1985, 24, 248–262. [Google Scholar] [CrossRef]
- Schmid, G.; Schütz, M. 1, 2-Azaborolyl complexes. Cobalt 1, 2-azaborolyl diene complexes. Organometallics 1992, 11, 1789–1792. [Google Scholar] [CrossRef]
- Koga, N.; Morokuma, K. Ab initio molecular orbital studies of catalytic elementary reactions and catalytic cycles of transition-metal complexes. Chem. Rev. 1991, 91, 823–842. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Veiros, L.F. Ring slippage in indenyl complexes: Structure and bonding. Coord. Chem. Rev. 1999, 185–186, 37–51. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Romão, C.C.; Veiros, L.F. The nature of the indenyl effect. Chem. Eur. J. 2002, 8, 868–875. [Google Scholar] [CrossRef]
- Basolo, F.; Pearson, R.G. Mechanisms of Inorganic Chemistry; John Wiley Sons Inc.: Hoboken, NJ, USA, 1967. [Google Scholar]
- Schuster-Woldan, H.G.; Basolo, F. Kinetics and Mechanism of Substitution Reactions of π-Cyclopentadienyldicarbonylrhodium. J. Am. Chem. Soc. 1966, 88, 1657–1663. [Google Scholar] [CrossRef]
- Dalla-Tiezza, M.; Bickelhaupt, F.M.; Orian, L. Half-Sandwich Metal-Catalyzed Alkyne [2+2+2] Cycloadditions and the Slippage Span Model. Chem. Open 2018, 143–154. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Diversi, P.; Ingrosso, G.; Lucherini, A.; Porzio, W.; Zocchi, M. Synthesis and x-ray structures of cobalta-, rhodia-, and iridiacycloalkanes. Observation of novel structural features in the metallocyclopentane rings. Inorg. Chem. 1980, 19, 3590–3597. [Google Scholar] [CrossRef]
- Borrini, A.; Diversi, P.; Ingrosso, G.; Lucherini, A.; Serra, G. Highly active rhodium catalysts for the [2+2+2] cycloaddition of acetylenes. J. Mol. Catal. 1985, 30, 181–195. [Google Scholar] [CrossRef]
- Abdulla, K.; Booth, B.L.; Stacey, C. Cyclotrimerization of acetylenes catalyzed by (η5-cyclopentadienyl)rhodium complexes. J. Organomet. Chem. 1985, 293, 103–114. [Google Scholar] [CrossRef]
- Hart-Davis, A.J.; Mawby, R.J. Reactions of π-indenyl complexes of transition metals. Part I. Kinetics and mechanisms of reactions of tricarbonyl-π-indenylmethylmolybdenum with phosphorus(III) ligands. J. Am. Chem. Soc. 1969, 0, 2403–2407. [Google Scholar] [CrossRef]
- Rerek, M.E.; Ji, L.-N.; Basolo, F. The indenyl ligand effect on the rate of substitution reactions of Rh(η-C9H7)(CO)2 and Mn(η-C9H7)(CO)3. J. Chem. Soc. Chem. Commun. 1983, 1208–1209. [Google Scholar] [CrossRef]
- Cioni, P.; Diversi, P.; Ingrosso, G.; Lucherini, A.; Ronca, P. Rhodium-catalyzed synthesis of pyridines from alkynes and nitriles. J. Mol. Catal. 1987, 40, 337–357. [Google Scholar] [CrossRef]
- Diversi, P.; Ermini, L.; Ingrosso, G.; Lucherini, A. Electronic and steric effects in the rhodium-complex catalysed co-cyclization of alkynes and nitriles to pyridine derivatives. J. Organomet. Chem. 1993, 447, 291–298. [Google Scholar] [CrossRef]
- Dahy, A.A.; Yamada, K.; Koga, N. Theoretical Study on the Reaction Mechanism for the Formation of 2-Methylpyridine Cobalt(I) Complex from Cobaltacyclopentadiene and Acetonitrile. Organometallics 2009, 28, 3636–3649. [Google Scholar] [CrossRef]
- Trost, B.M.; Ryan, M.C. Indenylmetal Catalysis in Organic Synthesis. Angew. Chem. Int. Ed. 2017, 56, 2862–2879. [Google Scholar] [CrossRef]
- Bonifaci, C.; Carta, G.; Ceccon, A.; Gambaro, A.; Santi, S.; Venzo, A. Heterobimetallic Indenyl Complexes. Kinetics and Mechanism of Substitution and Exchange Reactions of trans -[Cr(CO) 3 -indenyl-Rh(CO) 2 ] with Olefins. Organometallics 1996, 15, 1630–1636. [Google Scholar] [CrossRef]
- Bonifaci, C.; Ceccon, A.; Gambaro, A.; Ganis, P.; Santi, S.; Valle, G.; Venzo, A. Heterobimetallic indenyl complexes. Synthesis and structure of cis-[Cr(CO)3(indenyl)RhL2 (L2 = norbornadiene, (CO)2). Organometallics 1993, 12, 4211–4214. [Google Scholar] [CrossRef]
- Ceccon, A.; Santi, S.; Orian, L.; Bisello, A. Electronic communication in heterobinuclear organometallic complexes through unsaturated hydrocarbon bridges. Coord. Chem. Rev. 2004, 248, 683–724. [Google Scholar] [CrossRef]
- Ceccon, A.; Gambaro, A.; Santi, S.; Venzo, A. On different chemical and catalytic behavior of (η-indenyl)-Rh(η4-COD) and Cr(CO)3(μ-η:η-indenyl)Rh(η4-COD) complexes. J. Mol. Catal. 1991, 69, L1–L6. [Google Scholar] [CrossRef]
- Hardesty, J.H.; Koerner, J.B.; Albright, T.A.; Lee, G.-Y. Theoretical Study of the Acetylene Trimerization with CpCo. J. Am. Chem. Soc. 1999, 121, 6055–6067. [Google Scholar] [CrossRef]
- Kirchner, K.; Calhorda, M.J.; Schmid, R.; Veiros, L.F. Mechanism for the Cyclotrimerization of Alkynes and Related Reactions Catalyzed by CpRuCl. J. Am. Chem. Soc. 2003, 125, 11721–11729. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Costa, P.J.; Kirchner, K.A. Benzene and heterocyclic rings formation in cycloaddition reactions catalyzed by RuCp derivatives: DFT studies. Inorganica Chim. Acta 2011, 374, 24–35. [Google Scholar] [CrossRef]
- Dazinger, G.; Torres-Rodrigues, M.; Kirchner, K.; Calhorda, M.J.; Costa, P.J. Formation of pyridine from acetylenes and nitriles catalyzed by RuCpCl, CoCp, and RhCp derivatives—A computational mechanistic study. J. Organomet. Chem. 2006, 691, 4434–4445. [Google Scholar] [CrossRef]
- Orian, L.; van Stralen, J.N.P.; Bickelhaupt, F.M. Cyclotrimerization Reactions Catalyzed by Rhodium(I) Half-Sandwich Complexes: A Mechanistic Density Functional Study. Organometallics 2007, 26, 3816–3830. [Google Scholar] [CrossRef]
- Orian, L.; van Zeist, W.J.; Bickelhaupt, F.M. Linkage Isomerism of Nitriles in Rhodium Half-Sandwich Metallacycles. Organometallics 2008, 27, 4028–4030. [Google Scholar] [CrossRef]
- Orian, L.; Swart, M.; Bickelhaupt, F.M. Indenyl Effect Due to Metal Slippage? Computational Exploration of Rhodium-Catalyzed Acetylene [2+2+2] Cyclotrimerization. Chem. Phys. Chem. 2014, 15, 219–228. [Google Scholar] [CrossRef]
- Orian, L.; Wolters, L.P.; Bickelhaupt, F.M. In Silico Design of Heteroaromatic Half-Sandwich Rh(I) Catalysts for Acetylene [2+2+2] Cyclotrimerization: Evidence of a Reverse Indenyl Effect. Chem. Eur. J. 2013, 19, 13337–13347. [Google Scholar] [CrossRef]
- Dahy, A.A.; Suresh, C.H.; Koga, N. Theoretical Study of the Formation of a Benzene Cobalt Complex from Cobaltacyclopentadiene and Acetylene. Bull. Chem. Soc. Jpn. 2005, 78, 792–803. [Google Scholar] [CrossRef]
- Dahy, A.A.; Koga, N. Trimerization of Alkynes in the Presence of a Hydrotris(pyrazolyl)borate Iridium Catalyst and the Effect of Substituent Groups on the Reaction Mechanism: A Computational Study. Organometallics 2015, 34, 4965–4974. [Google Scholar] [CrossRef]
- Albright, T.A.; Hofmann, P.; Hoffmann, R.; Lillya, C.P.; Dobosh, P.A. Haptotropic rearrangements of polyene-MLn complexes. 2. Bicyclic polyene-MCp, M(CO)3 systems. J. Am. Chem. Soc. 1983, 105, 3396–3411. [Google Scholar] [CrossRef]
- Schore, N.E. Transition metal-mediated cycloaddition reactions of alkynes in organic synthesis. Chem. Rev. 1988, 88, 1081–1119. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Orian, L. Group 9 Metallacyclopentadienes as Key-Intermediates in [2+2+2] Alkyne Cyclotrimerizations. Insight from Activation Strain Analyses. Chem. Phys. Chem. 2018, 19, 1766–1773. [Google Scholar] [CrossRef]
- Bonifaci, C.; Ceccon, A.; Gambaro, A.; Ganis, P.; Mantovani, L.; Santi, S.; Venzo, A. Heterobimetallic heptamethylindenyl complexes of Cr0 and RhI: Trans-[Cr(CO)3-indenyl-RhL2] (L2 = COD, L = CO). J. Organomet. Chem. 1994, 475, 267–276. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Rerek, M.E.; Basolo, F. Kinetics and mechanism of substitution reactions of eta 5-cyclopentadienyldicarbonylrhodium (I) derivatives. Rate enhancement of associative substitution in cyclopentadienylmetal compounds. J. Am. Chem. Soc. 1984, 106, 5908–5912. [Google Scholar] [CrossRef]
- Fernández, I.; Bickelhaupt, F.M.; Cossío, F.P. Type-I Dyotropic Reactions: Understanding Trends in Barriers. Chem. Eur. J. 2012, 18, 12395–12403. [Google Scholar] [CrossRef]
- Bonifaci, C.; Ceccon, A.; Santi, S.; Mealli, C.; Zoellner, R.W. Cofacial and antarafacial indenyl bimetallic isomers: A descriptive MO picture and implications for the indenyl effect on ligand substitution reactions. Inorganica Chim. Acta 1995, 240, 541–549. [Google Scholar] [CrossRef]
- Kozuch, S. Steady State Kinetics of Any Catalytic Network: Graph Theory, the Energy Span Model, the Analogy between Catalysis and Electrical Circuits, and the Meaning of “Mechanism”. ACS Catal. 2015, 5, 5242–5255. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Bashford, D.; Baseggio, O.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerritger, P.M.; et al. ADF2016 SCM. In Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; International series of monographs on chemistry; 1. issue; Oxford University Press: New York, NY, USA, 1994; ISBN 978-0-19-509276-9. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.G.; Gill, P.M.W.; Pople, J.A. The performance of a family of density functional methods. J. Chem. Phys. 1993, 98, 5612–5626. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; Volume 15, pp. 1–86. ISBN 978-0-470-12592-2. [Google Scholar]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef]
Scheme 1.
(a) Different coordination modes (hapticities) of a metal (M) to a Cp ring. (b) Labelling scheme used in the definition of Δ and LISP (Equations (1) and (2)). (c). Definition of θ angle for LISP calculation.
Scheme 1.
(a) Different coordination modes (hapticities) of a metal (M) to a Cp ring. (b) Labelling scheme used in the definition of Δ and LISP (Equations (1) and (2)). (c). Definition of θ angle for LISP calculation.

Scheme 2.
Anti-(a) and syn-(b) [Cr(CO)3IndRhL2].

Scheme 3.
Mechanism of acetylene [2+2+2] cycloaddition to benzene catalyzed by ZRh (Z = Cp, Ind and anti or syn-Ind) (Path I) (a) and by CO-ZRh fragments (Z = Cp, Ind and anti-Ind; L = CO) (Path II) (b).
Scheme 3.
Mechanism of acetylene [2+2+2] cycloaddition to benzene catalyzed by ZRh (Z = Cp, Ind and anti or syn-Ind) (Path I) (a) and by CO-ZRh fragments (Z = Cp, Ind and anti-Ind; L = CO) (Path II) (b).

Figure 1.
Optimized structures with selected interatomic distances (Å) and angles (deg) of the intermediates and transition states located on the PES of the anti-IndRh catalyzed acetylene [2+2+2] cycloaddition to benzene (Path I, Scheme 2a). Level of theory: ZORA-BLYP/TZ2P.
Figure 1.
Optimized structures with selected interatomic distances (Å) and angles (deg) of the intermediates and transition states located on the PES of the anti-IndRh catalyzed acetylene [2+2+2] cycloaddition to benzene (Path I, Scheme 2a). Level of theory: ZORA-BLYP/TZ2P.


Figure 2.
(a) Energy profiles of acetylene [2+2+2] cycloaddition to benzene catalyzed by CpRh (black), IndRh (red), and bimetallic anti-IndRh (blue) (Scheme 3a, Path I). (b) Profiles of the slippage parameters Δ (dashed line) and (solid line) for the acetylene [2+2+2] cycloaddition cycles catalyzed by CpRh (black), IndRh (red), and bimetallic anti-IndRh (blue) along Path I (Scheme 3a). Level of theory: ZORA-BLYP/TZ2P.
Figure 2.
(a) Energy profiles of acetylene [2+2+2] cycloaddition to benzene catalyzed by CpRh (black), IndRh (red), and bimetallic anti-IndRh (blue) (Scheme 3a, Path I). (b) Profiles of the slippage parameters Δ (dashed line) and (solid line) for the acetylene [2+2+2] cycloaddition cycles catalyzed by CpRh (black), IndRh (red), and bimetallic anti-IndRh (blue) along Path I (Scheme 3a). Level of theory: ZORA-BLYP/TZ2P.

Figure 3.
Kohn-Sham HOMOs of Cp-1, Ind-1, and anti-Ind-1; level of theory: ZORA-BLYP/TZ2P. The isodensity value is 0.03.
Figure 3.
Kohn-Sham HOMOs of Cp-1, Ind-1, and anti-Ind-1; level of theory: ZORA-BLYP/TZ2P. The isodensity value is 0.03.

Figure 4.
(a) Energy profile of acetylene [2+2+2] cycloaddition to benzene catalyzed by syn-IndRh (Scheme 3a, Path I). (b) Profiles of the slippage parameters (dashed line) and (solid line) along the acetylene [2+2+2] cycloaddition cycle catalyzed by syn-IndRh (Scheme 3a, Path I). Level of theory: ZORA-BLYP/TZ2P.
Figure 4.
(a) Energy profile of acetylene [2+2+2] cycloaddition to benzene catalyzed by syn-IndRh (Scheme 3a, Path I). (b) Profiles of the slippage parameters (dashed line) and (solid line) along the acetylene [2+2+2] cycloaddition cycle catalyzed by syn-IndRh (Scheme 3a, Path I). Level of theory: ZORA-BLYP/TZ2P.

Figure 5.
HOMO-3 of syn-Ind-1; the isodensity value is 0.03.

Figure 6.
(a) Energy profiles of acetylene [2+2+2] cycloaddition to benzene catalyzed by CpRh(CO) (black), IndRh(CO) (red), and anti-IndRh(CO) (blue) (Scheme 3b, Path II). (b) Profiles of the slippage parameters Δ (dashed line) and (solid line) along the acetylene [2+2+2] cycloaddition cycles catalyzed by CpRh(CO) (black), IndRh(CO) (red), and anti-IndRh(CO) (blue) (Scheme 3b, Path II). Level of theory: ZORA-BLYP/TZ2P.
Figure 6.
(a) Energy profiles of acetylene [2+2+2] cycloaddition to benzene catalyzed by CpRh(CO) (black), IndRh(CO) (red), and anti-IndRh(CO) (blue) (Scheme 3b, Path II). (b) Profiles of the slippage parameters Δ (dashed line) and (solid line) along the acetylene [2+2+2] cycloaddition cycles catalyzed by CpRh(CO) (black), IndRh(CO) (red), and anti-IndRh(CO) (blue) (Scheme 3b, Path II). Level of theory: ZORA-BLYP/TZ2P.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Activation strain analysis for the oxidative coupling Z-1 → Z-2 (Path I); all values are in kcal mol−1. The fragments are Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C4H4 moiety (the reference are two acetylene molecules).
Table 1.
Activation strain analysis for the oxidative coupling Z-1 → Z-2 (Path I); all values are in kcal mol−1. The fragments are Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C4H4 moiety (the reference are two acetylene molecules).
| 2(C2H2) | Cp’Rh | Total | |||
|---|---|---|---|---|---|
| Cp-1/Cp-TS (1,2) | 33.21 | 2.36 | 35.57 | −23.16 | 12.41 |
| Ind-1/Ind-TS (1,2) | 37.02 | 2.66 | 39.68 | −24.59 | 15.09 |
| anti-Ind-1/anti-Ind-TS (1,2) | 37.31 | 2.72 | 40.03 | −24.52 | 15.51 |
| syn-Ind-1/syn-Ind-TS (1,2) | 39.67 | 46.98 | 86.65 | −71.47 | 15.18 |
Table 2.
Activation strain analysis for Cp-4, Ind-4, and anti-Ind-4; all values are in kcal mol−1. The fragments are Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and C6H6 moiety (the reference is benzene).
Table 2.
Activation strain analysis for Cp-4, Ind-4, and anti-Ind-4; all values are in kcal mol−1. The fragments are Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and C6H6 moiety (the reference is benzene).
| C6H6 | Cp’Rh | Total | |||
|---|---|---|---|---|---|
| Cp-4 | 39.14 | 4.95 | 44.09 | −83.44 | −39.35 |
| Ind-4 | 1.86 | 1.45 | 3.31 | −30.44 | −27.13 |
| anti-Ind-4 | 1.64 | 1.26 | 2.90 | −31.60 | −28.70 |
Table 3.
Activation strain analysis (ASA) of all the intermediates and transition states of the anti-IndRh catalyzed cycle (Path I); all the values are in kcal mol−1. The fragments are Cr(CO)3 and IndRhXi.
Table 3.
Activation strain analysis (ASA) of all the intermediates and transition states of the anti-IndRh catalyzed cycle (Path I); all the values are in kcal mol−1. The fragments are Cr(CO)3 and IndRhXi.
| Cr(CO)3 | IndRhXi | Total | ||||||
|---|---|---|---|---|---|---|---|---|
| anti-Ind-1 | 1.23 | 2.29 | 3.52 | −76.47 | 129.17 | −101.20 | −48.50 | −44.98 |
| anti-Ind-TS(1,2) | 1.26 | 17.17 | 18.43 | −73.37 | 124.90 | −99.39 | −47.86 | −29.43 |
| anti-Ind-2 | 1.67 | 1.78 | 3.45 | −73.03 | 130.93 | −105.13 | −47.24 | −43.79 |
| anti-Ind-3 | 1.36 | 1.85 | 3.21 | −73.99 | 129.02 | −102.09 | −47.06 | −43.85 |
| anti-Ind-TS(3,4) | 1.28 | 7.77 | 9.05 | −73.38 | 125.83 | −100.03 | −47.58 | −38.53 |
| anti-Ind-4 | 1.23 | 1.17 | 2.40 | −80.45 | 128.03 | −101.74 | −54.16 | −51.76 |
| anti-Ind-5 | 1.25 | 1.65 | 2.90 | −73.39 | 123.39 | −97.75 | −47.75 | −44.85 |
Table 4.
Activation strain analysis (ASA) of all the intermediates and transition states along the syn-IndRh catalyzed cycle (Path I); all the values are in kcal mol−1. The fragments are Cr(CO)3 and IndRhXi.
Table 4.
Activation strain analysis (ASA) of all the intermediates and transition states along the syn-IndRh catalyzed cycle (Path I); all the values are in kcal mol−1. The fragments are Cr(CO)3 and IndRhXi.
| ΔE | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cr(CO)3 | IndRhXi | Total | ΔEoi | |||||
| syn-Ind-1 | 1.65 | 5.02 | 6.67 | −80.49 | 132.33 | −102.88 | −51.04 | −44.37 |
| syn-Ind-TS (1,2) | 1.62 | 20.29 | 21.91 | −79.52 | 130.84 | −102.42 | −51.10 | −29.19 |
| syn-Ind-2 | 2.25 | 4.31 | 6.56 | −117.69 | 195.36 | −133.21 | −55.54 | −48.98 |
| syn-Ind-TZ (2,3) | 2.57 | 7.56 | 10.13 | −117.3 | 188.69 | −129.83 | −58.45 | −48.32 |
| syn-Ind-3 | 2.52 | 11.78 | 14.30 | −87.65 | 141.15 | −109.59 | −56.09 | −41.79 |
| syn-Ind-TS (3,4) | 1.82 | 12.54 | 14.36 | −81.06 | 135.13 | −105.64 | −51.57 | −37.21 |
| syn-Ind-4 | 1.15 | 4.50 | 5.65 | −79.61 | 129.57 | −99.01 | −49.04 | −43.39 |
| syn-Ind-5 | 1.56 | 5.28 | 6.84 | −78.6 | 128.78 | −99.76 | −49.58 | −42.74 |
Table 5.
Activation strain analysis (ASA) of CO-Cp-2, CO-Ind-2, and CO-anti-Ind-2; all values are in kcal mol−1. The fragments are: CO-Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C4H4 moiety (the reference are two acetylene molecules).
Table 5.
Activation strain analysis (ASA) of CO-Cp-2, CO-Ind-2, and CO-anti-Ind-2; all values are in kcal mol−1. The fragments are: CO-Cp’Rh (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and the C4H4 moiety (the reference are two acetylene molecules).
| C4H4 | CO-Cp’RhLn | Total | |||
|---|---|---|---|---|---|
| CO-Cp-2 | 50.63 | 6.08 | 56.71 | −138.15 | −81.44 |
| CO-Ind-2 | 50.96 | 34.04 | 85.00 | −153.65 | −68.65 |
| CO-anti-Ind-2 | 50.89 | 36.06 | 86.95 | −149.23 | −62.28 |
Table 6.
Activation strain analysis (ASA) of the transition states CO-Cp-TS (2,3), CO-Ind-TS (2,3) and CO-anti-Ind-TS (2,3); all values are in kcal mol−1. The fragments are CO-Cp’RhLn (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and acetylene.
Table 6.
Activation strain analysis (ASA) of the transition states CO-Cp-TS (2,3), CO-Ind-TS (2,3) and CO-anti-Ind-TS (2,3); all values are in kcal mol−1. The fragments are CO-Cp’RhLn (Cp’ = Cp, Ind, and anti-[Cr(CO)3Ind]) and acetylene.
| C2H2 | CO-Cp’RhLn | Total | |||
|---|---|---|---|---|---|
| CO-Cp-TS (2,3) | 0.18 | 43.96 | 44.14 | −0.50 | 43.64 |
| CO-Ind-TS (2,3) | 0.06 | 28.83 | 28.89 | −0.34 | 28.55 |
| CO-anti-Ind-TS (2,3) | 0.12 | 14.27 | 14.39 | −4.58 | 9.81 |
Table 7.
Calculated (s−1) and slippage span (Å) for the catalytic Path I and II of the [2+2+2] cycloaddition of acetylene to benzene.
Table 7.
Calculated (s−1) and slippage span (Å) for the catalytic Path I and II of the [2+2+2] cycloaddition of acetylene to benzene.
| ΔLISP* | |||||
|---|---|---|---|---|---|
| Path I | |||||
| CpRh | 4.83 × 103 | 1.82 × 102 | 6.66 × 105 | 5.79 × 101 | 0.85 |
| IndRh | 5.23 × 10 | 1.97 | 2.00 × 104 | 1.74 | 1.75 |
| anti-IndRh | 2.66 × 10 | 1 | 1.15 × 104 | 1 | 1.81 |
| syn-IndRh | 4.57 × 10 | _ | 1.78 × 104 | _ | 3.42 |
| Path II | |||||
| CpRh(CO) | 6.10 × 10−20 | 1 | 1.07 × 10−12 | 1 | 15.59 |
| IndRh(CO) | 6.41 × 10−8 | 1.05 × 1012 | 2.35 × 10−3 | 2.20 × 109 | 14.11 |
| anti-IndRh(CO) | 3.97 × 102 | 6.51 × 1021 | 9.55 × 104 | 8.93 × 1016 | 10.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Masood Ahmad, S.; Dalla Tiezza, M.; Orian, L. In Silico Acetylene [2+2+2] Cycloadditions Catalyzed by Rh/Cr Indenyl Fragments. Catalysts 2019, 9, 679. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9080679
AMA Style
Masood Ahmad S, Dalla Tiezza M, Orian L. In Silico Acetylene [2+2+2] Cycloadditions Catalyzed by Rh/Cr Indenyl Fragments. Catalysts. 2019; 9(8):679. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9080679
Chicago/Turabian StyleMasood Ahmad, Shah, Marco Dalla Tiezza, and Laura Orian. 2019. "In Silico Acetylene [2+2+2] Cycloadditions Catalyzed by Rh/Cr Indenyl Fragments" Catalysts 9, no. 8: 679. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9080679
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.