Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations





1
Unitat de Polimorfisme i Calorimetria, Centres Científics i Tecnològics, Universitat de Barcelona, Baldiri Reixac 10, 08028 Barcelona, Spain
2
Center for Intelligent Research in Crystal Engineering S.L., Parc Científic de Barcelona, Baldiri Reixac, 4-8, 08028 Barcelona, Spain
3
ALBA Synchrotron Light Source, 08220 Cerdanyola del Vallès, 08028 Barcelona, Spain
4
Departament de Química, Universitat de les Illes Balears, Crta. de Valldemossa, km 7.5, 07122 Palma (Baleares), Spain
*
Authors to whom correspondence should be addressed.
Crystals 2020, 10(12), 1126; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10121126
Submission received: 14 November 2020
/
Revised: 2 December 2020
/
Accepted: 7 December 2020
/
Published: 10 December 2020
(This article belongs to the Special Issue σ- and π-Hole Interactions (Volume II))
Abstract
:Herein, the X-ray powder diffraction (XRPD) crystal structure of a new Sildenafil cocrystal is reported, where resorcinol has been used as the coformer. The crystal structure has been solved by means of direct space methods used in combination with density functional theory (DFT) calculations. In the structure, the Sildenafil and resorcinol molecules form cooperative hydrogen bond (HB) and π-stacking interactions that have been analyzed using DFT calculations, the molecular electrostatic potential (MEP) surface, and noncovalent interaction plot (NCI plot). The formation of O–H⋯N H-bonds between resorcinol and Sildenafil increases the dipole moment and enhances the antiparallel π-stacking interaction.
1. Introduction
Sildenafil is a drug used in male patients with erectile dysfunction that acts by selectively inhibiting the cyclic guanosine monophosphate phosphodiesterase type 5 (Figure 1) [1,2]. The X-ray structures of Sildenafil and its citrate monohydrate salt have been previously reported [3,4]. Moreover, the solid-state structure of many salts and cocrystals is also available [5,6]. In addition, the cocrystallization of Sildenafil with acetylsalicylic acid [7] and also in the form of salicylate salt [8] has been used to mix a drug able to prevent strokes and myocardial infarctions with Sildenafil, which is not advised for patients with heart diseases. Moreover, we have previously discovered and analyzed, through a combined virtual/experimental cocrystal screen, new hybrid salt–cocrystal forms of Sildenafil [9].
Significant research has also been performed to synthesize and analyze a variety of Sildenafil solvates [5,10,11,12]. From a pharmaceutical point of view, these studies are very relevant because the presence of solvent molecules in the solid state may modify the solubility and the bioavailability of the drug [13,14,15,16,17]. Moreover, for drugs that are administered as solids like Sildenafil, the performance of the final product can change by the presence of solvates, which may have significant commercial interest in the pharmaceutical industry [15].
Herein, the X-ray powder diffraction (XRPD) crystal structure of a multicomponent solid form composed by Sildenafil free base and resorcinol in a (1:2) stoichiometry is reported. The cocrystal was previously discovered by some of us [9], although the crystal structure remained elusive until now. We focused this study on the analysis of the intermolecular H-bond between the resorcinol and Sildenafil molecules and its influence on the antiparallel stacking interaction between the pyrazolo[3,4-d]pyrimidine rings. For this purpose, this manuscript reports a complete theoretical study that consists of DFT calculations, molecular electrostatic potential maps, quantum theory of “atoms-in-molecules” (QTAIM) [18], and the noncovalent interaction plot (NCI plot) [19] analyses.
2. Materials, Experimental, and Theoretical Methods
2.1. Synthesis
Sildenafil (163 mg, 0.343 mmol, purchased from Polpharma, Warszawa, Poland) and resorcinol (76 mg, 0.690 mmol, purchased from Sigma-Aldrich, Darmstadt, Germany) were mixed and stirred in xylene (0.8 mL) during 4 days at 25 °C. The resulting suspension was filtered and dried under vacuum.
2.2. XRPD Analysis
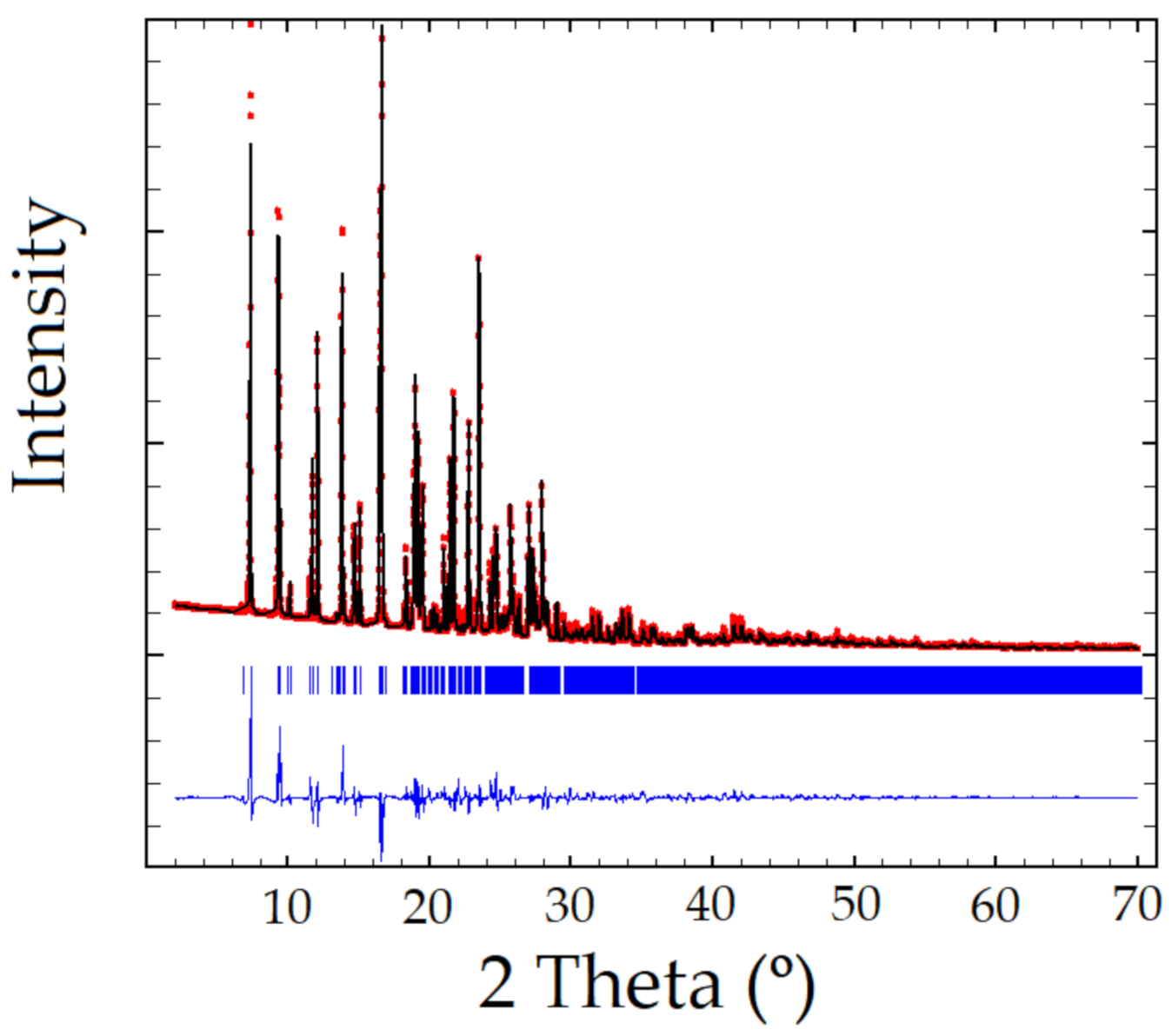
XRPD pattern of the Sildenafil-resorcinol cocrystal was obtained on a PANalytical X’Pert PRO MPD diffractometer (see Supplementary Materials for details). The XRPD was indexed to a monoclinic cell of approximately 3530 Å3 using Dicvol04, [20] (Figures of Merit: M = 72, F = 236), with the number of impurities equal to zero. The space group was assigned to P21/a by using the systematic absences. The crystal structure was solved by the direct space strategy implemented in TALP [21], showing in the asymmetric unit one independent molecule of Sildenafil and two independent molecules of resorcinol (Z = 4). Previously reported single-crystal structures of both Sildenafil and resorcinol molecules have been used to set the soft molecular restraints for their geometry. The Rietveld method was used for the final refinement of the crystal structure. The FullProf in combination with DFT calculations (see ESI for further details, Figures S1–S3) was used in order to improve through geometry optimization the planarity of the aromatic rings and to locate the hydrogen atomic coordinates (Figure 2 depicts the final Rietveld plot). Relevant crystal data of refinement parameters are given in Table 1.
2.3. Theoretical Methods
The Gaussian-16 program [22] was used for the calculations (PBE0 functional [23] and def2-TZVP basis set [24,25]). Moreover, the D3 correction [26] has been applied. The energies are BSSE-corrected [27]. The same level has been used for QTAIM and NCI plot analyses by means of the AIMAll package [28]. The MEP surface has been plotted using the 0.001 a.u. isosurface. For the calculations, we have used the experimental coordinates.
3. Results and Discussion
3.1. Description of the Cocrystal
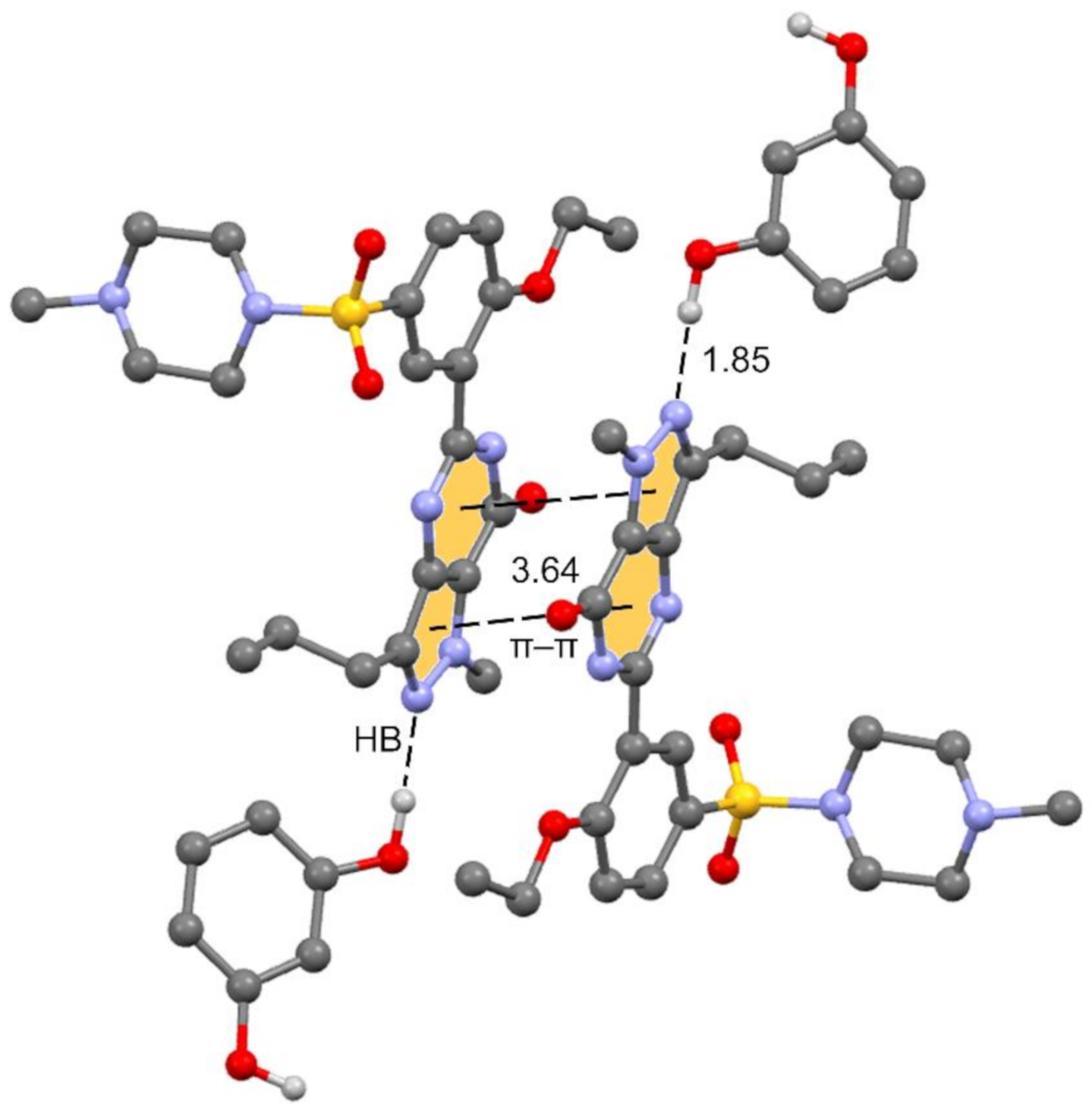
The unit cell contains 4 Sildenafil and 8 resorcinol molecules. In the crystal structure, Sildenafil forms an interesting hydrogen bond (HB)/π–π/HB assembly where the pyrazolo[3,4-d]pyrimidine rings are π-stacked in an antiparallel binding mode to maximize the dipole⋯dipole interaction. Simultaneously, an N-atom of the pyrazole ring interacts with one OH group of the resorcinol molecule. The assembly is represented in Figure 3, and in the following section, we further analyze the interplay between the H-bond and π-stacking interaction and how the H-bond polarizes the π-system, increasing the dipole moment and concomitantly reinforcing the π–π stacking.
3.2. Theoretical Study
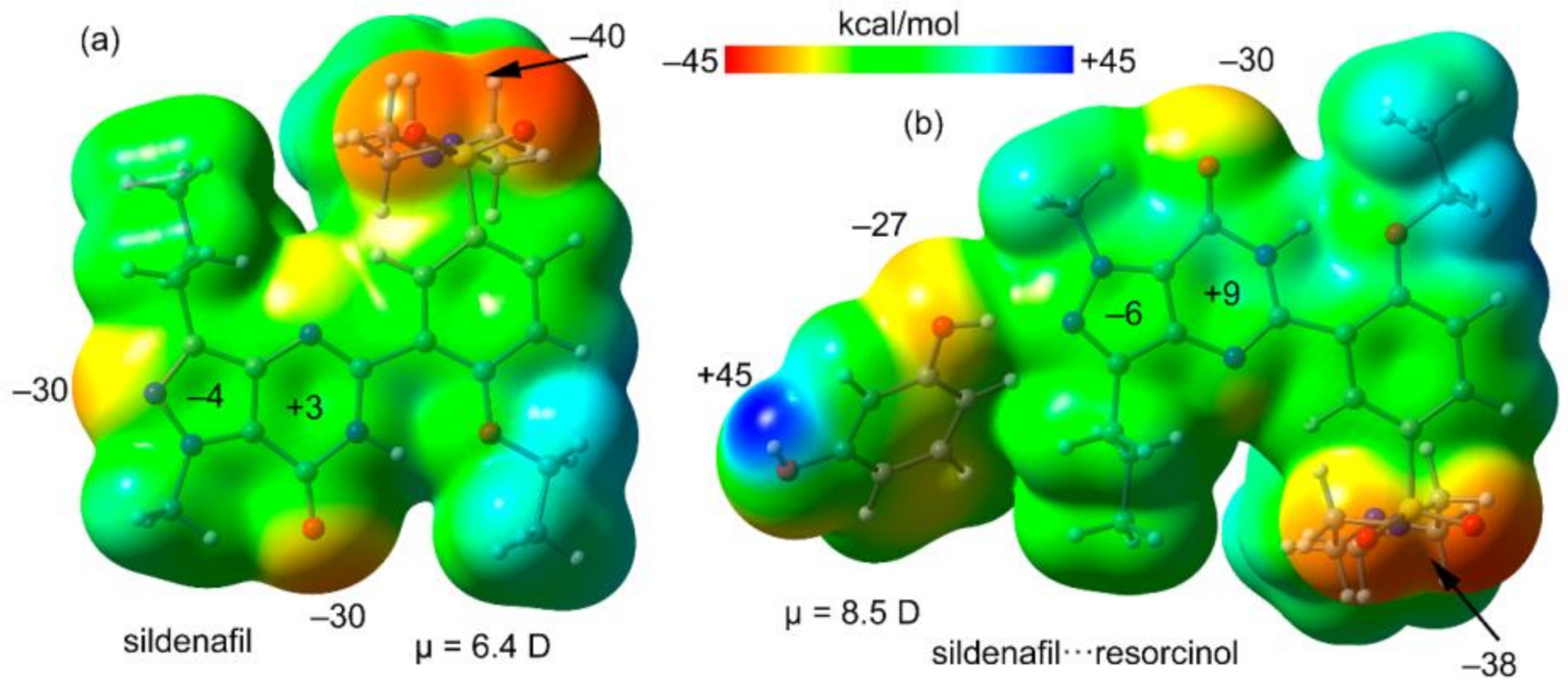
First, the MEP surfaces of Sildenafil and the H-bonded complex Sildenafil⋯resorcinol have been computed and compared. Both surfaces are given in Figure 4, showing that the minimum is located at the O-atom of the sulfonamide group. Both the N- and O-atoms of the pyrazolo[3,4-d]pyrimidine moiety also present large and negative MEP values (−30 kcal/mol). The values over the six- and five-membered rings of the pyrazolo[3,4-d]pyrimidine moiety are small and of opposite sign, thus favoring the antiparallel stacking. In Figure 4b, the MEP surface of the H-bonded complex is represented, showing that the π-system of the pyrazolo[3,4-d]pyrimidine moiety is more polarized than that of isolated Sildenafil. It also evidences that the maximum MEP value corresponds to the H-atom of the phenol group (+45 kcal/mol). The dipole moments are also indicated in Figure 4, confirming that the dipole moment of the H-bonded dimer (8.5 D) is significantly larger than that of Sildenafil (6.4 D), thus anticipating a larger ability to form dipole⋯dipole interaction in the dimer.
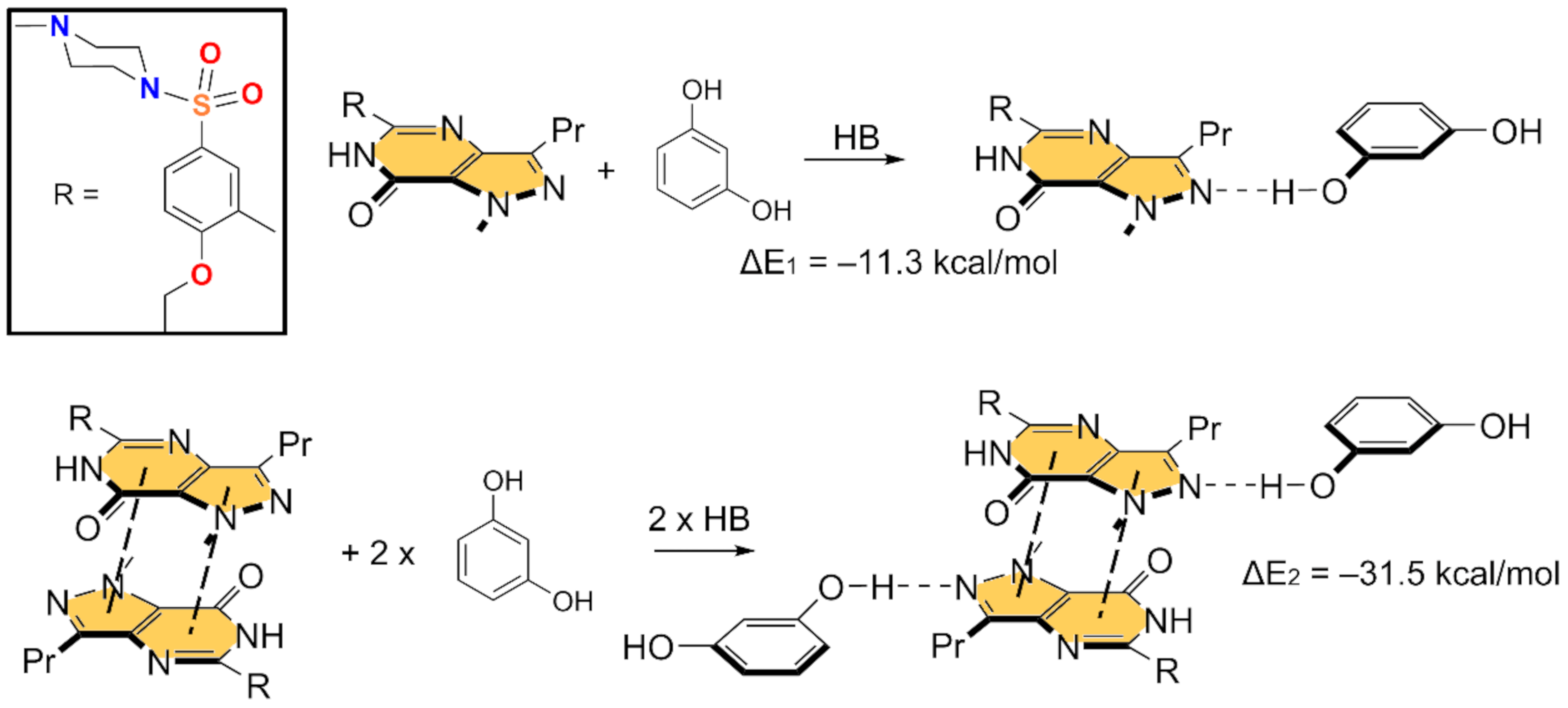
In Figure 5 and Figure 6, we show the different reactions used to evaluate the mutual influence of H-bonds and π-stacking interactions that are likely important for the formation of the HB/π–π/HB assembly shown in Figure 3. In Figure 5 (top), the interaction energy of the H-bonded dimer is shown, evidencing a very strong interaction (ΔE1 = −11.3 kcal/mol). If the π–π stacked dimer is used as a whole entity and starting point for the computation of the formation of the tetrameric assembly (Figure 5, bottom), the resulting formation energy (ΔE2 = −31.5 kcal/mol) is much greater than twice the H-bonding energy of the dimer (ΔE1), thus suggesting that the π stacking interaction reinforces the H-bond. Figure 6 (top) shows the formation energy of the π-stacked dimer, which is ΔE3 = −16.7 kcal/mol. In the case the H-bonded dimer is used to generate the tetrameric assembly (Figure 6, bottom), the formation energy is ΔE4 = −25.7 kcal/mol, thus suggesting that the H-bond also reinforces the π-stacking. This large reinforcement is attributed to the stronger dipole⋯dipole interaction, and also additional interactions that are established between the resorcinol and the Sildenafil upon complexation, as further discussed below.
We have used the QTAIM and NCI plot analyses to further characterize the H-bonded and π-stacking dimers observed in the structure and to rationalize the large dimerization energies. The results are shown in Figure 7. For the H-bonded dimer, the O–H⋯N H-bond is characterized by a bond critical point (CP) and bond path connecting the H and O-atoms. The NCI plot shows an intense blue isosurface, which indicates a very strong interaction, in agreement with the strong dimerization energy. Moreover, the QTAIM/NCI plot analysis also evidences the existence of an additional C–H⋯N H-bond involving the C–H bond adjacent to the interacting OH. The color of the NCI plot surface that characterizes this C–H⋯N bond is green, thus revealing a much weaker interaction (see Figure 7a). We have evaluated the individual contribution of each H-bond in this dimer by using the potential energy density at the bond CP (Vr). The energy of the H-bond can be calculated by using the equation proposed by Espinosa et al. (E = ½Vr) [29], and recently used by us to analyze similar interactions [30,31,32,33,34,35]. By using this formula, the contribution of the O–H⋯O bond is −9.32 kcal/mol (Vr = −0.0297 a.u.) and that of the C–H⋯N bond is −1.31 kcal/mol (Vr = −0.0042 a.u.). The sum of both contributions (−10.63 kcal/mol) is very similar to the dimerization energy, (−11.3 kcal/mol), thus giving reliability to the H-bond energy predictor.
Figure 7b shows the combined QTAIM/NCI plot analysis of the π-stacked dimer. The dimerization energy is very large due to the antiparallel orientation of the dipole and also the existence of secondary interactions, as revealed by the QTAIM distribution of CPs. The π–π stacking interaction is characterized by four CPs and bond paths interconnecting atoms of the pyrazolo[3,4-d]pyrimidine moieties. The QTAIM analysis reveals the existence of two symmetrically equivalent C–H⋯π interactions involving the methyl groups attached to the pyrazole rings and the phenyl rings. Moreover, two equivalent C–H⋯O interactions are also present involving one H-atom of the propyl group and the exocyclic O-atom of the pyrimidine ring.
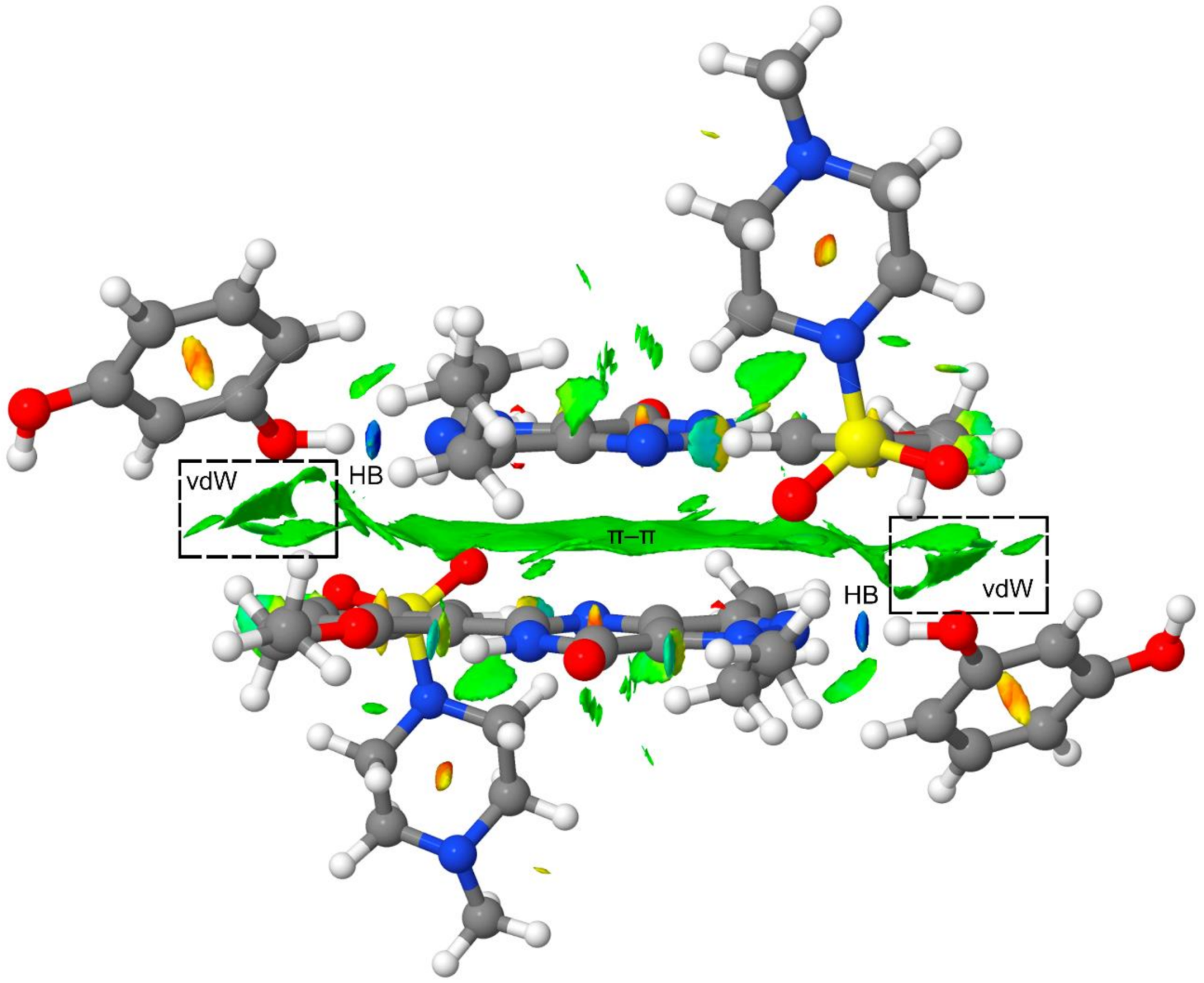
Finally, to further analyze the secondary interactions in the tetrameric assembly, we have represented the NCI plot in Figure 8. It can be observed that the π–π interaction is characterized by an extended green isosurface and the H-bonds by small and intense blue isosurfaces. The NCI plot also shows several green isosurfaces that are located between the resorcinol ring and the Sildenafil molecule, thus evidencing the existence of additional van der Waals contacts apart from the H-bonds and π-stacking interaction upon the formation of the tetramer (see dashed rectangles in Figure 8). These interactions also contribute to the enhancement of the π-stacking as a consequence of the H-bonds, and vice versa. Therefore, the cooperativity effects evidenced by the reactions gathered in Figure 5 and Figure 6 are also influenced by the long-range interactions between the resorcinol and the Sildenafil molecular fragments not directly H-bonded.
4. Concluding Remarks
We have determined the crystal structure of a Sildenafil-resorcinol cocrystal from X-ray powder diffraction data by means of direct space methods and analyzed its structural features through DFT calculations. The formation of strong H-bonds and π-stacking interactions has been studied and interpreted in terms of cooperativity effects between both interactions to generate the HB/π-π/HB assembly that is based on the enhanced dipole⋯dipole interaction. The energetic features of the assembly have been studied using DFT calculations and characterized by the QTAIM and NCI plot computational tools, showing that the π–π stacking interaction is reinforced by the H-bonding and vice versa.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4352/10/12/1126/s1, Figure S1: prf plot for the crystal structure refinement of Sildenafil–resorcinol cocrystal determined by TALP. The plot shows the experimental powder XRD profile (red marks), the calculated powder XRD profile (black solid line), and the difference profile (blue, lower line). Tick marks indicate peak positions, Figure S2: Comparative plot of the deformed resorcinol molecule: (a) After TALP crystal structure determination; (b) after DFT calculations, Figure S3: (a) Overlapping of Sildenafil-resorcinol cocrystal structure initially determined by TALP (blue) and final structure refined by the Rietveld method (red). The computed root-mean-square distance (RMSD) is 0.0003 Å. (b) Detail of the overlapped resorcinol molecules.
Author Contributions
Conceptualization, A.F. and R.P.; methodology, R.B., R.P. and A.F.; software, R.P., A.F. and O.V.; validation, R.P., R.B. and V.K.; formal analysis, R.B. and O.V.; investigation, A.F., R.P. and R.B.; resources, R.P. and A.F.; data curation, R.B.; writing—original draft preparation, A.F. and R.P.; writing—review and editing, A.F. and R.P.; supervision, A.F. and R.P.; project administration, A.F. and R.P.; funding acquisition, A.F. and R.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICIU/AEI from Spain, CTQ2017-85821-R, FEDER funds.
Acknowledgments
We thank the “centre de tecnologies de la informació” (CTI) at the University of the Balearic Islands for computational facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boolell, M.; Allen, M.J.; Ballard, S.A.; Gepi-Attee, S.; Muirhead, G.J.; Naylor, A.M.; Osterloh, I.O.; Gingell, C. Sildenafil: An orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [Google Scholar]
- Ballard, S.A.; Gingell, C.J.; Tang, K.; Turner, L.A.; Price, M.E.; Naylor, A.M. Effects of Sildenafil on the relaxation of human corpus cavernosum tissue in vitro and on the activities of cyclic nucleotide phosphodiesterase isozymes. J. Urol. 1998, 159, 2164–2171. [Google Scholar] [CrossRef]
- Yathirajan, H.S.; Nagaraj, B.; Nagaraja, P.; Bolte, M. Sildenafil citrate monohydrate. Acta Crystallogr. Sect. E: Struct. Rep. Online 2005, 61, 489–491. [Google Scholar] [CrossRef]
- Stepanovs, D.; Mishnev, A. Molecular and crystal structure of Sildenafil base. Z. Naturforsch. B: J. Chem. Sci. 2012, 67, 491–494. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Desiraju, G.R. Solvates of Sildenafil Saccharinate. A New Host Material. Cryst. Growth Des. 2006, 6, 1468–1478. [Google Scholar] [CrossRef]
- Sanphui, P.; Tothadi, S.; Ganguly, S.; Desiraju, G.R. Salt and Cocrystals of Sildenafil with Dicarboxylic Acids: Solubility and Pharmacokinetic Advantage of the Glutarate Salt. Mol. Pharm. 2013, 10, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Žegarac, M.; Lekšic, E.; Šket, P.; Plavec, J.; Bogdanovic, M.D.; Bucar, D.-K.; Dumic, M.; Meštrovic, E.A. Sildenafil cocrystal based on acetylsalicylic acid exhibits an enhanced intrinsic dissolution rate. CrystEngComm 2014, 16, 32–35. [Google Scholar] [CrossRef]
- Stepanovs, D.; Jureb, M.; Mishnev, A. Preparation and crystal structure of Sildenafil salicylate. Mendeleev Commun. 2015, 25, 49–50. [Google Scholar] [CrossRef]
- Barbas, R.; Font-Bardia, M.; Paradkar, A.; Hunter, C.A.; Prohens, R. Combined Virtual/Experimental Multicomponent Solid Forms Screening of Sildenafil: New Salts, Cocrystals, and Hybrid Salt−Cocrystals. Cryst. Growth Des. 2018, 18, 7618–7627. [Google Scholar] [CrossRef]
- Barbas, R.; Font-Bardia, M.; Prohens, R. Polymorphism of Sildenafil: A New Metastable Desolvate. Cryst. Growth Des. 2018, 18, 3740–3746. [Google Scholar] [CrossRef]
- Barbas, R.; Prohens, R.; Font-Bardía, M.; Bauzá, A.; Frontera, A. Hydrogen bonding versus π-interactions: Their key competition in Sildenafil solvates. CrystEngComm 2018, 20, 4526–4530. [Google Scholar] [CrossRef]
- Wang, C.; Perumalla, S.R.; Sun, C.C. Anion Exchange Reaction for Preparing Acesulfame Solid Forms. Cryst. Growth Des. 2018, 18, 4215–4219. [Google Scholar] [CrossRef]
- Byrn, S.R.; Zografi, G.; Chen, X. Solid-State Properties of Pharmaceutical Materials, Chapter 3: Solvates and Hydrates; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Rodríguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodríguez-Hornedo, N. General principles of pharmaceutical solid polymorphism: A supramolecular perspective. Adv. Drug Delivery Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Griesser, U.J. Polymorphism in the Pharmaceutical Industry; Hilfiker, R., von Raumer, M., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2006. [Google Scholar]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs; SSCI Inc.: West Lafayette, IN, USA, 1999. [Google Scholar]
- Morissette, S.L.; Almarsson, O.; Peterson, M.L.; Remenar, J.F.; Read, M.J.; Lemmo, A.V.; Ellis, S.; Cima, M.J.; Gardner, C.R. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Delivery Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Boultif, A.; Louër, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- Vallcorba, O.; Rius, J.; Frontera, C.; Miravitlles, C. TALP: A multisolution direct-space strategy for solving molecular crystals from powder diffraction data based on restrained least squares. J. Appl. Crystallogr. 2012, 45, 1270–1277. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. J. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Todd, A.; Keith, T.K. AIMAll; Version 13.05.06; Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Efimenko, Z.M.; Eliseeva, A.A.; Ivanov, D.M.; Galmes, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Bifurcated μ2-I⋯(N,O) Halogen Bonding: The Case of (Nitrosoguanidinate)NiII Cocrystals with Iodine(I)-Based σ-Hole Donors. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Ivanov, D.M.; Sadykov, E.K.; Bokach, N.A.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Semicoordination Bond Breaking and Halogen Bond Making Change the Supramolecular Architecture of Metal-Containing Aggregates. Cryst. Growth Des. 2020, 20, 6956–6965. [Google Scholar] [CrossRef]
- Soldatova, N.S.; Postnikov, P.S.; Suslonov, V.V.; Kissler, T.Y.; Ivanov, D.M.; Yusubov, M.S.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Diaryliodonium as a double σ-hole donor: The dichotomy of thiocyanate halogen bonding provides divergent solid state arylation by diaryliodonium cations. Org. Chem. Front. 2020, 7, 2230–2242. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl)I⋯dz2 [PtII] Halogen Bonds. Chem. Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Eliseeva, A.A.; Baykov, S.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. One-Pot Route to X-perfluoroarenes (X = Br, I) Based on FeIII-Assisted C–F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding. Cryst. Growth Des. 2020, 20, 5908–5921. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole⋯dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar] [CrossRef]
Figure 1.
Structure of Sildenafil.

Figure 2.
Rietveld plot of the Sildenafil–resorcinol cocrystal, Rwp = 8.61%, Chi2 = 107. Experimental powder XRD profile in red, the calculated one in black, and the difference in blue. Peak positions (in blue) are indicated using tick marks.
Figure 2.
Rietveld plot of the Sildenafil–resorcinol cocrystal, Rwp = 8.61%, Chi2 = 107. Experimental powder XRD profile in red, the calculated one in black, and the difference in blue. Peak positions (in blue) are indicated using tick marks.

Figure 3.
Partial view of the cocrystal structure showing the antiparallel π stacking and the Sildenafil⋯resorcinol H-bonds.
Figure 3.
Partial view of the cocrystal structure showing the antiparallel π stacking and the Sildenafil⋯resorcinol H-bonds.

Figure 4.
Molecular electrostatic potential (MEP) surface of Sildenafil (a) and its H-bonded complex with resorcinol (b) at the PBE0/def2-TZVP level of theory. Isosurface 0.001 a.u. Energies at selected points in kcal/mol.
Figure 4.
Molecular electrostatic potential (MEP) surface of Sildenafil (a) and its H-bonded complex with resorcinol (b) at the PBE0/def2-TZVP level of theory. Isosurface 0.001 a.u. Energies at selected points in kcal/mol.

Figure 5.
Reactions employed to measure the H-bonds in the dimer (top) and tetramer (bottom).

Figure 6.
Reactions employed to measure the π-stacking in the dimer (top) and tetramer (bottom).

Figure 7.
Combined QTAIM (bond, ring, and cage critical points (CPs) represented as red, yellow, and blue spheres, respectively) and noncovalent interaction (NCI) plot analysis (−0.04 < sign(λ2) ρ < 0.05) of the H-bonded dimer (a) and π-stacking dimer (b). NCI plot isosurface = 0.5 a.u.
Figure 7.
Combined QTAIM (bond, ring, and cage critical points (CPs) represented as red, yellow, and blue spheres, respectively) and noncovalent interaction (NCI) plot analysis (−0.04 < sign(λ2) ρ < 0.05) of the H-bonded dimer (a) and π-stacking dimer (b). NCI plot isosurface = 0.5 a.u.

Figure 8.
NCI plot of the tetrameric assembly at the PBE0-D3/def2-TZVP level of theory. Range: −0.04 < sign(λ2)ρ < 0.05 and RDG isosurface = 0.5 a.u.
Figure 8.
NCI plot of the tetrameric assembly at the PBE0-D3/def2-TZVP level of theory. Range: −0.04 < sign(λ2)ρ < 0.05 and RDG isosurface = 0.5 a.u.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal data and structure refinement parameters for Sildenafil resorcinol cocrystal.
| Crystal Structure Data | |
|---|---|
| Empirical formula | C34H42N6O8S |
| Formula Weight | 694.80 |
| Temperature (K) | 298(2) |
| Wavelength (Å) | 1.5406 |
| Crystal system | Monoclinic |
| space group | P 21/a |
| a, b, c (Å) | 14.27040(15) 26.0868(3) 9.99412(12) |
| α, β, γ (°) | 90 108.3991(7) 90 |
| Volume (Å3) | 3530.31(7) |
| Z, Density (calc.) (mg/m3) | 4, 1.307 |
| θ range for data collection (°) | 2.0 to 70 step 0.013 (2θ) |
| Refinement method | Rietveld |
| Data/restraints/parameters | 3477/52/77 |
| Final R indices [I > 2σ(I)] | Rwp = 8.61 Chi2 = 107 (Rexp = 0.834) |
| CCDC | 2044269 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barbas, R.; Kumar, V.; Vallcorba, O.; Prohens, R.; Frontera, A. Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations. Crystals 2020, 10, 1126. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10121126
AMA Style
Barbas R, Kumar V, Vallcorba O, Prohens R, Frontera A. Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations. Crystals. 2020; 10(12):1126. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10121126
Chicago/Turabian StyleBarbas, Rafael, Vineet Kumar, Oriol Vallcorba, Rafel Prohens, and Antonio Frontera. 2020. "Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations" Crystals 10, no. 12: 1126. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10121126
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.