
LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target

1
Hunan Key Laboratory of Oral Health Research & Hunan 3D Printing Engineering Research Center of Oral Care & Hunan Clinical Research Center of Oral Major Diseases and Oral Health & Academician Workstation for Oral-Maxillofacial and Regenerative Medicine & Xiangya School of Stomatology, Central South University, Changsha 410008, China
2
Xiangya School of Pharmaceutical Sciences, Central South University, Changsha 410013, China
*
Author to whom correspondence should be addressed.
Crystals 2021, 11(3), 249; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030249
Submission received: 14 February 2021
/
Revised: 24 February 2021
/
Accepted: 25 February 2021
/
Published: 28 February 2021
(This article belongs to the Special Issue Crystalline Micro- and Nano-Materials for Medical and Other Biochemical Applications)
Abstract
:Due to the lack of an effective method for the treatment of fibrosis, there are numerous patients suffering from the effects of fibrosis. Severe fibrosis can cause dysfunction of relevant organs characterized by excessive deposition of extracellular matrix components. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a long non-coding RNA that is widely expressed and highly conserved in human tissues. It can regulate gene expression at various molecular levels, involved in the fibrosis of the liver, heart, lung, and kidney. In this review, we first described the pathogenesis by which MALAT1 promotes fibrosis. Furthermore, we summarized current studies of MALAT1 in the fibrosis of various organs. Hope this review will contribute to a better understanding of the molecular mechanism of fibrosis and the potential of MALAT1 as a novel therapeutic target for fibrosis.
1. Introduction
Fibrosis is a complex biological process characterized by abnormal inflammatory damage and excessive accumulation of the extracellular matrix (ECM) [1], which may involve a variety of tissues and organs, such as the heart, liver, kidney and lung [2,3,4,5]. Chronic stimulation resulting from persistent infections, hormonal changes, and long-term mechanochemical stimuli, as well as acute stimulation caused by sudden changes in hemodynamics can lead to fibrosis [6,7]. Although connective tissue deposition is a physiological phenomenon [8], excessive or progressive fibrosis leads to the destruction of tissue structure, resulting in organ dysfunction and even organ failure [9,10,11,12]. Therefore, fibrosis is considered a major reason for the function deterioration of various human organ functions [13,14,15]. At present, the number of people affected by fibrosis-related diseases has reached nearly 1/4 of the total global population [16], resulting in a heavy burden and tremendous challenge for the global medical system.
Many studies have focused on the pathogenesis of fibrosis to identify methods that can be used to slow down, stop, or even reverse fibrotic progress. The occurrence of fibrosis has been found to be related to the renin-angiotensin-aldosterone system, inflammation, oxidative stress, transduction of the transforming growth factor-β1 (TGF-β1)/SMAD signal, transduction of the Wnt/β-catenin signal, and lipid metabolism [17,18,19,20]. At present, there are very few anti-fibrosis drugs on the market, and their efficacy is not ideal. In addition, most treatment methods are still in the preclinical evaluation stage, and systemic inhibition of fibrosis-related signals will inevitably produce serious adverse reactions. Therefore, in the treatment of fibrosis, it is particularly important to find therapeutic targets with high efficiency, low toxicity and side effects.
LncRNAs are a class of non-coding RNAs with a length of more than 200 nt, which are involved in a variety of disease processes. The role of lncRNAs in regulating fibrosis has received wide attention [21,22,23]. Especially in recent years, the discovery of lncRNAs in body fluids, such as serum and urine, has indicated that circulating lncRNAs can be used as minimally invasive or even non-invasive disease markers [24]. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), an important lncRNA, was first discovered in non-small cell lung cancer in 2003 [25]. MALAT1, which is found on human chromosome 11q13.1, is widely expressed and highly conserved in normal tissues. It is abnormally expressed in lung cancer, esophageal cancer, gastric cancer and other malignant human tumors, and it regulates cell proliferation, invasion and metastasis through specific mechanisms in different tumors [26,27], indicating the potentially important biological function of MALAT1. An increasing number of studies have shown that MALAT1 plays an important role in the fibrosis of a variety of organs and tissues. Therefore, studies on the mechanism by which MALAT1 is involved in fibrosis will not only help us to further understand the pathogenesis of fibrosis but will also provide new ideas and methods for identifying the diagnostic and therapeutic targets of fibrosis.
2. Association between MALAT1 and the Pathogenesis of Fibrosis
Fibrosis, a pathological process driven by multiple factors, involves processes such as fibroblast proliferation and activation, ECM accumulation, epithelial cell to fibroblast transformation, inflammation, multiple cytokines and signaling cascades [28]. As a functional molecule, MALAT1 is also involved in epithelial-mesenchymal transformation, as well as inflammation regulation and metabolic regulation of various physiological and pathological processes [29,30]. The potential connection between MALAT1 and the pathogenesis of fibrosis is shown in Figure 1.
2.1. MALAT1 Can Stimulate Fibroblast Proliferation and Affect ECM Balance
MALAT1 plays an essential role in G1/S and mitotic processes by regulating the expression of cell cycle transcription factors and/or pre-gene processing, thereby promoting the proliferation of fibroblasts [31]. On one hand, MALAT1 can promote the secretion and synthesis of a variety of ECM components (such as I, II, III collagen and fibronectin). On the other hand, it can also restrain ECM degradation. Through the inhibition of the activity of enzymes that degrade ECM components (such as matrix metalloproteinases), MALAT1 affects the dynamic balance of ECM [32,33]. In addition, MALAT1 can also activate α-smooth muscle actin (α-SMA) [34]. After α-SMA has been activated, it can facilitate the transformation of cells into a myofibroblast (MFB) phenotype [35]. MFBs are able to synthesize and secrete the major components of ECM, such as fibrin and collagen [35].
2.2. The Role of MALAT1 in EMT
Epithelial-mesenchymal transformation (EMT) refers to the process in which differentiated mature epithelial cells, stimulated by external factors, acquire certain characteristics of mesenchymal cells (such as the loss of polarity, cytoskeletal protein changes and a fibroblast-like appearance), to exert stronger activity and migration. Cells undergoing EMT showed down-regulation of E-cadherin expression levels and increased N-cadherin, vimentin, and fibronectin expression levels at the molecular level [36]. The EMT process is a key event in the fibrosis of various tissues and organs [37,38,39]. It has been found that MALAT1 may be directly or indirectly associated with EMT. Xiang and colleagues investigated the role of MALAT1 in EMT and found that MALAT1 promoted EMT induced by TGF-β1 by competitively binding to miR-145 in endothelial progenitor cells [40]. Similarly, MALAT1 expression significantly increased in human retinal pigment epithelial cells involved in TGF-β1 induction, and MALAT1 exerts an enormous function on EMT, proliferation, and migration of cells [41]. In addition, MALAT1 can also participate in the regulation of EMT through the relevant signaling pathway axis. For example, the Yes-related protein 1/MALAT1/miR126-5p axis can regulate EMT in colorectal cancer [42]. Other studies have found that MALAT1 regulates Snail by acting as a competitive endogenous RNA of miR-22 [43]. Snail is a potent inducer of EMT, which can motivate renal fibrosis in mice [44]. Therefore, MALAT1 may indirectly induce EMT through a variety of pathways, such as Snail, and participate in the occurrence and development of fibrosis.
2.3. MALAT1 Is Involved in Inflammation
Usually, inflammation is the initial stage of fibrosis, which causes the damage of resident epithelial cells and endothelial cells [45]. By secreting inflammatory mediators, including cytokines and chemokines, the aggregated inflammatory cells induce the activation of effector cells, such as fibroblasts, thus driving the fiber formation process [45]. Various studies have shown that MALAT1 can act as an inflammatory regulator [46,47]. Zhang et al. [48] reported that the expression of MALAT1 was upregulated in a cerebral infarction model of mice, which achieved a protective effect by inhibiting the inflammatory response in cerebral ischemia injury. In addition, MALAT1 can also mediate inflammatory processes induced by hyperglycemia [49]. It is noteworthy that macrophages play an important role in interstitial fibrosis [50]. Gast et al. [51,52] found that the MALAT1-related small cytoplasmic RNA system can inhibit immune and inflammatory responses by inhibiting the phagocytosis of macrophages. The team observed a significant increase in the levels of interferon 1 (INF-γ), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in MALAT1 knockout mice, and it was found that the ablation of the MALAT1-related small cytoplasmic RNA system significantly affects the expression of inflammatory mediators. In LPS-activated macrophages. MALAT1 can also interact with the nuclear factor-B light chain enhancer (NF-KB) in the nucleus to inhibit the binding of NF-kB DNA to the inflammatory cytokine gene promoter. This leads to a reduction in the production of TNF-α and IL-6 [53].
2.4. MALAT1 Interacts with TGF-β to Promote the Development of Fibrosis
TGF-β is a recognized EMT signal regulator and fibrosis driver, which has been identified in various types of fibrosis during recent years [19]. TGF-β was initially thought to be an indicator of the malignant transformation of embryonic renal fibroblasts [54]. As further research was conducted, TGF-β was also found to be overexpressed in fibrotic tissues, and was found to be able to induce the accumulation of extracellular matrix by enhancing collagen synthesis and inhibiting protease production both in vivo and in vitro [55]. Overexpression of TGF-β1 in the liver can even induce fibrotic diseases in multiple organs of transgenic mice [56]. TGF-β can induce the differentiation of epithelial cells into cells with a distinct myofibroblast morphology by activating both typical (SMAD-based) and atypical (non-SMAD) signaling pathways, leading to ECM overproduction [57,58]. An increasing number of studies have found that the interaction between multiple lncRNAs and the TGF-β/SMADs signaling pathway plays a crucial role in inflammation and fibrosis [59]. In hepatocytes, MALAT1 regulates the TGF-β/SMADs signaling pathway by forming lncRNA protein complexes that contain SMADs [60]. Knockdown of MALAT1 in TGF-β-treated human lung fibroblasts can improve cell viability and simultaneously inhibit the expression of mesenchymal protein and α-SMA [61]. These findings reveal that MALAT1 is involved in the pathophysiological process of fibrosis through the TGF-β signaling pathway.
In brief, the relationship between MALAT1, ECM, EMT, inflammation, TGF-β and other cytokines is complex, and their interactions with each other are jointly involved in the occurrence and development of fibrosis.
3. MALAT1 and Fibrotic Disease
Organs containing a large number of parenchymal cells, such as liver, heart, kidney, and lung, are prone to the formation of excess fibrous tissue during tissue repair, resulting in the development of fibrotic lesions [62]. The causes of fibrosis in these organs are different. Along with the increase of in-depth studies conducted on the mechanism by which MALAT1 is involved in fibrosis diseases, we concluded that MALAT1 participates in the activation of fibrotic effector cells and the generation of ECM through different signaling pathways. The role of MALAT1 in the fibrosis of different organs is discussed below.
3.1. MALAT1 and Liver Fibrosis
Liver fibrosis is a typical stage in the development of liver diseases, such as chronic hepatitis and hepatocellular carcinoma. It usually results from multiple causes, such as viral infections, alcoholism and metabolic diseases [63,64] and has become a health concern worldwide. During the process of fibrosis, the injury of hepatocytes or bile duct cells trigger an inflammatory response, which subsequently activates hepatic stellate cells (HSCs) and promotes the accumulation of ECM [65] (Table 1).
HSC is a key factor in the pathogenesis of liver fibrosis, which can be activated by a variety of cell signal transduction pathways and cytokines [66], which transform cells into myofibroblasts that secrete pro-fibrotic mediators, which induce fibrosis [67]. TGF-β is an important signal that induces HSC activation [68]. Wu et al. found for the first time that the expression level of MALAT1 increased in liver fibrosis models in vivo and in vitro. Silencing of MALAT1 can restore the expression of Silent Information Regulator 1 (SIRT1) protein and decrease the expression of myofibroblast marker α-SMA [69]. As the most effective deacetylase, SIRT1 can induce the deacetylation of SMAD3, a downstream molecule of the TGF-β signaling pathway, Significantly weakened its ability to bind to the promoter of fibroblast genes [70,71], which promotes HSCs to return to their resting state or promotes their apoptosis. Therefore, MALAT1 can promote the activation of HSC by blocking the TGF-β signaling pathway mediated by SIRT1, thus accelerating the process of fibrosis. At the same time, Yu et al. [72] also found that MALAT1 was overexpressed in fibrosis, and knockdown of MALAT1 could significantly decrease collagen deposition in vivo and inhibit HSC activation in vitro. The study also found that there was a negative feedback loop between levels of MALAT1 and miR-101b. Downregulation of MALAT1 can decrease the level of Ras-related C3 botulinum toxin substrate1 (Rac1)-GTP protein. Rac1 is an important pathogenic factor involved in the pathogenesis of liver fibrosis [73]. The results revealed that MALAT1 acts as a competitive endogenous RNA of miR-101b to influence HSC activation, proliferation, and the cell cycle by regulating Rac1 expression, which leads to liver fibrosis [72].
Damaged hepatocytes or hepatic sinusoidal endothelial cells will release exosomes in the early stage of liver fibrosis [74]. By transferring its contents, including active proteins, mRNA, miRNA, lncRNA, lipids, etc., it is internalized by neighboring HSCs, and HSCs are induced to transdifferentiate from resting to activated myofibroblasts, thus initiating and encouraging the liver fibrosis process [74]. In recent study, they found that MALAT1 can be transported into HSCs via exosomes of arsenic-treated hepatocytes to regulate type I α2 collagen (COL1A2), which promotes the activation of HSCs by downregulating miR-26b [75]. It can be seen that HSCs and other cells in the liver can secrete extracellular vesicles in an autocrine or paracrine manner to achieve cell-to-cell communication, thereby playing an important regulatory role. In the future, it may be possible to use extracellular vesicles as drug carriers to achieve the goal of preventing the progression of liver fibrosis by sufficient means. One after another, researchers have found that exosomes derived from hepatocytes rich in lncRNA also affect the proliferation and activation of HSCs [76,77]. In addition, MALAT1 expression of the circulating exosomes was upregulated in individuals exposed to arsenite [75]. These findings suggested that exosomal MALAT1 is involved in cell-to-cell communication during arsenite induced liver fibrosis and may serve as a potential biomarker.
Non-alcoholic steatohepatitis (NASH) is a serious clinical phenotype of non-alcoholic fatty liver disease (NAFLD) and is a significant cause of liver cirrhosis and liver failure [78]. NASH is characterized by significant liver inflammation and liver fibrosis [78]. Among NASH patients, liver fibrosis accounts for about 40.76% of the total [79]. Leti et al. found that MALAT1 can promote NASH-related fibrosis by increasing the expressions of inflammatory chemokines, such as C-X-C motif chemokine ligand 5 (CXCL5), and is regulated by hyperglycemia and insulin in vitro, even though its mechanism of action and influence is not yet clear [80]. We speculate that MALAT1 may be involved in the development of liver fibrosis in fatty liver patients in a cytokine-mediated manner. Sookoian et al. [81] proposed that MALAT1 can be used as a common molecular driver for the pathogenesis of NASH and chronic immune-mediated liver injury, and the abundance of liver MALAT1 was significantly related to the severity of NASH histology. Accordingly, MALAT1 may also play a similar role in the pathogenesis of convergent pathological phenotypes, such as in inflammation and fibrosis.
3.2. MALAT1 and Cardiac Fibrosis
Heart cells have a weak regeneration ability compared with liver cells. Various pathophysiological conditions, such as ischemic cardiomyopathy, diabetes and hypertension, can all be fibrotic [82]. Cardiac fibrosis is the core link between the occurrence and development of heart diseases and an important cause for the remodeling of heart structure. It can even lead to cardiac systolic and diastolic dysfunction, which results in arrhythmias and even heart failure [83,84] (Table 2).
Myocardial infarction (MI) is a major cause of human death worldwide. It is characterized by the death of a large number of myocardial cells within a short period of time, increase in the cell activity of cardiac fibroblasts (CFs), degradation of normal intercellular substances, and excessive proliferation of new ECM, which gradually develop into myocardial fibrosis [85,86,87]. Huang et al. [88] found that MALAT1 expression was upregulated in mouse myocardial infarction models and myocardial fibroblasts treated with angiotensin II (Ang II). Silencing of MALAT1 can attenuate MI-induced fibrosis. Mechanism studies have found that MALAT1 can directly regulate the level of miR-145, while miR-145 can inhibit TGF-β1 activity by reducing Furin expression. Furin is a pre-protein converting enzyme that plays an important role in activating TGF-β1 to promote myocardial fibrosis [89]. Therefore, MALAT1 inhibits the expression of miR-145, thereby activating Furin and TGF-β1, and participates in the pathological process of myocardial fibrosis. The difference is that the study by Wu et al. [90] found through RNA-seq analysis that MALAT1 was more abundant in infarcted myocardium and cardiomyocytes treated with human pluripotent stem cells (hPSCs)-derived cardiovascular progenitor cells (CVPCs)-secreted extracellular vesicles (hCVPC-EVs), which was further enhanced under hypoxic (EV-H) conditions. MALAT1 plays a cardioprotective effect in the healing process of MI by targeting and regulating miRNAs.
Diabetic cardiomyopathy (DCM) is a common cardiovascular complication of diabetes [91]. It is caused by insulin resistance, compensatory hyperinsulinemia, and hyperinsulinemia in the metabolic tissues of the heart [91]. Cardiac remodeling, myocardial fibrosis, and even diastolic dysfunction are caused by metabolic disorders such as impaired insulin metabolism signals, excessive blood insulin, impaired glucose uptake, increased myocardial nonesterified fatty acid intake, and mitochondrial dysfunction [92]. Zhang et al. [93] found through a microarray analysis that MALAT1 was highly expressed in a DCM rat model. Silencing of MALAT1 for 12 weeks could reduce myocardial apoptosis and improve left ventricular systolic and diastolic functions. In addition, they also found that the levels of TNF-α, IL-1β and IL-6 were significantly decreased in DCM, while knockdown of MALAT1 could significantly decrease the concentration of inflammatory cytokines, suggesting that MALAT1 plays a certain role in the inflammatory process of DCM [94]. Che et al. [95] demonstrated that MALAT1 could significantly increase the protein levels of Nod-like receptor protein3 (NLRP3), cleaved Caspase-1, IL-1β, TGF-β1, p-SMAD2, and p-SMAD3, and participate in the formation of fibrosis in DCM by regulating its downstream target, miR-141-mediated NLRP3 inflammasomes and the TGF-β1/SMADs signaling pathway. Other scholars have found MALAT1 can increase the production of type I collagen products, inflammation, cell proliferation and migration, phosphorylation of MST1 and LATS1, and promote nuclear translocation of Yes-related protein (YAP) by binding to cAMP-response element binding protein 1 (CREB), while the silencing of MALAT1 could decrease the accumulation of collagen and inflammation in high glucose CFs and DCM mice through the Hippo/YAP and CREB pathways, thereby decreasing myocardial fibrosis and myocardial injury [96].
Hypertensive heart disease (HHD) is one of the manifestations of human target organ damage caused by hypertension, which results in high levels of morbidity and mortality [83]. Continuous pressure overload can cause changes to myocardial fibrosis and cardiovascular remodeling [83]. Li et al. [97] found that the overexpression of MALAT1 could enhance the activity of arterial smooth muscle cells (ASMCs) and myocardial fibrosis in spontaneously hypertensive rats (SHRs). It is well known that the myogenic determination gene (MyoD) can initiate and promote myogenesis of a variety of non-myogenic cells, including fibroblasts, and is an important transcription factor in myocardial reconstruction [98]. The study by Li et al. found that MALAT1 could recruit histone methyltransferase Suv39h1 to myo-binding loci, leading to the trimethylation of H3K9me3, thereby inhibiting the transcription of MyoD and promoting cell proliferation, myocardial fibrosis and cell cycle entry, which play an important role in the cardiac remodeling of hypertensive rats [97]. These results indicate the value of MALAT1 as a therapeutic target for hypertensive heart disease. Although Peters et al. [99] believed that MALAT1 played no obvious role in stress-induced cardiac remodeling in mice, the contradiction between this paper and other papers may be due to the transcriptional independent function or the compensatory mechanism of the MALAT1 locus.
3.3. MALAT1 and Renal Fibrosis
Renal fibrosis is the common final outcome of progressive chronic kidney disease (CKD) caused by various causes. The characteristic pathological changes are the activation of renal interstitial fibroblasts, the accumulation of excessive ECM in the renal interstitium, and the partial destruction of the normal renal tissue structure. Similar to that of other organs, renal fibrosis is mediated by cellular factors, such as inflammatory cells, and molecular elements, such as TGF-β1 [100,101,102] (Table 3).
Diabetic nephropathy (DN) is a microvascular complication frequently associated with the development of diabetes, and renal fibrosis is an important link between cause and damage. Early studies found that MALAT1 was significantly upregulated in retinal endothelial cells under high glucose culture conditions and in the retina of diabetic mice [46], which indicated that MALAT1 might be involved in the occurrence of diabetic microvascular complications. MALAT1 can participate in the hyperglycemic injury of endothelial cells, tubular epithelial cells, and podocytes. For example, in endothelial cells, MALAT1 induces the inflammatory response and promotes endothelial cell injury by activating inflammatory ligands-serum amyloid 3 (SAA3) and inflammatory cytokines [49]. In tubular epithelial cells, MALAT1 upregulates the expression of pyrolysis-associated proteins, ELAVL1, NLRP3 and caspase-1, as well as pro-inflammatory cytokines, such as IL-1, through the adsorption of miR-23c, resulting in the pyroptosis of tubular epithelial cells [103]. MALAT1 can also inhibit the binding of fork head box O1 (Foxo1) to the SIRT1 promoter by interacting with Foxo1, thereby inhibiting SIRT1 transcription and promoting hyperglycemic-induced tubular epithelial cell injury [104]. In podocytes, β-catenin regulates MALAT1 transcription via binding to its promoter; while MALAT1 in turn post-transcriptionally changes the pattern of the pre-mRNA processing of β-catenin, while high glucose disrupts the MALAT1/β-catenin loop, resulting in the destruction of the structural and functional integrity of podocytes [105]. β-catenin is known to be a key mediator of the Wnt signaling pathway, and its overreaction to injury may result in renal fibrosis [106,107]. In addition, MALAT1 facilitates the nuclear translocation of β-catenin by physically combining and alternatively splicing with serine/arginine splicing factor 1 (SRSF1), further proving its essential role in diabetic nephropathy and high glucose-related podocyte injury [105].
Damaged cells can be repaired by releasing a variety of fibrogenic cytokines, and their excessive release leads to abnormal transdifferentiation of the extracellular matrix (represented by EMT), makes a large number of collagen fibers deposited in the intercellular substance [108]. Therefore, EMT is a key factor in renal fibrosis. Liu et al. [33] reported that MALAT1 expression increased in DN mice and cell models, and that knockdown of MALAT1 significantly attenuated the downregulation of the epithelial marker, E-cadherin, which was caused by high-glucose and mesenchymal markers, such as vimentin, and the upregulation of mesenchymal markers, such as vimentin, Zinc finger E-box binding homeobox1 (ZEB1) and ZEB2. The expression of α-SMA, fibronectin and Col I was also downregulated. The study then proposed that MALAT1 acts as the ceRNA of miR-145, which can induce EMT and promote related molecular mechanisms of DN fibrosis by inhibiting the expression of the target gene, ZEB2. Recently, studies have confirmed that the MALAT1/MiR-145/focal adhesion kinase (FAK) pathway takes an important role in TGF-β1-induced renal fibrosis. Furthermore, N6-methyladenosine (m6A) modification was found to be involved in the upregulation of MALAT1 in renal fibrosis, and methyltransferase-like protein 3 (METTL3) has been shown to be the main m6A-modified methyltransferase on MALAT1 [109]. Li et al. [110] found that through the negative regulation of miR-100-3p, MALAT1 upregulated the expression of type IV collagen (COL4A1) and α-SMA, which induced EMT in renal tubular epithelial cells, and promoted renal fibrosis in mice with unilateral ureteral obstruction. Although Kölling et al. [111] detected increased expression levels of MALAT1 in the hypoxic/ischemic kidney injury model, and in vitro found that MALAT1 affects endothelial cell cycle progression, proliferation, migration ability, and apoptosis induction. The researchers then constructed a mouse model of renal ischemia-reperfusion (I/R) injury and found that interstitial fibrosis increased, but there was no significant difference in the expression level of Col1a2, Col III, and TGF-β between MALAT1 knockout mice and wild-type mice. Whole-genome expression analysis and small ribonucleic acid sequencing also revealed only minor differences between the two groups. The reason for this phenomenon may be due to the fact that during the mouse embryonic development stage, the researchers knocked out the MALAT1 gene, thereby activating other compensatory mechanisms in the body to cover up the effect of the knockout of the MALAT1 gene, or the role of MALAT1 is limited in related signal pathways in vivo. It is affirmative that MALAT1 is a viable biomarker of renal I/R-injury, but more research about targeted therapy for renal I/R-injury needs to be done to prove it.
3.4. MALAT1 and Pulmonary Fibrosis
Pulmonary fibrosis is a lung disease caused by the repair of persistent and pathological damage to alveolar epithelial cells, which is not only the common result of lung diseases caused by various internal and external pathogens, but is also important for the pathological basis of respiratory failure [112]. Factors and diseases that cause pulmonary fibrosis include long-term exposure to silica or bleomycin, idiopathic pulmonary fibrosis (IPF) and acute respiratory distress syndrome (ARDS), etc. [113]. Cao et al. first proved that there are significant changes in the expression many lncRNAs in lung tissues with bleomycin-induced fibrosis [114] (Table 4).
Silicosis is one of the most serious vocational diseases in the world [115]. It mainly manifests as chronic progressive pulmonary fibrosis. During the development of pulmonary fibrosis, silica particles stimulate epithelial cells and macrophages, secrete a large number of cytokines and inflammatory mediators, attract fibroblasts and promote EMT [116,117]. Yan et al. [118] found that in silica-induced human bronchial epithelial cells (HBE), the expression of MALAT1 was enhanced and that it competitively binds to miR-503, thereby activating the phosphatidylinositol-3-kinases (P13K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR)/Snail signaling pathway, which is closely related to EMT, to promote the development of silica-induced pulmonary fibrosis. Huang et al. [119] used gene chip technology to determine that MALAT1 is an upregulated gene that undergoes alternative splicing during silica damage, and a bioinformatics analysis showed that it was related to the EMT pathway. In addition, Cui et al. [120] found that the expression of MALAT1 in macrophages was significantly regulated under both polarization states. MALAT1 is upregulated in M1, while it is downregulated in M2. Inhibition of the expression of MALAT1 can attenuate M1 macrophage activation induced by bacterial LPS through a decrease in the expression of Clec16a, enhanced IL-4–activated M2 differentiation and the pro-fibrotic phenotype of macrophages. At the same time, LPS-induced systemic and pulmonary inflammation and injury decreased significantly in MALAT1 myeloid knockout mice, while bleomycin-induced fibrosis increased. These experiments showed that macrophage MALAT1 exerts an important blocking effect on the development of pulmonary fibrosis [120]. Some scholars have conducted research on the mechanism of pulmonary fibrosis caused by PM2.5 particles and have found that after PM2.5-treated HBE, the resulting change in MALAT1 expression was correlated with the process of fibrosis, and elevated levels of MALAT1 were positively associated with EMT processes and inflammatory factor, IL-6 and IL-8 expression, indicating that MALAT1 participates in the PM2.5-induced pulmonary fibrosis process by regulating inflammatory factors [121].
IPF is one of the most common and most serious interstitial lung diseases, characterized by diffuse alveolitis, massive proliferation and activation of lung fibroblast cells and myofibroblasts, and alveolar structural disorders [122]. The cause of IPF is unknown. It is currently generally considered to be the result of the interaction between heredity and environmental factors, which involves the abnormal activation of multiple pathways for wound repair, and the damage and abnormal repair of the alveolar epithelium are considered the initial process of pulmonary fibrosis [122]. EMT of alveolar epithelial cells may be an important pathogenic mechanism of idiopathic fibrosis. Wang et al. [123] found that there were distinguishable differences in the expression of MALAT1 in the peripheral blood of IPF patients, suggesting that MALAT1 may be a crucial regulatory factor involved in the pathogenesis of IPF, it is involved in the mesenchymal transition of alveolar epithelial cells, inflammation, myofibroblast differentiation, and collagen production in the pathogenesis of IPF. and the specific regulatory mechanism needs to be verified through further studies.
ARDS is a common clinical manifestation of severe lung injury and respiratory failure. In the United States, more than 190,000 people suffer from ARDS each year, with a fatality rate of 27% to 45% [124]. It is characterized by an excessive inflammatory reaction in a short period of time followed by rapid pulmonary fibrosis formation. Pathological biopsy of lung tissues of dead patients found excessive fibrosis, which was mainly composed of fibroblast aggregation, collagen deposition, and extracellular matrix deposition [125]. Wang et al. [126] found that MALAT1 overexpressed in the plasma and peripheral blood mononuclear cells (PBMCs) of ARDS patients. They reported that MALAT1 might upregulate the expression of phosphatase and the tensin homolog (PTEN) by adsorbing miR-425, and inducing apoptosis, thereby playing a role in the pathophysiological process of ARDS. In addition, Yao et al. [127] showed that the silencing of MALAT1 could inhibit the apoptosis of human pulmonary microvascular endothelial cells (HPMECs) through the miR-150-5p-mediated intercellular adhesion molecule-1 (ICAM-1) axis, and decrease the expression of pro-inflammatory cytokines (IL-6, IL-1, TNF-α) and E-selectin, thereby decreasing the level of lung injury in ARDS. Dai et al. [128] also mentioned the possible mechanism by which the knockdown of MALAT1 inhibits the LPS-induced inflammatory response of mice alveolar macrophages and mice alveolar epithelial cells by sponging miR-146a, and plays a role in the process of acute lung injury. Interestingly, Zhu [129] suggested that MALAT1 can alleviate the damage of LPS to lung fibroblasts. It blocks the phosphorylation of NF-κB p65 through negative feedback, thereby blocking the activation of NF-κB caused by LPS and inhibiting inflammation. As early as 2013, Gutschner et al. [130] found that subcutaneous injection of anti-MALAT1 antisense oligonucleotide (ASO) into mice could effectively block the metastasis of lung cancer, and indicated the feasibility of MALAT1 as a predictive marker and therapeutic target.
4. Conclusions and Prospects
Organ fibrosis is a complex dynamic process, involving multi-cell and multi-molecule participation and interaction. Based on research conducted on lncRNAs, this review focused on MALAT1, a key lncRNA that is extensively involved in the pathological mechanism of fibrosis in various organs.
The detection of lncRNA in body fluids and the report of the level of exosomal MALAT1 may be used as a biologically active carrier to achieve its targeted therapy on target tissues and cells. Although increasing studies have revealed the potential value of MALAT1, it is widely distributed in various tissues in the human body. Its mechanism of action involves a wide range, and it plays a role in both physiological and pathological tissues. Therefore, no suitable method has been found to inhibit or activate MALAT1. MALAT1 has a complex regulatory network. The effect of MALAT1 varied greatly depending on the tissue differentiation and the amount of expression. As a result, the function of MALAT1 is not only a positive regulation function as previously recognized. It may also have the opposite effect at the right time and the right amount. The function of MALAT1 in fibrosis may depend on the microenvironment, while the effect of MALAT1 on drug action has also provided a promising research direction for its clinical application.
In summary, MALAT1 plays a significant role in the process of organ fibrosis by regulating the expression of a variety of extracellular matrix components and inflammatory factors, and by participating in multiple signaling pathways related to fibrosis. At present, no targeted MALAT1 regimen is used for the treatment of fibrosis. This review can not only help us understand the partial mechanism of fibrosis, but also provide a scientific basis for MALAT1 as a valuable biomarker and potential fibrosis treatment target. In addition, related proteins and target genes that bind to MALAT1 may be potential new therapeutic targets, which will contribute to the exploration of new anti-fibrosis treatment therapies that will provide enormous potential benefits for patients with fibrosis.
Author Contributions
Writing—original draft preparation, Y.L.; resources, F.L.; writing—review and editing, Y.C., Y.Y. and Y.W.; funding acquisition, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by National Natural Science Foundation of China (No.: 81901020), Hunan Provincial Health and Family Planning Commission’s 2017 Scientific Research Plan Project (No.: B2017036), and Natural Science Foundation of Hunan Province, China (No.: 2020JJ4458).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Birbrair, A.; Zhang, T.; Files, D.C.; Mannava, S.; Smith, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, F.; Regolisti, G.; Delsante, M.; Cantaluppi, V.; Castellano, G.; Gesualdo, L.; Villa, G.; Fiaccadori, E. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J. Nephrol. 2018, 31, 351–359. [Google Scholar] [CrossRef]
- Fischereder, M.; Schroppel, B. The role of chemokines in acute renal allograft rejection and chronic allograft injury. Front. Biosci. 2009, 14, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Gomez, H.; Kellum, J.A.; Ronco, C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T.; et al. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.; Collett, J.A.; Gunst, S.J.; Basile, D.P. Th17 cells contribute to pulmonary fibrosis and inflammation during chronic kidney disease progression after acute ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R265–R273. [Google Scholar] [CrossRef] [PubMed]
- Popovic, B.; Sutic, I.; Skocibusic, N.; Ljubotina, A.; Diminic-Lisica, I.; Bukmir, L. Cholestasis and Inflammation of the Pancreas in Family Medicine. Acta Med. Croatica 2015, 69, 319–326. [Google Scholar]
- Cao, L.; Nicosia, J.; Larouche, J.; Zhang, Y.; Bachman, H.; Brown, A.C.; Holmgren, L.; Barker, T.H. Detection of an Integrin-Binding Mechanoswitch within Fibronectin during Tissue Formation and Fibrosis. ACS Nano 2017, 11, 7110–7117. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, P.; Jayakumar, C.; Ramesh, G. Proximal tubule-specific overexpression of netrin-1 suppresses acute kidney injury-induced interstitial fibrosis and glomerulosclerosis through suppression of IL-6/STAT3 signaling. Am. J. Physiol. Renal Physiol. 2013, 304, F1054–F1065. [Google Scholar] [CrossRef] [Green Version]
- Tewes, S.; Gueler, F.; Chen, R.; Gutberlet, M.; Jang, M.S.; Meier, M.; Mengel, M.; Hartung, D.; Wacker, F.; Rong, S.; et al. Functional MRI for characterization of renal perfusion impairment and edema formation due to acute kidney injury in different mouse strains. PLoS ONE 2017, 12, e0173248. [Google Scholar] [CrossRef]
- Xiao, Y.; Yang, N.; Zhang, Q.; Wang, Y.; Yang, S.; Liu, Z. Pentraxin 3 inhibits acute renal injury-induced interstitial fibrosis through suppression of IL-6/Stat3 pathway. Inflammation 2014, 37, 1895–1901. [Google Scholar] [CrossRef]
- Espindola, M.S.; Habiel, D.M.; Narayanan, R.; Jones, I.; Coelho, A.L.; Murray, L.A.; Jiang, D.; Noble, P.W.; Hogaboam, C.M. Targeting of TAM Receptors Ameliorates Fibrotic Mechanisms in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1443–1456. [Google Scholar] [CrossRef]
- Lemmer, A.; VanWagner, L.B.; Ganger, D. Assessment of Advanced Liver Fibrosis and the Risk for Hepatic Decompensation in Patients With Congestive Hepatopathy. Hepatology 2018, 68, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Jiang, S.; Yang, Y. Database Selection and Heterogeneity-More Details, More Credibility. JAMA Oncol. 2018, 4, 1295. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef]
- Hocher, B.; Adamski, J. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Cao, G.; Chen, H.; Liu, D.; Su, W.; Yu, X.Y.; Vaziri, N.D.; Liu, X.H.; Bai, X.; Zhang, L.; et al. Gene and protein expressions and metabolomics exhibit activated redox signaling and wnt/beta-catenin pathway are associated with metabolite dysfunction in patients with chronic kidney disease. Redox Biol. 2017, 12, 505–521. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Wang, H.L.; Cheng, X.L.; Wei, F.; Bai, X.; Lin, R.C.; Vaziri, N.D. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci. Rep. 2015, 5, 12936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; He, Y.; Li, J. MicroRNA-21: A central regulator of fibrotic diseases via various targets. Curr. Pharm. Des. 2015, 21, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tsitsiou, E.; Herrick, S.E.; Lindsay, M.A. MicroRNAs and the regulation of fibrosis. FEBS J. 2010, 277, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, F. Long noncoding RNA: A new contributor and potential therapeutic target in fibrosis. Epigenomics 2017, 9, 1233–1241. [Google Scholar] [CrossRef]
- Jiang, X.; Lei, R.; Ning, Q. Circulating long noncoding RNAs as novel biomarkers of human diseases. Biomark. Med. 2016, 10, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Guo, L.; Li, Y.; Zhou, Q.; Li, Z. MALAT1 is an oncogenic long non-coding RNA associated with tumor invasion in non-small cell lung cancer regulated by DNA methylation. Int. J. Clin. Exp. Pathol. 2015, 8, 15903–15910. [Google Scholar]
- Wang, W.; Zhu, Y.; Li, S.; Chen, X.; Jiang, G.; Shen, Z.; Qiao, Y.; Wang, L.; Zheng, P.; Zhang, Y. Long noncoding RNA MALAT1 promotes malignant development of esophageal squamous cell carcinoma by targeting beta-catenin via Ezh2. Oncotarget 2016, 7, 25668–25682. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, R.; Mayeda, A.; Yoshida, M.; Nakagawa, S. MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta 2016, 1859, 192–199. [Google Scholar] [CrossRef]
- Zhang, X.; Hamblin, M.H.; Yin, K.J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. [Google Scholar] [CrossRef]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013, 9, e1003368. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Chen, G.; He, R.; Yang, L. LncRNA MALAT1 promotes osteoarthritis by modulating miR-150-5p/AKT3 axis. Cell Biosci. 2019, 9, 54. [Google Scholar] [CrossRef]
- Liu, B.; Qiang, L.; Wang, G.D.; Duan, Q.; Liu, J. LncRNA MALAT1 facilities high glucose induced endothelial to mesenchymal transition and fibrosis via targeting miR-145/ZEB2 axis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3478–3486. [Google Scholar] [PubMed]
- Gong, Y.; Zhu, Y.; Zhu, B.; Si, X.; Heng, D.; Tang, Y.; Sun, X.; Lin, L. LncRNA MALAT1 is up-regulated in diabetic gastroparesis and involved in high-glucose-induced cellular processes in human gastric smooth muscle cells. Biochem. Biophys. Res. Commun. 2018, 496, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, T.; Liang, X.; Shu, S.; Xiang, X.; Zhang, W.; Guo, T.; Xie, W.; Deng, W.; Tang, X. lncRNA MALAT1 mediated high glucose-induced HK-2 cell epithelial-to-mesenchymal transition and injury. J. Physiol. Biochem. 2019, 75, 443–452. [Google Scholar] [CrossRef]
- Marlar, S.; Abdellatef, S.A.; Nakanishi, J. Reduced adhesive ligand density in engineered extracellular matrices induces an epithelial-mesenchymal-like transition. Acta Biomater. 2016, 39, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Syn, W.K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 2004, 82, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, Z.; Li, H.; Fan, J.; Yang, S.; Chen, C.; Wang, D.W. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell 2017, 16, 387–400. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, Y.; Tang, Y.; Li, Q. MALAT1 Modulates TGF-beta1-Induced Endothelial-to-Mesenchymal Transition through Downregulation of miR-145. Cell. Physiol. Biochem. 2017, 42, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yao, H.; Li, M.; Li, H.; Wang, F. Long Non-Coding RNA MALAT1 Mediates Transforming Growth Factor Beta1-Induced Epithelial-Mesenchymal Transition of Retinal Pigment Epithelial Cells. PLoS ONE 2016, 11, e0152687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Ou, C.; Liu, J.; Chen, C.; Zhou, Q.; Yang, S.; Li, G.; Wang, G.; Song, J.; Li, Z.; et al. YAP1-induced MALAT1 promotes epithelial-mesenchymal transition and angiogenesis by sponging miR-126-5p in colorectal cancer. Oncogene 2019, 38, 2627–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.M.; Thum, T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat. Rev. Nephrol. 2016, 12, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, X.; Liu, K.; Hamblin, M.H.; Yin, K.J. Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke. J. Neurosci. 2017, 37, 1797–1806. [Google Scholar] [CrossRef]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Schroen, B.; Voigt, A.; Haas, J.; Kuehl, U.; Lassner, D.; Skurk, C.; Escher, F.; Wang, X.; Kratzer, A.; et al. Long noncoding RNA MALAT1-derived mascRNA is involved in cardiovascular innate immunity. J. Mol. Cell. Biol. 2016, 8, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Bohm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE-/-mice. Cardiovasc Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016, 590, 2884–2895. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Anzano, M.A.; Lamb, L.C.; Smith, J.M.; Frolik, C.A.; Marquardt, H.; Todaro, G.J.; Sporn, M.B. Isolation from murine sarcoma cells of novel transforming growth factors potentiated by EGF. Nature 1982, 295, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-beta Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, N.; Factor, V.; Nagy, P.; Kopp, J.; Kondaiah, P.; Wakefield, L.; Roberts, A.B.; Sporn, M.B.; Thorgeirsson, S.S. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. USA 1995, 92, 2572–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chung, A.C.; Huang, X.R.; Dong, Y.; Yu, X.; Lan, H.Y. Identification of novel long noncoding RNAs associated with TGF-beta/SMAD3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 2014, 184, 409–417. [Google Scholar] [CrossRef]
- Zhang, J.; Han, C.; Song, K.; Chen, W.; Ungerleider, N.; Yao, L.; Ma, W.; Wu, T. The long-noncoding RNA MALAT1 regulates TGF-beta/SMAD signaling through formation of a lncRNA-protein complex with SMADs, SETD2 and PPM1A in hepatic cells. PLoS ONE 2020, 15, e0228160. [Google Scholar]
- Hu, T.J.; Huang, H.B.; Shen, H.B.; Chen, W.; Yang, Z.H. Role of long non-coding RNA MALAT1 in chronic obstructive pulmonary disease. Exp. Ther. Med. 2020, 20, 2691–2697. [Google Scholar] [PubMed]
- Zhang, S.S.; Gong, Z.J.; Xiong, W.; Wang, X.; Min, Q.; Luo, C.D.; Ling, T.Y. A rat model of oral submucous fibrosis induced by bleomycin. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 216–223. [Google Scholar] [CrossRef]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, N.; Zhao, S.X.; Kong, L.B.; Du, J.H.; Ren, W.G.; Han, F.; Zhang, Q.S.; Li, W.C.; Cui, P.; Wang, R.Q.; et al. LncRNA-ATB/microRNA-200a/beta-catenin regulatory axis involved in the progression of HCV-related hepatic fibrosis. Gene 2017, 618, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, G.; De Chiara, F.; Bottcher, K.; Levi, A.; Dhar, D.; Longato, L.; Mazza, G.; Zhang, Z.; Marrali, M.; Fernandez-Iglesias, A.; et al. The adenosine monophosphate-activated protein kinase-vacuolar adenosine triphosphatase-pH axis: A key regulator of the profibrogenic phenotype of human hepatic stellate cells. Hepatology 2018, 68, 1140–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Iredale, J.P. Liver fibrosis: Cellular mechanisms of progression and resolution. Clin. Sci. 2007, 112, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.W.; Hien, T.T.; Lim, S.C.; Jun, D.W.; Choi, H.S.; Yoon, J.H.; Cho, I.J.; Kang, K.W. Pin1 induction in the fibrotic liver and its roles in TGF-beta1 expression and SMAD2/3 phosphorylation. J. Hepatol. 2014, 60, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, X.; Zhou, Q.; Huang, C.; Meng, X.; Xu, F.; Li, J. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol. Appl. Pharmacol. 2015, 289, 163–176. [Google Scholar] [CrossRef]
- Sun, L.; Fan, Z.; Chen, J.; Tian, W.; Li, M.; Xu, H.; Wu, X.; Shao, J.; Bian, Y.; Fang, M.; et al. Transcriptional repression of SIRT1 by protein inhibitor of activated STAT 4 (PIAS4) in hepatic stellate cells contributes to liver fibrosis. Sci. Rep. 2016, 6, 28432. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ghosh, A.K.; Chu, H.; Fang, F.; Hinchcliff, M.E.; Wang, J.; Marangoni, R.G.; Varga, J. The Histone Deacetylase Sirtuin 1 Is Reduced in Systemic Sclerosis and Abrogates Fibrotic Responses by Targeting Transforming Growth Factor beta Signaling. Arthritis Rheumatol. 2015, 67, 1323–1334. [Google Scholar] [CrossRef]
- Yu, F.; Lu, Z.; Cai, J.; Huang, K.; Chen, B.; Li, G.; Dong, P.; Zheng, J. MALAT1 functions as a competing endogenous RNA to mediate Rac1 expression by sequestering miR-101b in liver fibrosis. Cell Cycle 2015, 14, 3885–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Witek, R.P.; Yang, L.; Omenetti, A.; Syn, W.K.; Moylan, C.A.; Jung, Y.; Karaca, G.F.; Teaberry, V.S.; Pereira, T.A.; et al. Activation of Rac1 promotes hedgehog-mediated acquisition of the myofibroblastic phenotype in rat and human hepatic stellate cells. Hepatology 2010, 52, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Dai, X.; Chen, C.; Xue, J.; Xiao, T.; Mostofa, G.; Wang, D.; Chen, X.; Xu, H.; Sun, Q.; Li, J.; et al. Exosomal MALAT1 derived from hepatic cells is involved in the activation of hepatic stellate cells via miRNA-26b in fibrosis induced by arsenite. Toxicol. Lett. 2019, 316, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Huang, Z.; Gurley, E.C.; Wang, X.; Wang, J.; He, H.; Yang, H.; Lai, G.; Zhang, L.; et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology 2018, 68, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Leti, F.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; Gerhard, G.S.; DiStefano, J.K. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl. Res. 2017, 190, 25–39.e21. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; San Martino, J.; Castano, G.O.; Pirola, C.J. Metastasis-associated lung adenocarcinoma transcript 1 as a common molecular driver in the pathogenesis of nonalcoholic steatohepatitis and chronic immune-mediated liver damage. Hepatol. Commun. 2018, 2, 654–665. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P. Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, L.; Song, J.; Wang, Z.; Huang, X.; Guo, Z.; Chen, F.; Zhao, X. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019, 234, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, J.; Tan, W.L.W.; Jiang, Y.; Wang, S.; Li, Q.; Yu, X.; Tan, J.; Liu, S.; Zhang, P.; et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef]
- Adeghate, E.; Singh, J. Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail. Rev. 2014, 19, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gu, H.; Xu, W.; Zhou, X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int. J. Cardiol. 2016, 203, 214–216. [Google Scholar] [CrossRef]
- Zhang, M.; Gu, H.; Chen, J.; Zhou, X. Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int. J. Cardiol. 2016, 202, 753–755. [Google Scholar] [CrossRef]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/SMADs signaling in diabetic cardiomyopathy. FASEB J. 2020, 34, 5282–5298. [Google Scholar] [CrossRef]
- Liu, J.; Xu, L.; Zhan, X. LncRNA MALAT1 regulates diabetic cardiac fibroblasts through the Hippo/YAP signaling pathway. Biochem. Cell Biol. 2020, 98, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, C.; Li, J.; Che, J.; Yang, X.; Xian, Y.; Li, X.; Cao, C. Long non-coding RNA MALAT1 promotes cardiac remodeling in hypertensive rats by inhibiting the transcription of MyoD. Aging 2019, 11, 8792–8809. [Google Scholar] [CrossRef]
- Murry, C.E.; Kay, M.A.; Bartosek, T.; Hauschka, S.D.; Schwartz, S.M. Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with MyoD. J. Clin. Investig. 1996, 98, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Hermans-Beijnsberger, S.; Beqqali, A.; Bitsch, N.; Nakagawa, S.; Prasanth, K.V.; de Windt, L.J.; van Oort, R.J.; Heymans, S.; Schroen, B. Long Non-Coding RNA Malat-1 Is Dispensable during Pressure Overload-Induced Cardiac Remodeling and Failure in Mice. PLoS ONE 2016, 11, e0150236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Kaissling, B.; Lehir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 2013, 1832, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zeng, L.; Cao, C.; Lu, C.; Lian, W.; Han, J.; Zhang, X.; Zhang, J.; Tang, T.; Li, M. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp. Cell Res. 2017, 350, 327–335. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, D.Y.; Sha, W.G.; Shen, L.; Lu, G.Y. Long non-coding RNA MALAT1 interacts with transcription factor Foxo1 to regulate SIRT1 transcription in high glucose-induced HK-2cells injury. Biochem. Biophys. Res. Commun. 2018, 503, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, R.; Li, X.; Fan, M.; Lin, J.; Zhen, J.; Chen, L.; Lv, Z. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with beta-catenin. J. Cell. Mol. Med. 2017, 21, 2732–2747. [Google Scholar] [CrossRef]
- Monga, S.P. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef] [Green Version]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanthakumar, C.B.; Hatley, R.J.; Lemma, S.; Gauldie, J.; Marshall, R.P.; Macdonald, S.J. Dissecting fibrosis: Therapeutic insights from the small-molecule toolbox. Nat. Rev. Drug Discov. 2015, 14, 693–720. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, B.; Chen, Z.; He, Y.; Du, Y.; Liu, Y.; Chen, X. m(6)A-induced lncRNA MALAT1 aggravates renal fibrogenesis in obstructive nephropathy through the miR-145/FAK pathway. Aging 2020, 12, 5280–5299. [Google Scholar] [CrossRef]
- Li, H.; He, L.; Lou, W.; Wang, H.; Li, Y. Metastasis-related lung adenocarcinoma transcript 1 (MALAT1) promotes hypoxia-induced transdifferentiation of renal tubular mesothelial cells. Chin. J. Cell. Mol. Immunol. 2018, 11, 1007–1008. (In Chinese) [Google Scholar]
- Kolling, M.; Genschel, C.; Kaucsar, T.; Hubner, A.; Rong, S.; Schmitt, R.; Sorensen-Zender, I.; Haddad, G.; Kistler, A.; Seeger, H.; et al. Hypoxia-induced long non-coding RNA Malat1 is dispensable for renal ischemia/reperfusion-injury. Sci. Rep. 2018, 8, 3438. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundarakrishnan, A.; Chen, Y.; Black, L.D.; Aldridge, B.B.; Kaplan, D.L. Engineered cell and tissue models of pulmonary fibrosis. Adv. Drug Deliv. Rev. 2018, 129, 78–94. [Google Scholar] [CrossRef]
- Cao, G.; Zhang, J.; Wang, M.; Song, X.; Liu, W.; Mao, C.; Lv, C. Differential expression of long non-coding RNAs in bleomycin-induced lung fibrosis. Int. J. Mol. Med. 2013, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, P.J.; Clevelario, A.L.; Padilha, G.A.; Silva, J.D.; Kitoko, J.Z.; Olsen, P.C.; Capelozzi, V.L.; Rocco, P.R.; Cruz, F.F. Bosutinib Therapy Ameliorates Lung Inflammation and Fibrosis in Experimental Silicosis. Front. Physiol. 2017, 8, 159. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Wu, Q.; Yao, W.; Li, Y.; Liu, Y.; Yuan, J.; Han, R.; Yang, J.; Ji, X.; Ni, C. MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K p85 and is sponged by lncRNA MALAT1. Sci. Rep. 2017, 7, 11313. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Liu, X.; Li, H.; Ning, H.; Zhou, P.K. PRKCSH Alternative Splicing Involves in Silica-Induced Expression of Epithelial-Mesenchymal Transition Markers and Cell Proliferation. Dose Response 2020, 18, 1559325820923825. [Google Scholar] [CrossRef]
- Cui, H.; Banerjee, S.; Guo, S.; Xie, N.; Ge, J.; Jiang, D.; Zornig, M.; Thannickal, V.J.; Liu, G. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 2019, 4, e124522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Chi, Y.; Deng, J.; Liu, Y.; Lu, Y.; Chen, J.; Dong, S. Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA. Sci. Rep. 2017, 7, 9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Wang, F.; Li, P.; Li, F.S. Integrated Analysis of a Gene Correlation Network Identifies Critical Regulation of Fibrosis by lncRNAs and TFs in Idiopathic Pulmonary Fibrosis. Biomed. Res. Int. 2020, 2020, 6537462. [Google Scholar] [CrossRef] [PubMed]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Bulpa, P.A.; Dive, A.M.; Mertens, L.; Delos, M.A.; Jamart, J.; Evrard, P.A.; Gonzalez, M.R.; Installe, E.J. Combined bronchoalveolar lavage and transbronchial lung biopsy: Safety and yield in ventilated patients. Eur. Respir. J. 2003, 21, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Xie, W.; Li, G.; Yao, L.; Zhang, R.; Xu, B. Overexpression of MALAT1 Relates to Lung Injury through Sponging miR-425 and Promoting Cell Apoptosis during ARDS. Can. Respir. J. 2019, 2019, 1871394. [Google Scholar] [CrossRef]
- Yao, M.Y.; Zhang, W.H.; Ma, W.T.; Liu, Q.H.; Xing, L.H.; Zhao, G.F. Long non-coding RNA MALAT1 exacerbates acute respiratory distress syndrome by upregulating ICAM-1 expression via microRNA-150-5p downregulation. Aging 2020, 12, 6570–6585. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, G.; Cheng, Z.; Wang, X.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect. Tissue Res. 2018, 59, 581–592. [Google Scholar] [CrossRef]
- Zhu, W.; Men, X. Negative feedback of NF-kappaB signaling by long noncoding RNA MALAT1 controls lipopolysaccharide-induced inflammation injury in human lung fibroblasts WI-38. J. Cell. Biochem. 2020, 121, 1945–1952. [Google Scholar] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
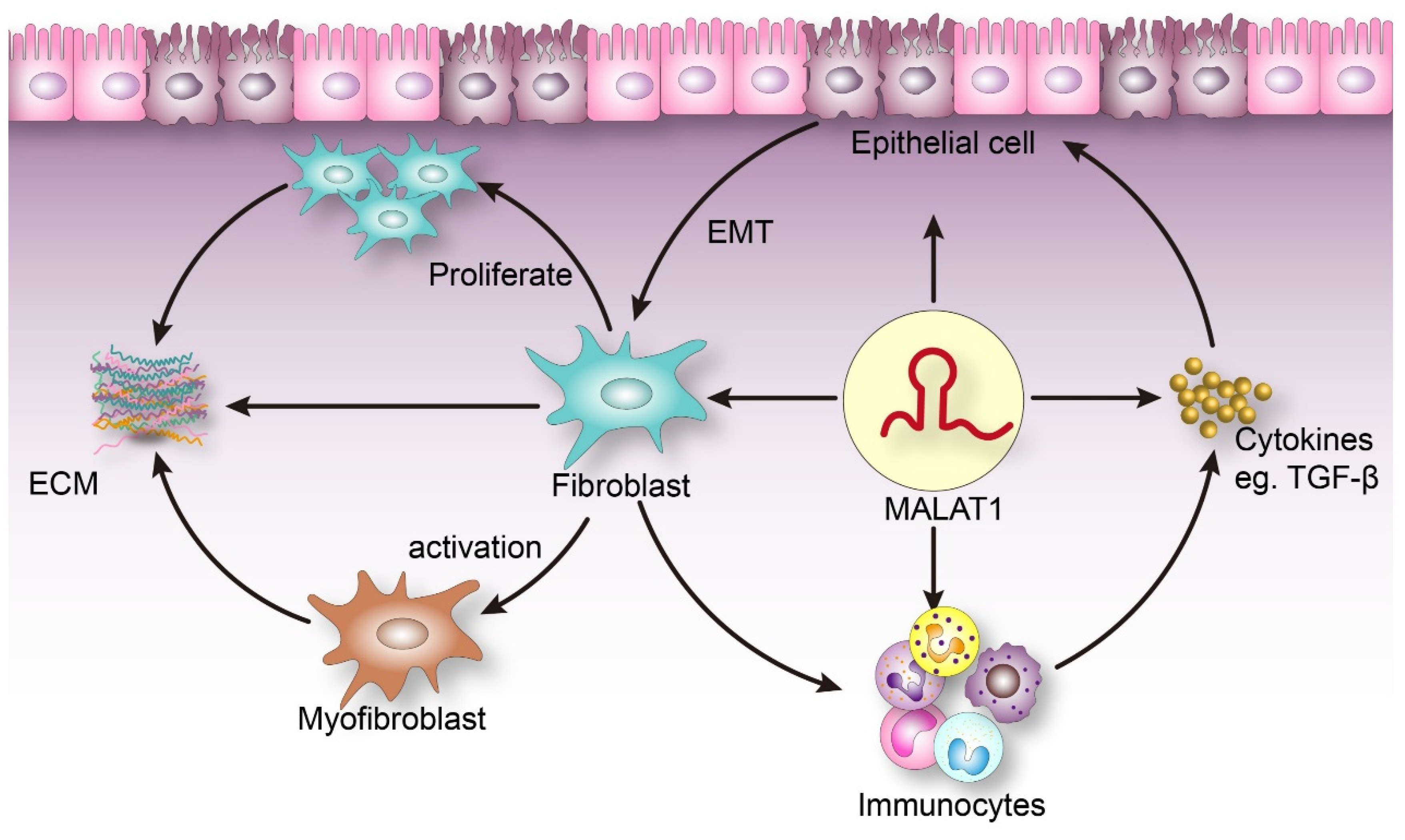
Figure 1.
Injured parenchymal cells can undergo an epithelial–-mesenchymal transformation and adsorb immune cells. Immune cells release cytokines, such as transforming growth factor-β1 (TGF-β), and fibroblasts proliferate and transdif-ferentiate into myofibroblasts, leading to extracellular matrix (ECM) deposition. Metastasis-associated lung adenocarci-noma transcript 1 (MALAT1) plays different roles in regulating each of the above links.
Figure 1.
Injured parenchymal cells can undergo an epithelial–-mesenchymal transformation and adsorb immune cells. Immune cells release cytokines, such as transforming growth factor-β1 (TGF-β), and fibroblasts proliferate and transdif-ferentiate into myofibroblasts, leading to extracellular matrix (ECM) deposition. Metastasis-associated lung adenocarci-noma transcript 1 (MALAT1) plays different roles in regulating each of the above links.


{kind=link}
Table 1.
The mechanism of MALAT1 in hepatic fibrosis.
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Rat liver tissues of CCl4-induced liver fibrosis. TGF-β1activated HSCs. | SIRT1 | HSCs activation | [69] |
| Up | Rat liver tissues of CCl4 and bile duct ligation (BDL) induced liver fibrosis. Activated HSCs and hepatocytes. | miR-101b/Rac1 | HSCs activation and proliferation | [72] |
| Up | Activated HSCs. | CXCL5 | NASH mechanisms of inflammatory chemokines | [80] |
| Up | NAFLD patients. | unknown | NASH | [81] |
| UP | The liver of arsenite induces Mice. Activated HSCs co-culture with arsenite treated L-02 cells. The circulating exosomal of people exposed to arsenite. | miR-26b | HSCs activation | [75] |
Table 2.
The mechanism of MALAT1 in cardiac fibrosis.
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Peri-infarct zone of MI mice hearts. in Ang II-induced CFs. | miR-145 | Cardiac fibrosis after infarction | [88] |
| Up | Infarcted myocardium and cardiomyocytes treated with hCVPC-EVs. | miR-497 | Cardioprotective | [90] |
| UP | High-glucose CFs. DCM mice. | miR-141/NLRP3 | Cardiac fibrosis of DCM | [95] |
| Up | High-glucose CFs. DCM mouse model. | Hippo/YAP CREB | CFs proliferation and invasion Inflammation and collagen accumulation Cardiac fibrosis of DCM | [96] |
| Up | Myocardial tissues and thoracic aortic vascular tissues of SHRs. | MyoD | ASMCs activity Myocardial fibrosis in SHRs | [97] |
| Not change | Pressure overload-induced cardiac remodeling and failure in mice. | Unknown | Unknown | [99] |
Table 3.
The mechanism of MALAT1 in renal fibrosis.
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Renal tissues of db/db mice. High glucose-stimulated HK-2 cells | miR-145/ZEB2 | EMT of HK-2 cells Renal fibrosis | [33] |
| Up | Renal fibrosis in patients with obstructive nephropathy. TGF-β1-treated HK2 cells | miR-145/FAK | viability, proliferation and migration of HK2 cells. Renal fibrosis | [109] |
| UP | Renal tissues of unilateral ureteral obstruction mice. Hypoxia-induced HK-2 cells. | miR-100-3p/COL4A1 | EMT of HK-2 cells Renal fibrosis | [110] |
| Up | Hypoxic/ischemic kidney injury in humans, mice, and cells. | Unknown | Unknown | [111] |
Table 4.
The mechanism of MALAT1 in pulmonary fibrosis.
| Levels of MALAT1 | Models | Targets | Functions | Reference |
|---|---|---|---|---|
| Up | Silica-treated HBE | miR503 PI3K/Akt/mTOR/Snail | Silica-induced pulmonary fibrosis | [118] |
| UP | LPS-treated macrophages | Clec16a | Macrophage activation and bleomycin-induced pulmonary fibrosis | [120] |
| Up | PM2.5-treated HBE | Unknown | Inflammation and EMT process | [121] |
| Up | Peripheral blood of IPF patients | Unknown | Idiopathic pulmonary fibrosis | [123] |
| Up | ARDS patient plasma and PBMCs. | miR-425/PTEN | Cell apoptosis and ARDS | [126] |
| Up | ARDS patients and LPS-treated HPMECs | miR-150-5p/ICAM-1 | Cell apoptosis and lung injury in ARDS | [127] |
| Up | LPS-induced acute lung injury rat model | miR-146a | Inflammatory response | [128] |
| Up | WI-38 cells after treatment by LPS | NF-κB p65 | LPS-induced inflammation injury | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Y.; Liu, F.; Cai, Y.; Yang, Y.; Wang, Y. LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target. Crystals 2021, 11, 249. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030249
AMA Style
Li Y, Liu F, Cai Y, Yang Y, Wang Y. LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target. Crystals. 2021; 11(3):249. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030249
Chicago/Turabian StyleLi, Yijie, Fenglin Liu, Yunzhou Cai, Yanqing Yang, and Yuehong Wang. 2021. "LncRNA MALAT1: A Potential Fibrosis Biomarker and Therapeutic Target" Crystals 11, no. 3: 249. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11030249
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.