
1,4-Dibromo-2,5-bis(phenylalkoxy)benzene Derivatives: C–Br...π(arene) Versus C–H...Br and Br...Br Interactions in the Solid State


Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
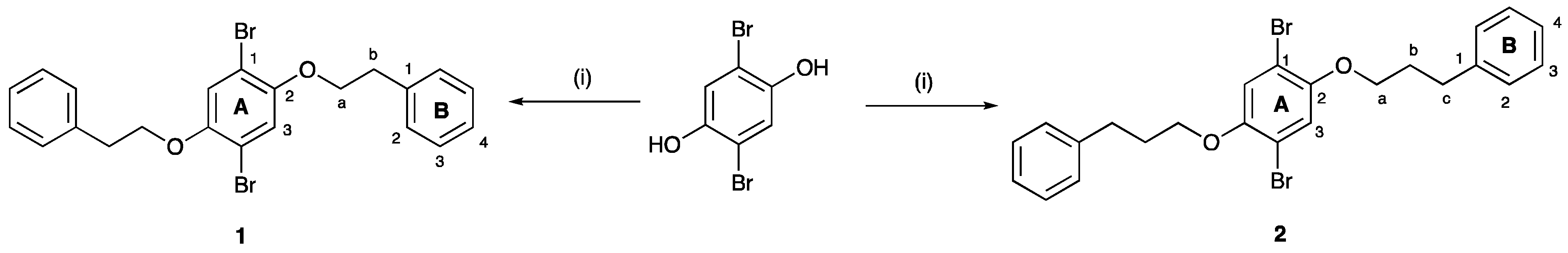
2.2. 1,4-Dibromo-2,5-bis(2-phenylethoxy)benzene, 1
2.3. 1,4-Dibromo-2,5-bis(3-phenylpropoxy)benzene, 2
2.4. Crystallography
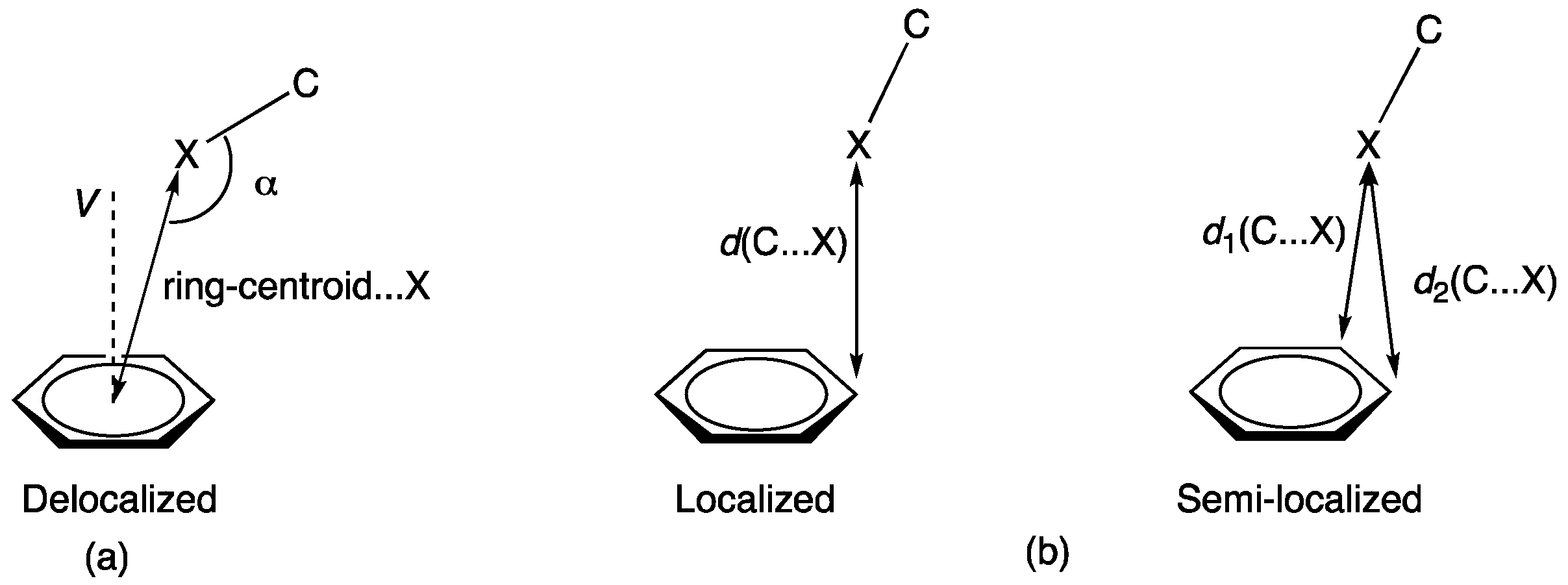
3. Results and Discussion
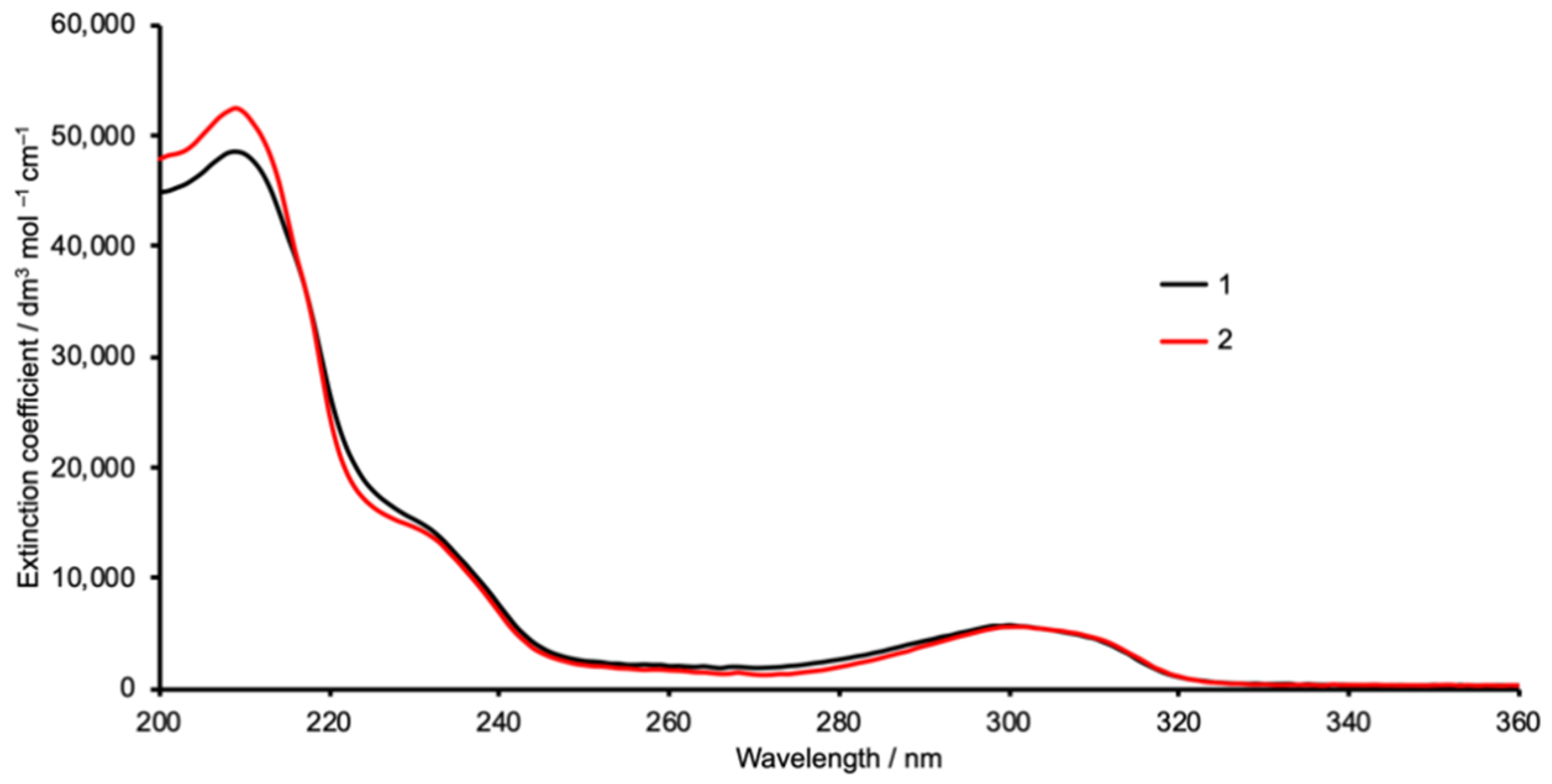
3.1. Synthesis and Characterization
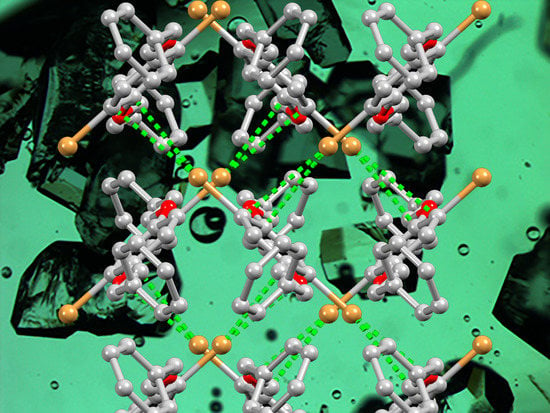
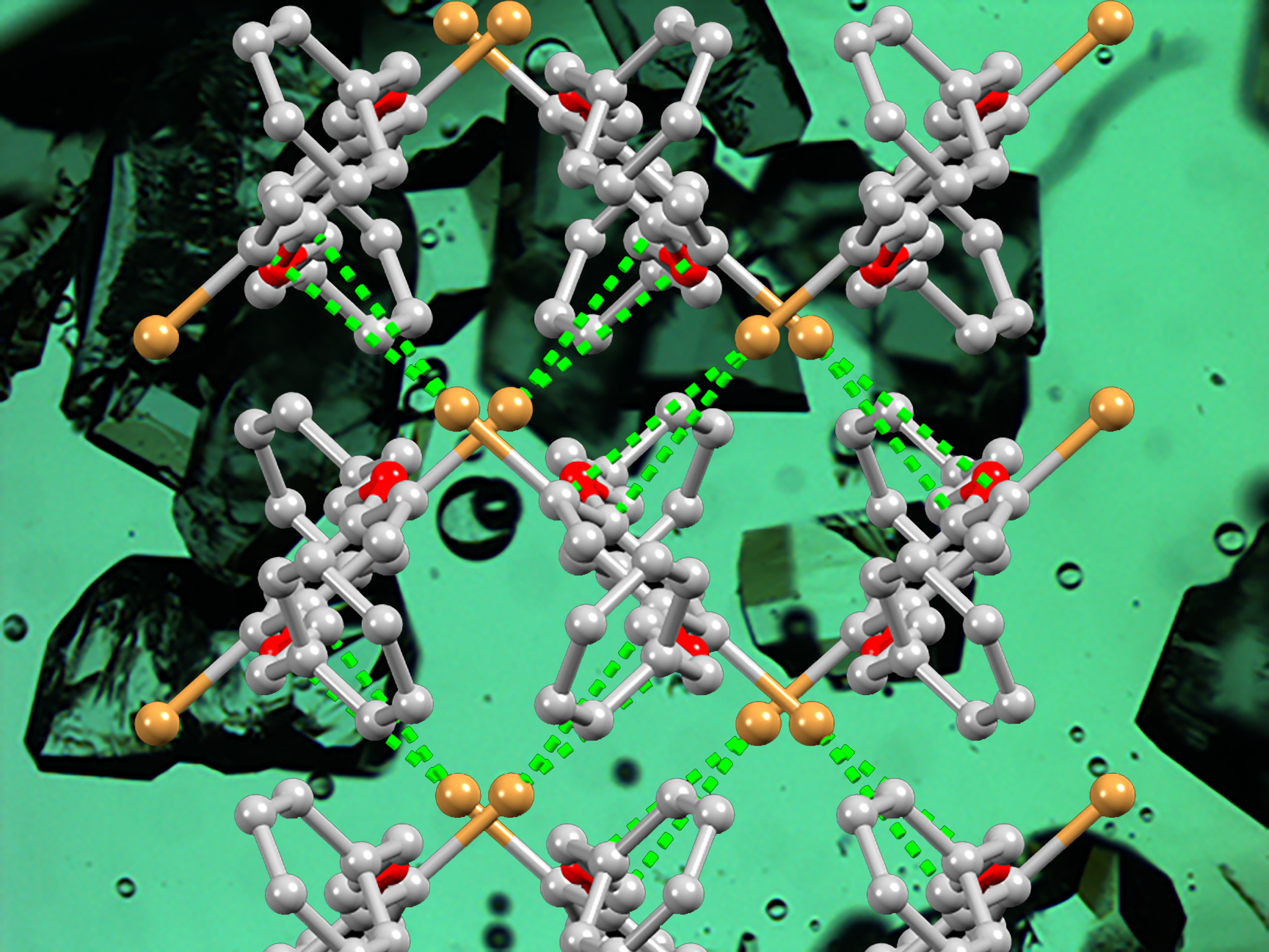
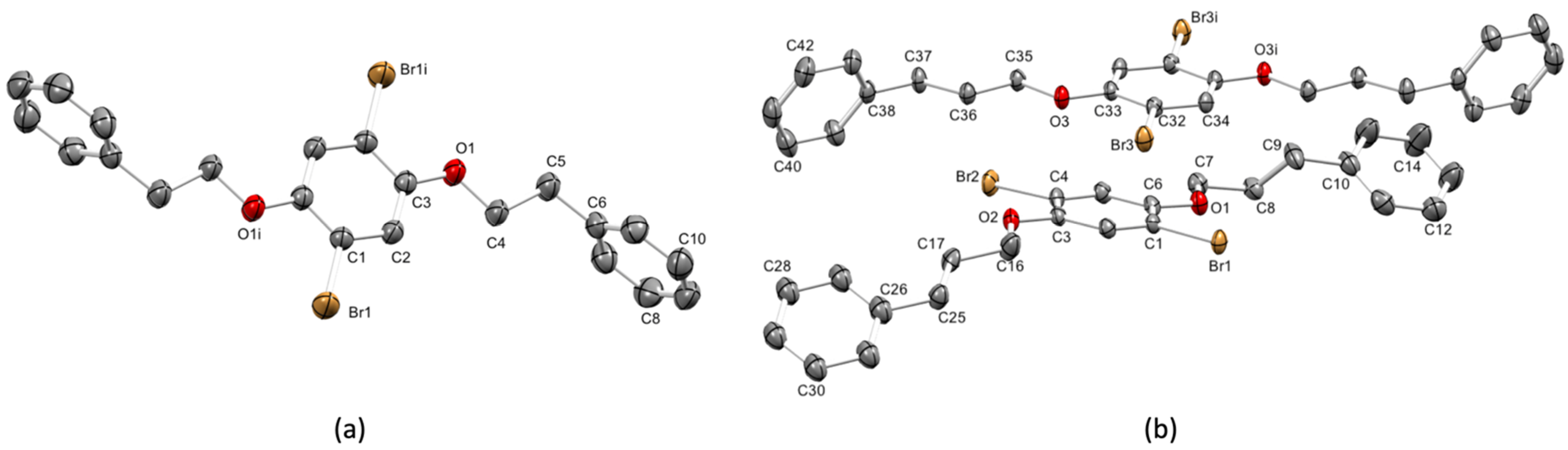
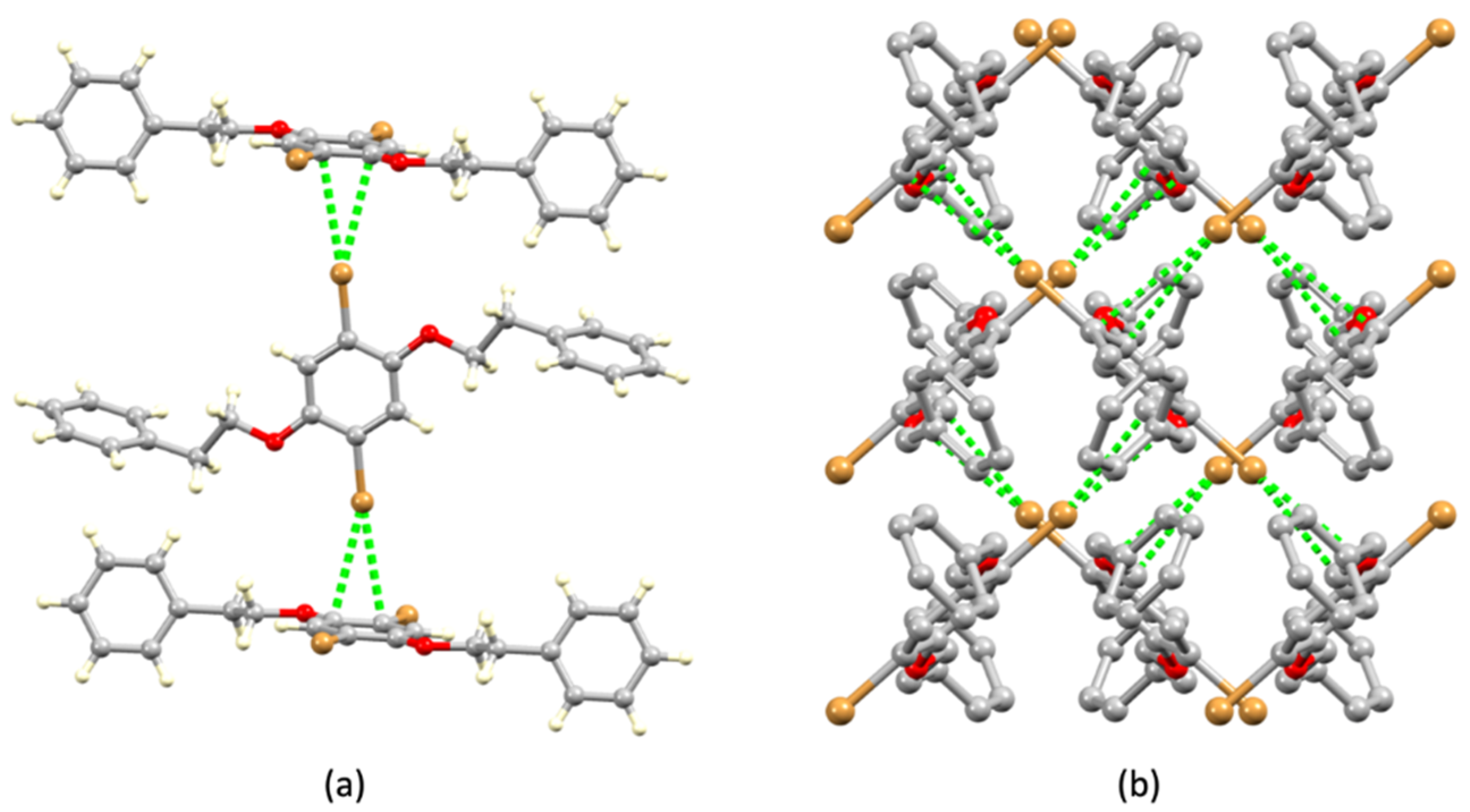
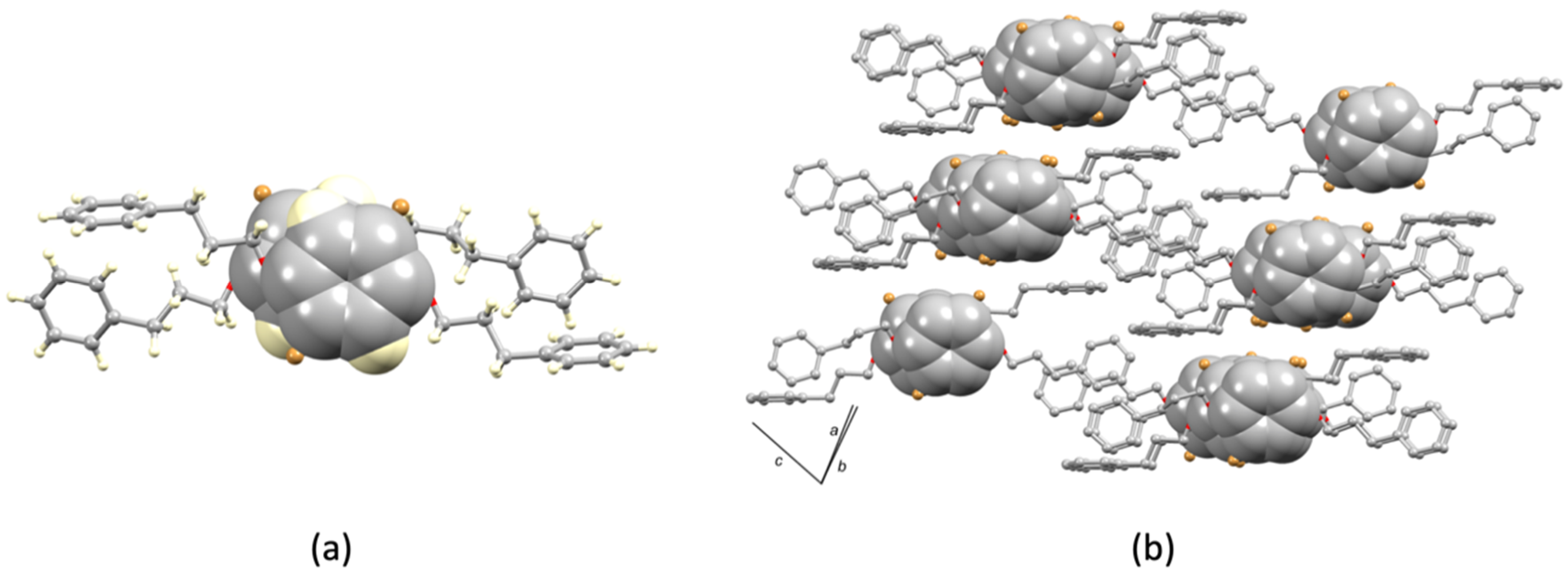
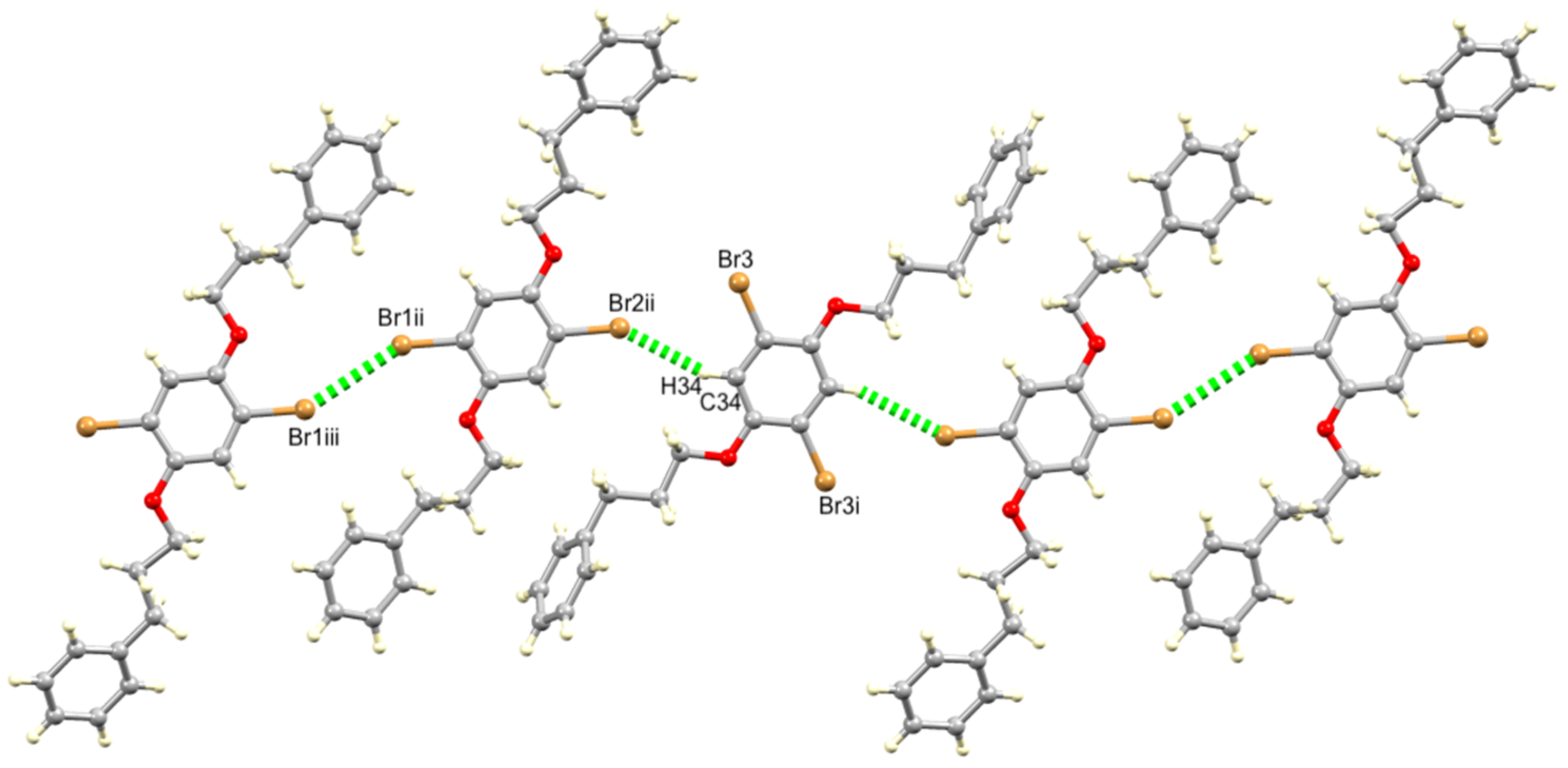
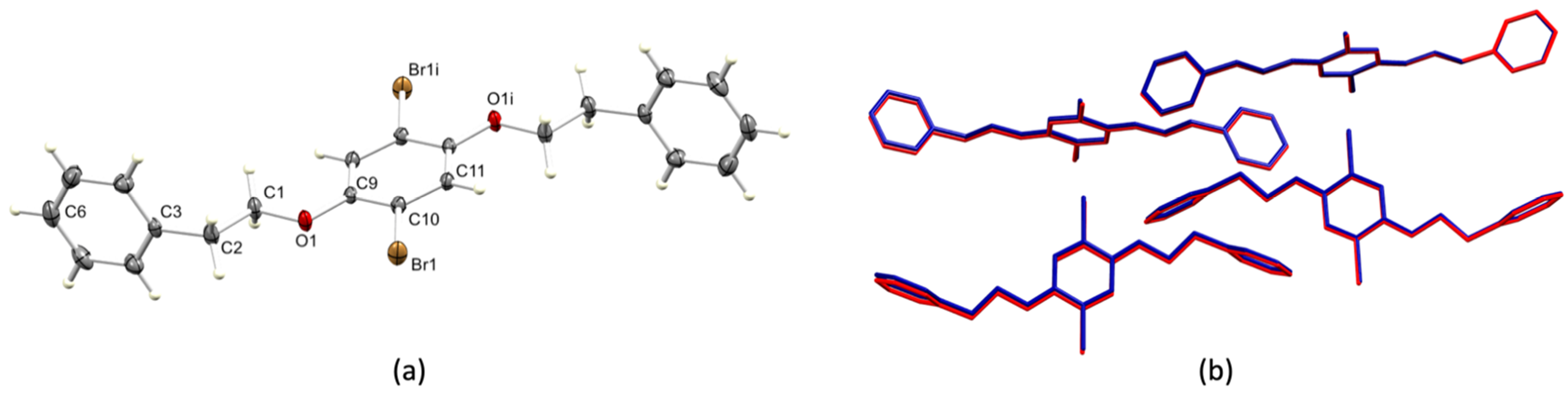
3.2. Single Crystal Structures of 1 (Polymorph I) and 2
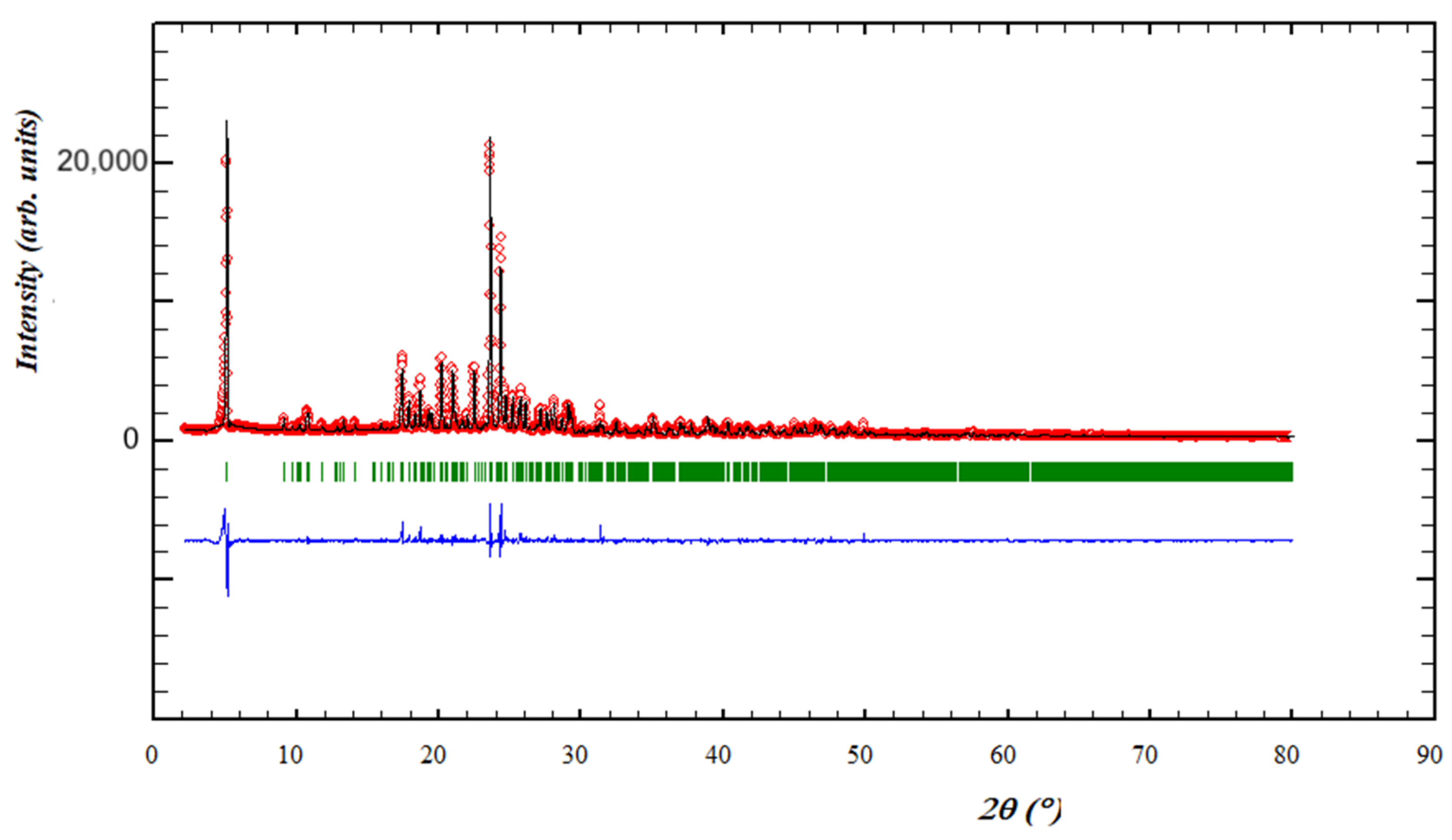
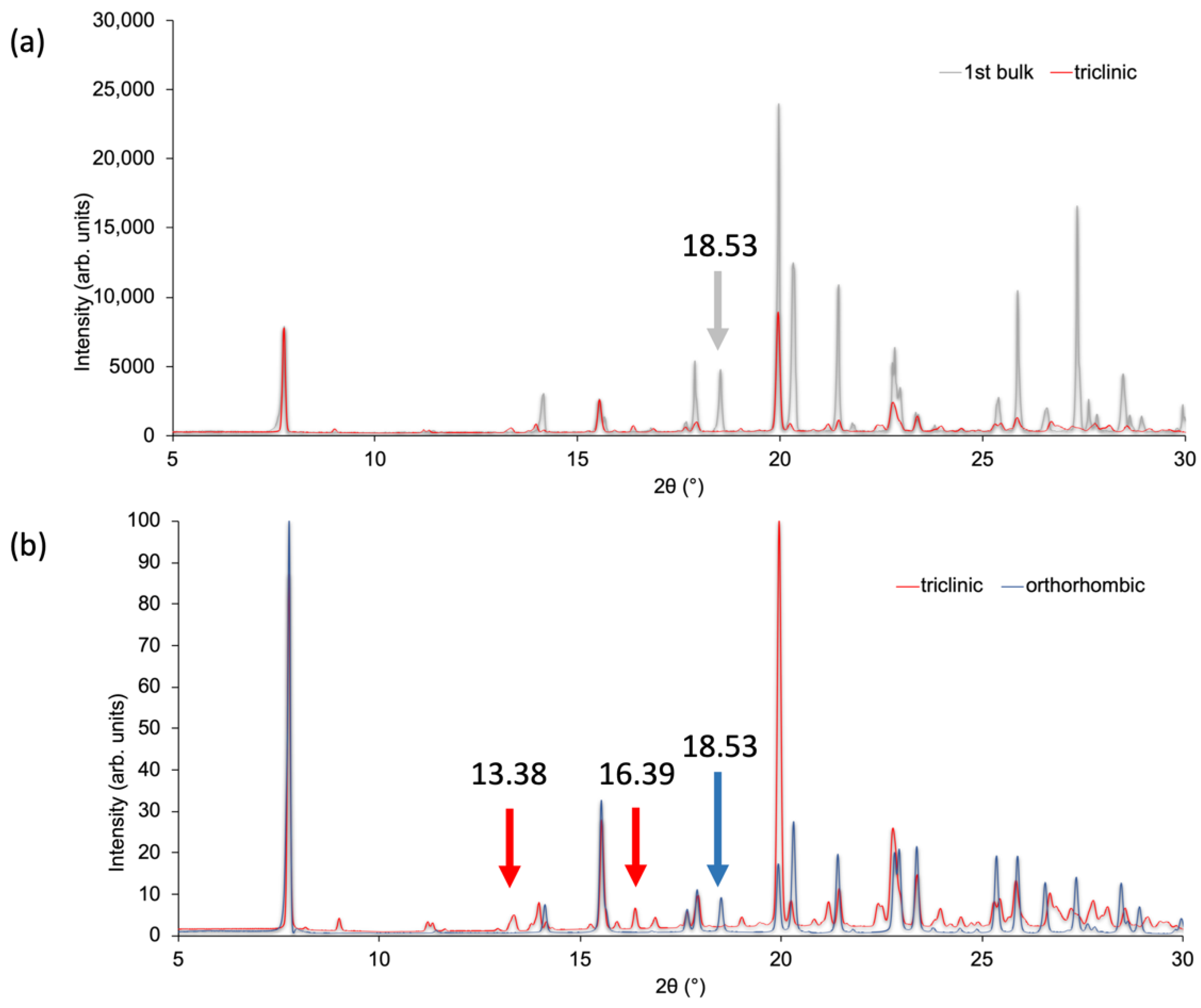
3.3. Powder XRD of Bulk Materials and a Second Polymorph of Compound 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Berger, G.; Fragville, P.; Meyer, F. Halogen bonding for molecular recognition: New developments in materials and biological sciences. Chem. Commun. 2020, 56, 4970–4981. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L.; Espallargas, G.M.; Libri, S. Combining metals with halogen bonds. CrystEngComm 2008, 10, 1712–1727. [Google Scholar] [CrossRef]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coord. Chem. Rev. 2020, 416, 213281. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.-C.C.; Rappé, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Info. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: A Paradigm in Supramolecular Chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Scholfield, M.R.; Vander Zanden, C.M.; Carter, M.; Ho, P.M. Halogen bonding (X-bonding): A biological perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef]
- Spilfogel, T.S.; Titi, H.M.; Friščić, T. Database Investigation of Halogen Bonding and Halogen...Halogen Interactions between Porphyrins: Emergence of Robust Supamolecular Motifs and Frameworks. Cryst. Growth Des. 2021, 21, 1810–1832. [Google Scholar] [CrossRef]
- Teyssandier, J.; Mali, K.S.; De Feyter, S. Halogen Bonding in Two-Dimensional Crystal Engineering. ChemistryOpen 2020, 9, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Pedireddi, V.R.; Reddy, D.S.; Goud, B.S.; Craig, D.C.; Rae, A.D.; Desiraju, G.R. The Nature of Halogen...Halogen Interactions and the Crystal Structure of 1,3,5,7-Tetraiodoadamantane. J. Chem. Soc. Perkin Trans. 1994, 2, 2353–2360. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular architectures sustained by delocalised C–I...π(arene) interactions in molecular crystals and the propensity of their formation. CrystEngComm 2021, 23, 904–928. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Schollmeyer, D.; Shishkin, O.V.; Rühl, T.; Vysotsky, M.O. OH–π and halogen–π interactions as driving forces in the crystal organisations of tri-bromo and tri-iodo trityl alcohols. CrystEngComm 2008, 10, 715–723. [Google Scholar] [CrossRef]
- Shishkin, O.V. Evaluation of true energy of halogen bonding in crystals of halogen derivatives of trityl alcohol. Chem. Phys. Lett. 2008, 458, 96–100. [Google Scholar] [CrossRef]
- Prasanna, M.D.; Row, T.N.G. C–halogen··· π interactions and their influence on molecular conformation and crystal packing: A database study. Cryst. Eng. 2000, 3, 135–154. [Google Scholar] [CrossRef]
- Saraogi, I.; Vijay, V.G.; Das, S.; Sekar, K.; Row, T.N.G. C–halogen…π interactions in proteins: A database study. Cryst. Eng. 2003, 6, 69–77. [Google Scholar] [CrossRef]
- Robertson, C.C.; Perutz, R.N.; Brammer, L.; Hunter, C.A. A solvent-resistent halogen bond. Chem. Sci. 2014, 5, 4179–4183. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. Superflip—A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Cryst. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
- Bruker. Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems; Bruker axs: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- LeBail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Physica B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodríguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction patterns analysis Materials Science Forum. In Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), Barcelona, Spain, 20–23 May 2000; pp. 118–123. [Google Scholar]
- Klein, Y.M.; Prescimone, A.; Neuburger, M.; Constable, E.C.; Housecroft, C.E. What a difference a tail makes: 2D→2D parallel interpenetration of sheets to interpenetrated nbo networks using ditopic-4,2′:6′,4″-terpyridine ligands. CrystEngComm 2017, 19, 2894–2902. [Google Scholar] [CrossRef]
- Moy, C.L.; Kaliappan, R.; McNeil, A.J. Aryl Trihydroxyborate Salts: Thermally Unstable Species with Unusual Gelation Abilities. J. Org. Chem. 2011, 76, 8501–8507. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
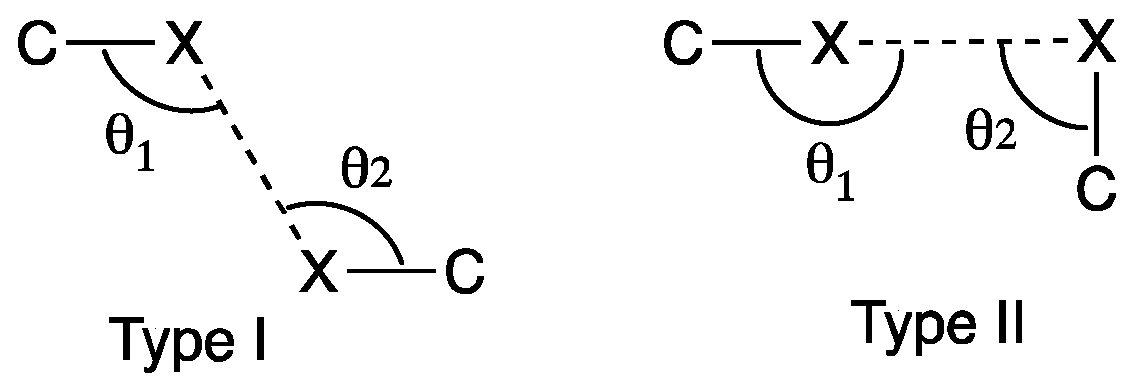{kind=link}
{kind=link}
{kind=link}
| Bond Parameter | 1 | 2 Molecule A | 2 Molecule B |
|---|---|---|---|
| C–Br/Å | 1.891(2) a | 1.889(3), 1.891(3) | 1.892(3) |
| Carene–O/Å | 1.361(3) a | 1.358(4), 1.363(4) | 1.365(4) |
| Calkyl–O/Å | 1.422(3) a | 1.434(4), 1.418(5) | 1.433(4) |
| Carene–O–Calkyl/° | 118.51(19) a | 117.7(3), 118.4(3) | 116.5(2) |
| Angle between planes of phenyl and central arene rings/° | 82.6 b | 29.3, 38.1 | 73.3 |
| Crystal | a/Å | b/Å | c/Å | α/Deg | β/Deg | γ/Deg |
|---|---|---|---|---|---|---|
| Polymorph I (P–1) a | 8.2806(13) | 10.6327(16) | 22.885(4) | 91.027(14) | 91.663(14) | 89.806(13) |
| Recrystallized bulk, crystal 1 b | 8.23 c | 10.55 | 22.84 | 89.88 | 89.81 | 89.93 |
| Recrystallized bulk, crystal 2 a | 8.22 c | 10.52 | 22.78 | 89.97 | 89.92 | 89.99 |
| Recrystallized bulk, crystal 3 a | 8.22 c | 10.52 | 22.76 | 89.99 | 89.98 | 90.00 |
| Recrystallized bulk, crystal 4 a | 8.21 c | 10.52 | 22.77 | 90.07 | 90.07 | 90.05 |
| Polymorph II (Pbca) a | 10.5176(7) | 8.2178(5) | 22.7853(14) | 90 | 90 | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manfroni, G.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. 1,4-Dibromo-2,5-bis(phenylalkoxy)benzene Derivatives: C–Br...π(arene) Versus C–H...Br and Br...Br Interactions in the Solid State. Crystals 2021, 11, 325. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11040325
Manfroni G, Prescimone A, Constable EC, Housecroft CE. 1,4-Dibromo-2,5-bis(phenylalkoxy)benzene Derivatives: C–Br...π(arene) Versus C–H...Br and Br...Br Interactions in the Solid State. Crystals. 2021; 11(4):325. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11040325
Chicago/Turabian StyleManfroni, Giacomo, Alessandro Prescimone, Edwin C. Constable, and Catherine E. Housecroft. 2021. "1,4-Dibromo-2,5-bis(phenylalkoxy)benzene Derivatives: C–Br...π(arene) Versus C–H...Br and Br...Br Interactions in the Solid State" Crystals 11, no. 4: 325. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11040325