Co-Crystal Structures of Furosemide:Urea and Carbamazepine:Indomethacin Determined from Powder X-Ray Diffraction Data

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
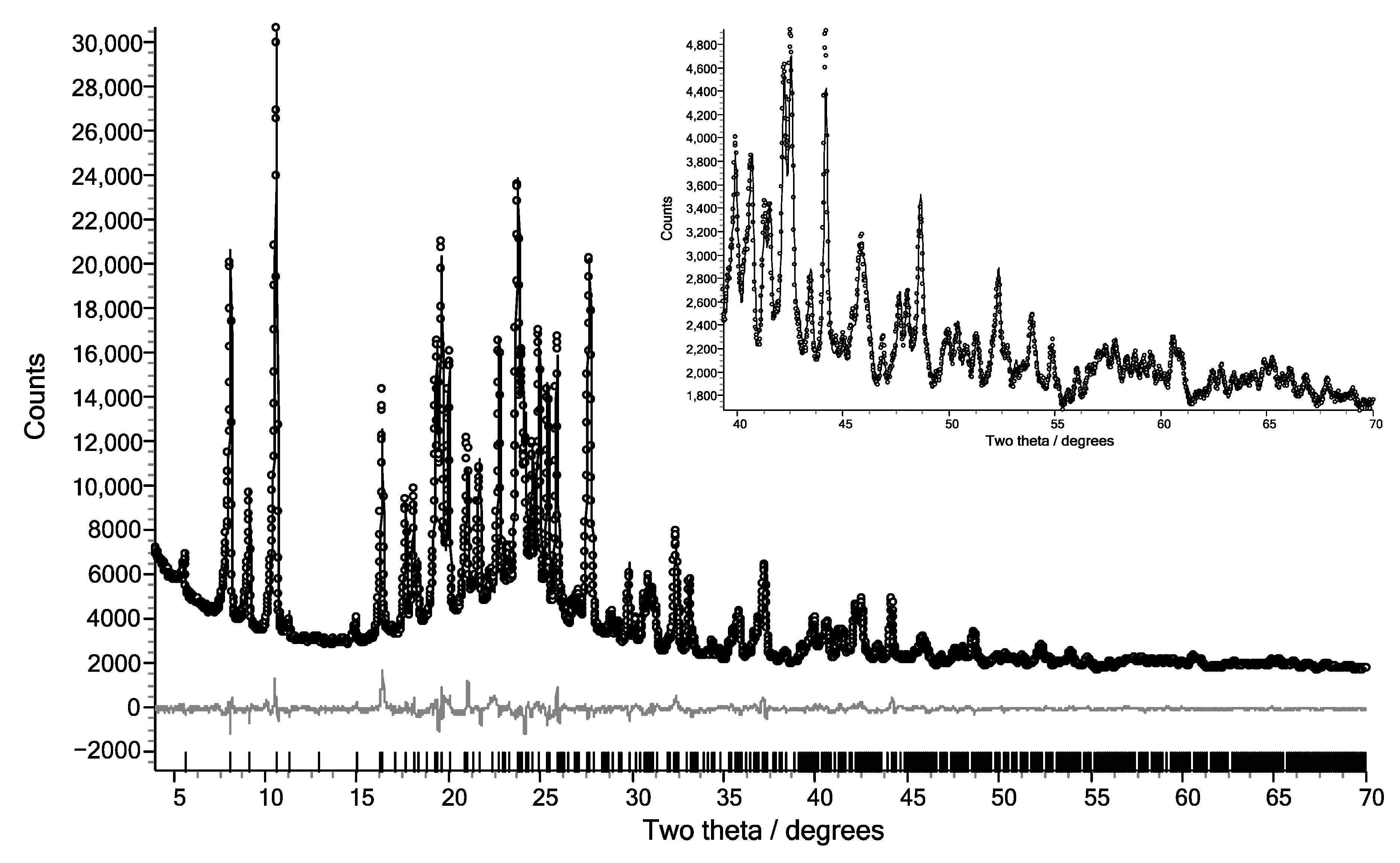
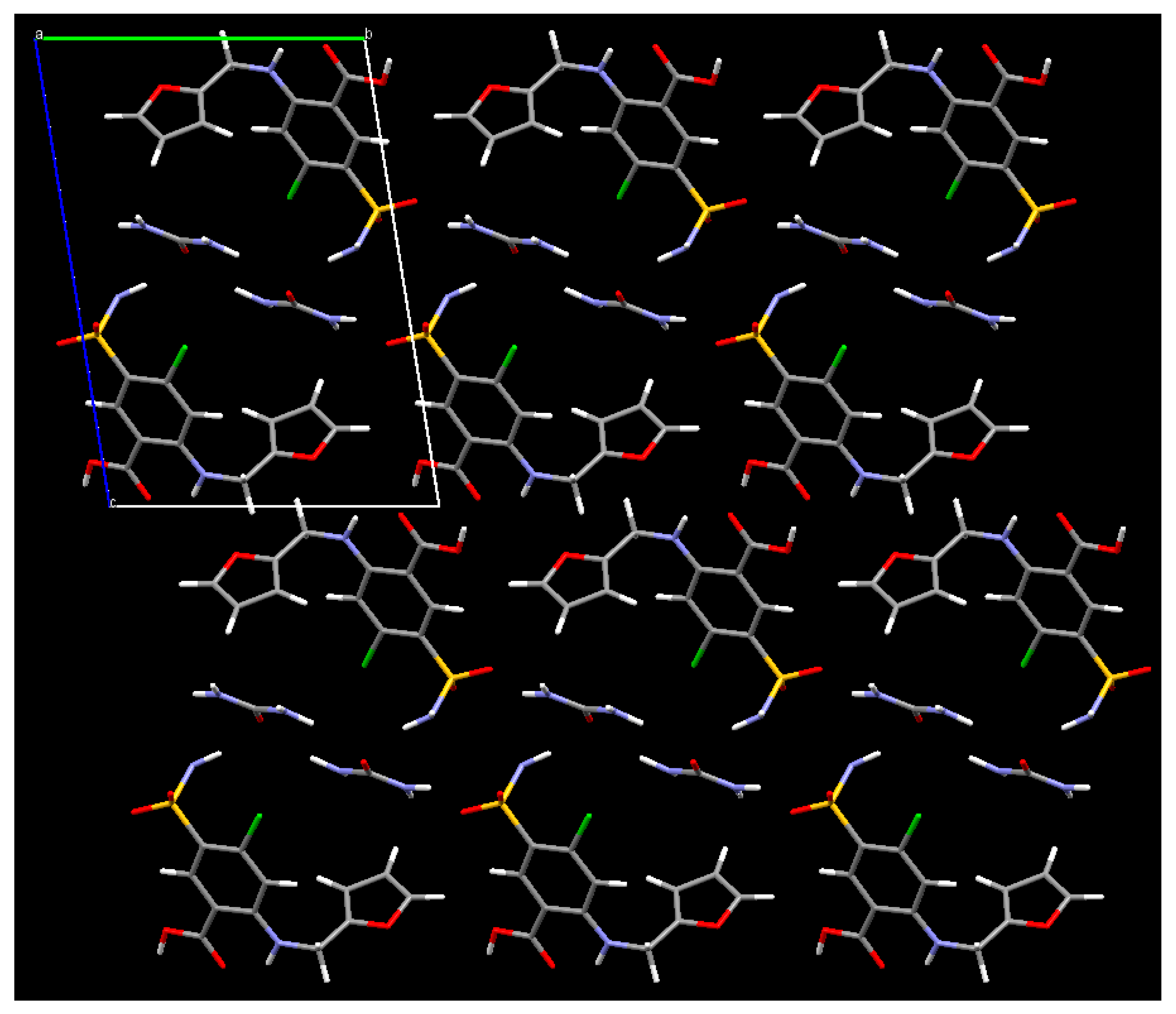
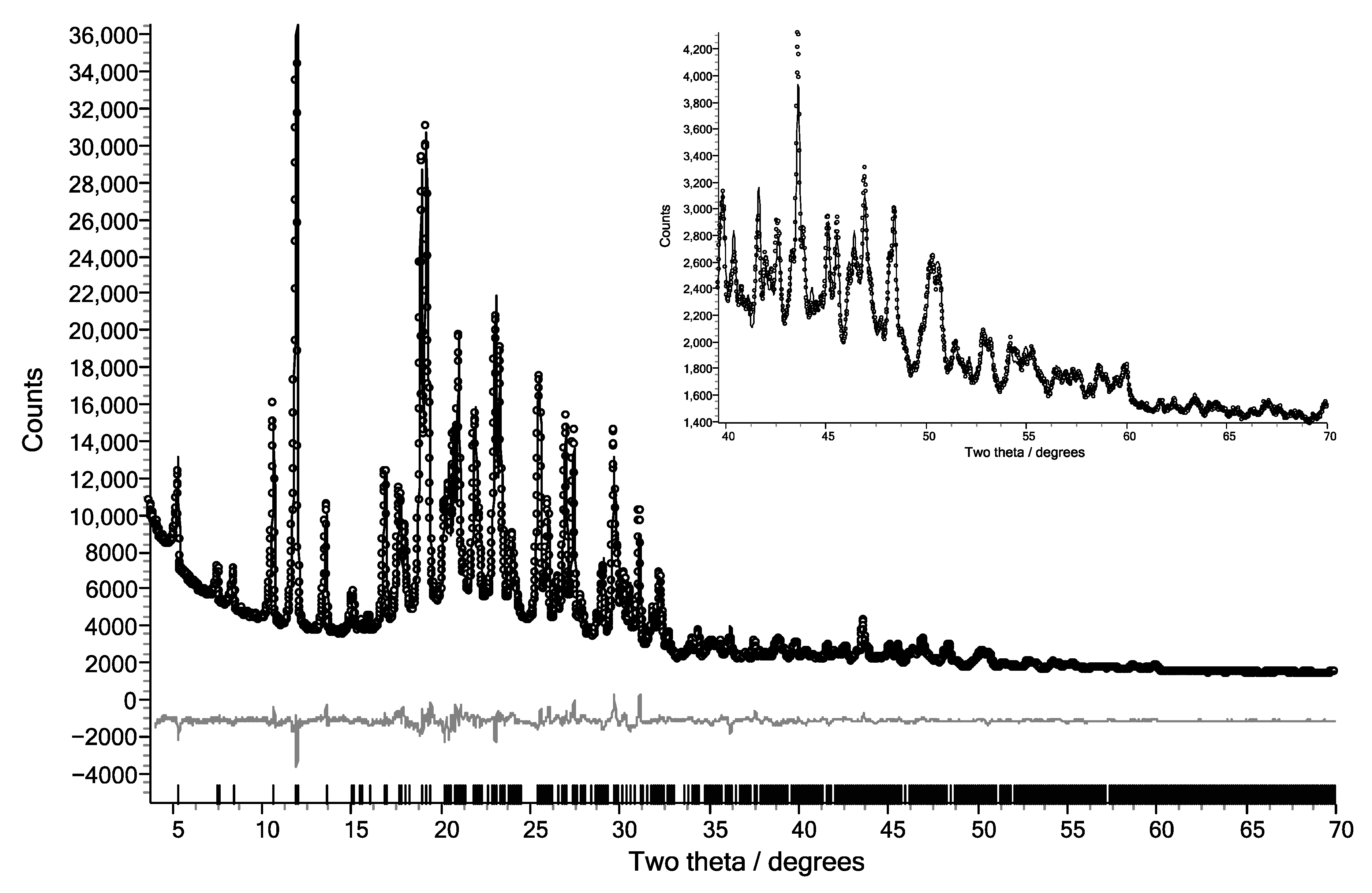
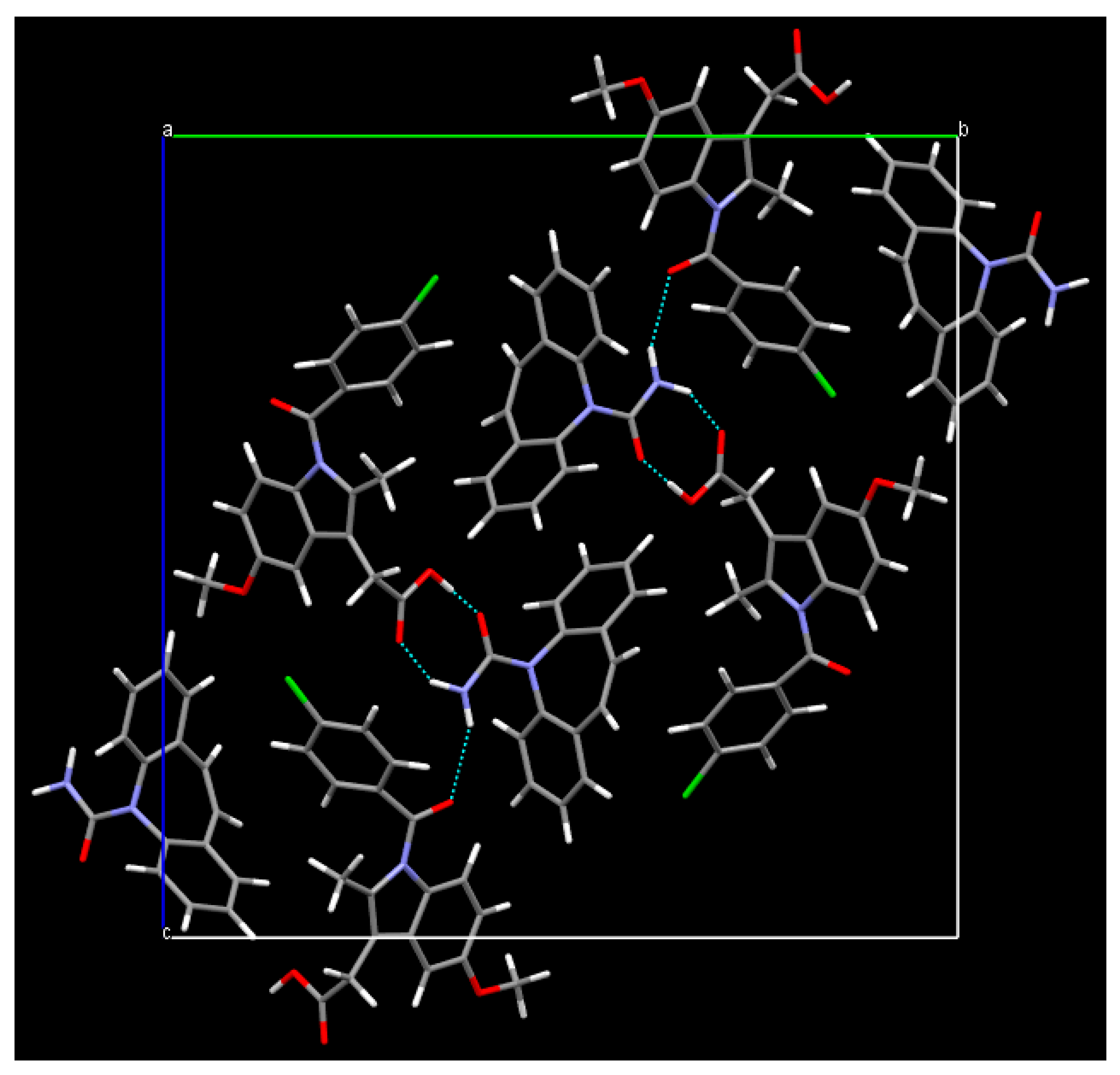
3. Results
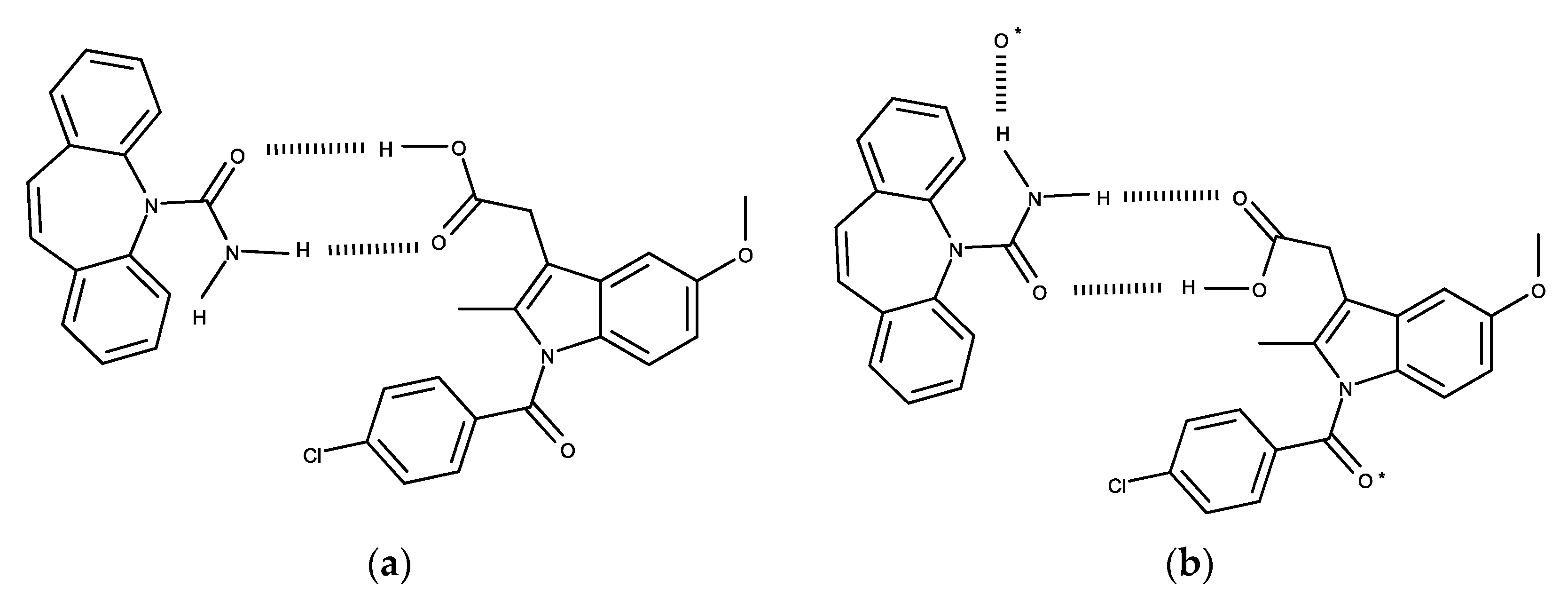
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R. Crystal engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- FDA. Regulatory Classification of Pharmaceutical Co-Crystals. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/regulatory-classification-pharmaceutical-co-crystals (accessed on 19 December 2019).
- Wood, P.A.; Feeder, N.; Furlow, M.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Knowledge-based approaches to co-crystal design. CrystEngComm 2014, 16, 5839–5848. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Braga, D.; Giaffreda, S.L.; Grepioni, F.; Pettersen, A.; Maini, L.; Curzi, M.; Polito, M. Mechanochemical preparation of molecular and supramolecular organometallic materials and coordination networks. Dalton Trans. 2006, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Greco, K.; Bogner, R. Solution-mediated phase transformation: Significance during dissolution and implications for bioavailability. J. Pharm. Sci. 2012, 101, 2996–3018. [Google Scholar] [CrossRef] [PubMed]
- Cerny, R. Crystal Structures from Powder Diffraction: Principles, Difficulties and Progress. Crystals 2017, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- David, W.I.F.; Shankland, K. Structure determination from powder diffraction data. Acta Crystallogr. Sect. A 2008, 64, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankland, K.; Spillman, M.J.; Kabova, E.A.; Edgeley, D.S.; Shankland, N. The principles underlying the use of powder diffraction data in solving pharmaceutical crystal structures. Acta Crystallogr. Sect. C Struct. Chem. 2013, 69, 1251–1259. [Google Scholar] [CrossRef]
- Shankland, K. An overview of currently used structure determination methods for powder diffraction data. In International Tables for Crystallography Volume H: Powder Diffraction; Gilmore, C.J., Kaduk, J.A., Schenk, H., Eds.; Wiley: Hoboken, NJ, USA, 2019; pp. 386–394. [Google Scholar]
- Goud, N.R.; Gangavaram, S.; Suresh, K.; Pal, S.; Manjunatha, S.G.; Nambiar, S.; Nangia, A. Novel furosemide cocrystals and selection of high solubility drug forms. J. Pharm. Sci. 2012, 101, 664–680. [Google Scholar] [CrossRef]
- Shankland, K.; David, W.I.F.; Sivia, D.S. Routine ab initio structure determination of chlorothiazide by X-ray powder diffraction using optimised data collection and analysis strategies. J. Mater. Chem. 1997, 7, 569–572. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Kabova, E.A.; Cole, J.C.; Korb, O.; Lopez-Ibanez, M.; Williams, A.C.; Shankland, K. Improved performance of crystal structure solution from powder diffraction data through parameter tuning of a simulated annealing algorithm. J. Appl. Crystallogr. 2017, 50, 1411–1420. [Google Scholar] [CrossRef]
- Kabova, E.A.; Cole, J.C.; Korb, O.; Williams, A.C.; Shankland, K. Improved crystal structure solution from powder diffraction data by the use of conformational information. J. Appl. Crystallogr. 2017, 50, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci.Cryst. Eng. Mat. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, H. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys. Condes. Matter 2017, 29, 30. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condes. Matter 2009, 21, 19. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- van de Streek, J.; Neumann, M.A. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mat. 2014, 70, 1020–1032. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Majumder, M.; Buckton, G.; Rawlinson-Malone, C.; Williams, A.C.; Spillman, M.J.; Shankland, N.; Shankland, K. A carbamazepine-indomethacin (1:1) cocrystal produced by milling. CrystEngComm 2011, 13, 6327–6328. [Google Scholar] [CrossRef] [Green Version]
- Majumder, M. Exploring the Role of the Amorphous State in Pharmaceutical Co-Crystal Production. Ph.D. Thesis, University of Reading, Reading, UK, 2013. [Google Scholar]
- Mnguni, M.J.; Michael, J.P.; Lemmerer, A. Binary polymorphic cocrystals: An update on the available literature in the Cambridge Structural Database, including a new polymorph of the pharmaceutical 1:1 cocrystal theophylline-3,4-dihydroxybenzoic acid. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Butler, Z.R.; Kaduk, J.A.; Gindhart, A.M.; Blanton, T.N. Crystal structure of prednicarbate, C27H36O8. Powder Diffr. 2019, 34, 368–373. [Google Scholar] [CrossRef]
- Kaduk, J.A.; Gindhart, A.M.; Blanton, T.N. Crystal structure of metolazone, C16H16ClN3O3S. Powder Diffr. 2019, 34, 361–367. [Google Scholar] [CrossRef]
- Kaduk, J.A.; Gindhart, A.M.; Blanton, T.N. Crystal structure of cloxacillin sodium monohydrate, C19H17ClN3O5SNa(H2O). Powder Diffr. 2019, 34, 374–378. [Google Scholar] [CrossRef]
- van de Streek, J. Structure of Pigment Yellow 181 dimethylsulfoxide N-methyl-2-pyrrolidone (1:1:1) solvate from XRPD plus DFT-D. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mat. 2015, 71, 89–94. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2θ Start/° | 2θ End/° | Time/s | Step Size/° |
|---|---|---|---|
| 4.0 | 20.5 | 5 | 0.017 |
| 20.5 | 37.0 | 10 | 0.017 |
| 37.0 | 53.5 | 16 | 0.017 |
| 53.5 | 70.0 | 25 | 0.017 |
| Compound | Structure | REFCODE | Torsional DoF 1 |
|---|---|---|---|
| Furosemide |  | FURSEM16 | 5 |
| Urea |  | UREAXX23 | 0 |
| Carbamazepine |  | CBMZPN01 | 1 |
| Indomethacin |  | INDMET03 | 5 |
| Crystallographic Data | FUR:URE | CBZ:IND Form II |
|---|---|---|
| Empirical formula | C12H11ClN2O5S⋅CH4N2O | C15H12N2O⋅C19H16ClNO4 |
| Temperature (K) | 298 | 298 |
| Sample formulation | Powder | Powder |
| Molar mass, g mol−1 | 390.80 | 594.06 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P21/n |
| a (Å) | 4.87321(17) | 5.1257(2) |
| b (Å) | 11.0689(3) | 23.3627(16) |
| c (Å) | 15.8709(4) | 23.5774(13) |
| α (°) | 80.4004(18) | 90 |
| β (°) | 84.737(2) | 91.212(5) |
| γ (°) | 83.213(3) | 90 |
| Volume (Å3) | 835.92(4) | 2822.8(3) |
| Zero point (°) | 0.0149(5) | 0.0045(8) |
| Z, Z’ | 2, 1 | 4, 1 |
| 2θ range (°) | 4 to 70 | 4 to 70 |
| No. of unique reflections | 744 | 1232 |
| No. of parameters | 62 | 43 |
| R factors | Rwp (Pawley) = 1.62% | Rwp (Pawley) = 1.29% |
| Rwp (Rietveld) = 2.84% | Rwp (Rietveld) = 2.92% | |
| CCDC Deposition Number * | 1970312 | 1970317 |
| Atom | CBZ:IND Form II | INDMET03 |
|---|---|---|
| Cl 1 | 0.118 Å | 0.058 Å |
| C 9 | 0.234 Å | 0.119 Å |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Rahal, O.; Majumder, M.; Spillman, M.J.; van de Streek, J.; Shankland, K. Co-Crystal Structures of Furosemide:Urea and Carbamazepine:Indomethacin Determined from Powder X-Ray Diffraction Data. Crystals 2020, 10, 42. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10010042
Al Rahal O, Majumder M, Spillman MJ, van de Streek J, Shankland K. Co-Crystal Structures of Furosemide:Urea and Carbamazepine:Indomethacin Determined from Powder X-Ray Diffraction Data. Crystals. 2020; 10(1):42. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10010042
Chicago/Turabian StyleAl Rahal, Okba, Mridul Majumder, Mark J. Spillman, Jacco van de Streek, and Kenneth Shankland. 2020. "Co-Crystal Structures of Furosemide:Urea and Carbamazepine:Indomethacin Determined from Powder X-Ray Diffraction Data" Crystals 10, no. 1: 42. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10010042