
A Systematic Study of Compositionally Dependent Dielectric Tensors of SnSxSe1-x Alloys by Spectroscopic Ellipsometry


 , ,
, ,
Abstract
:1. Introduction
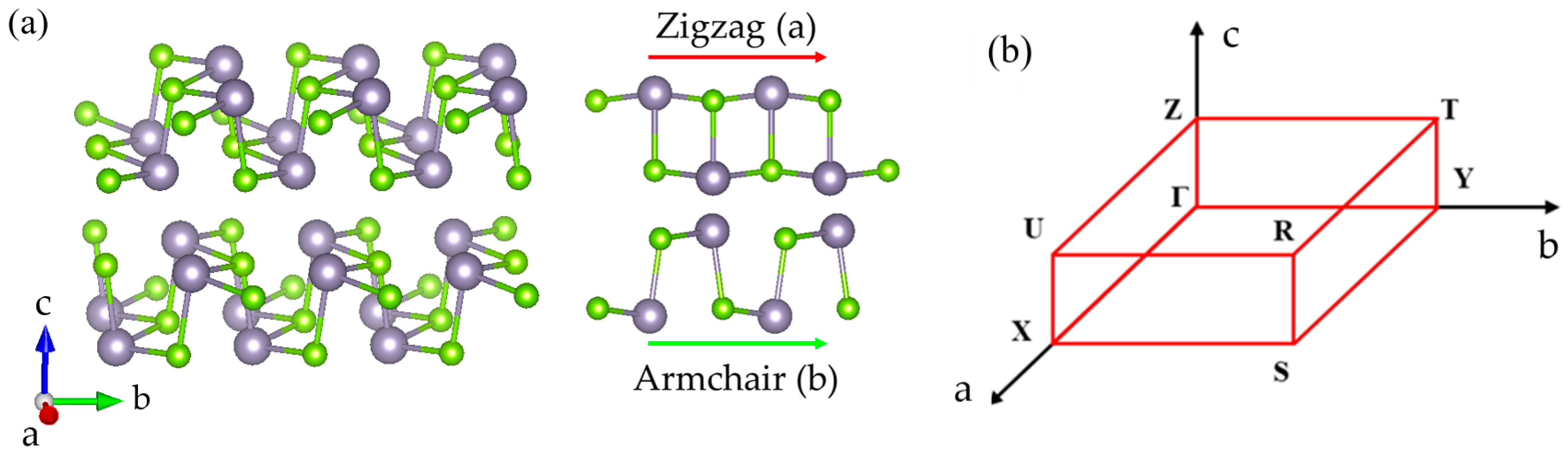
2. Experimental Methods and ab Initio Calculations
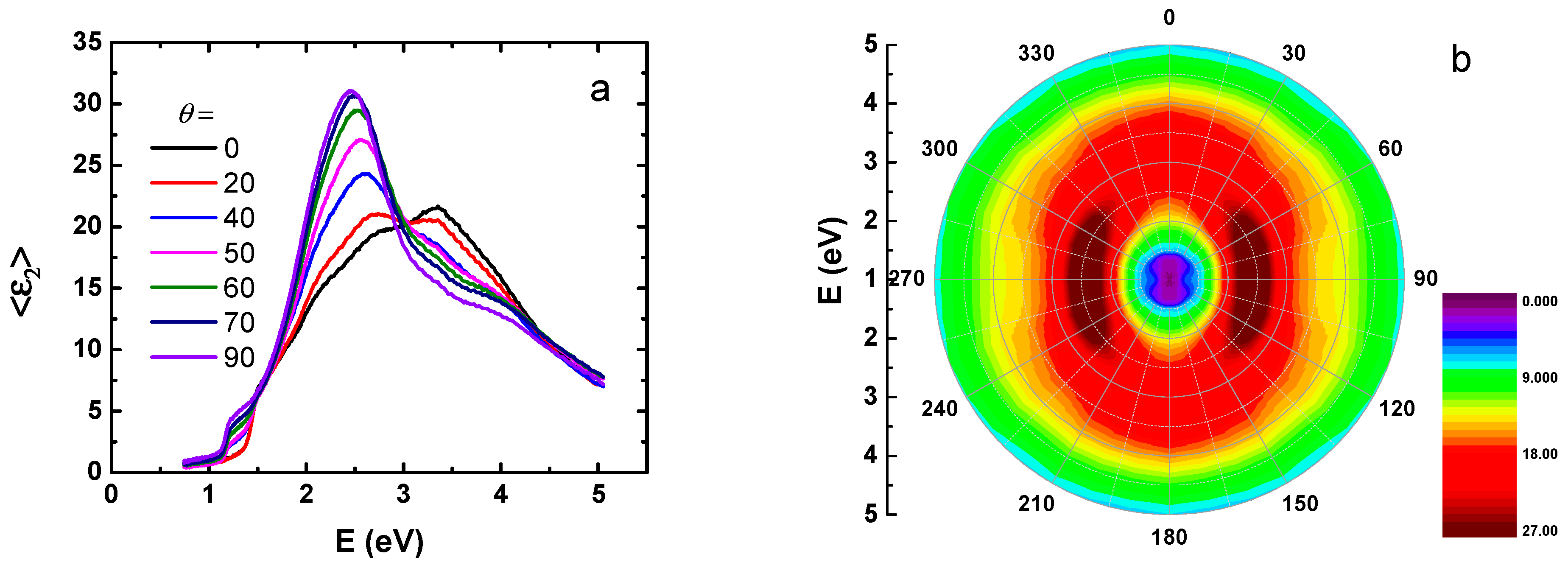
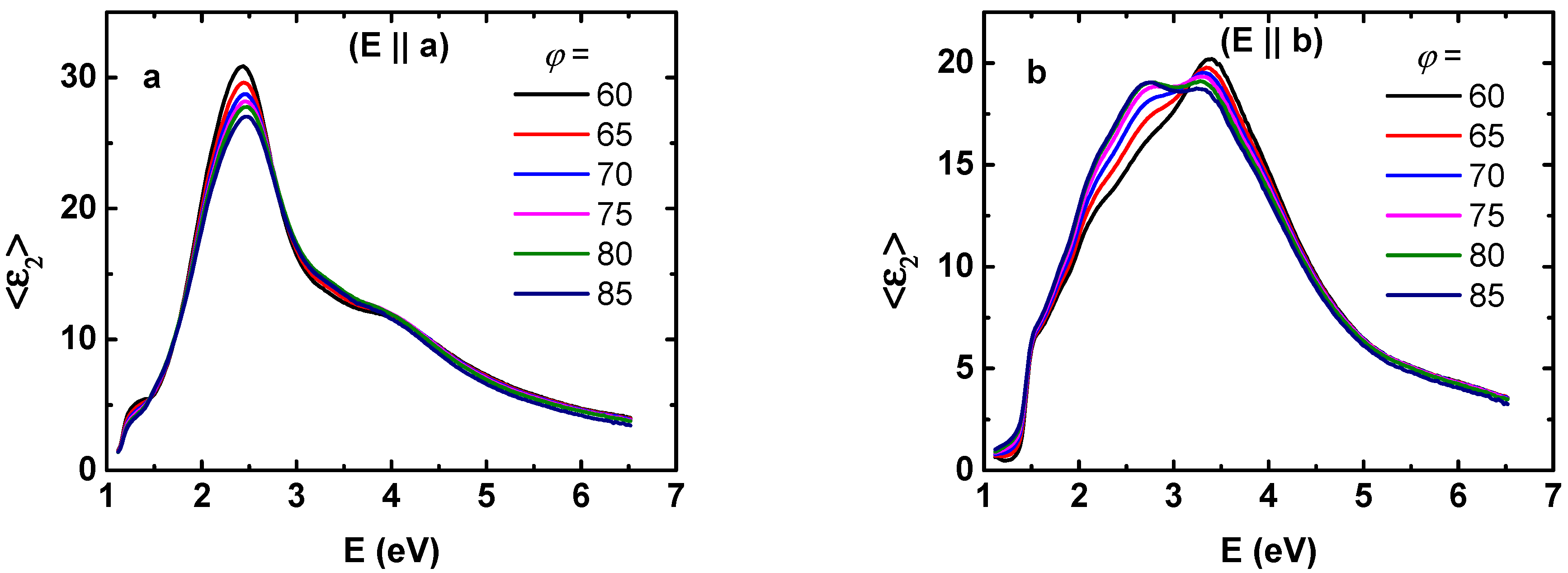
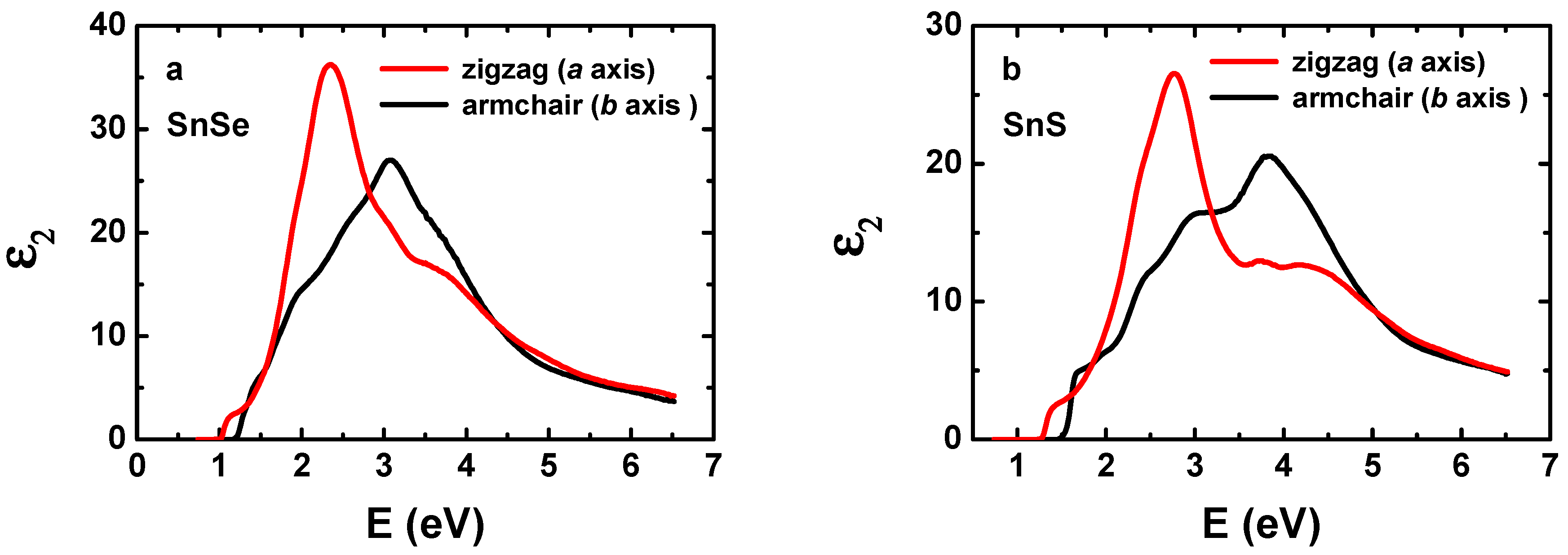
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ai, J.; Zhao, X.; Lei, Y.; Yang, S.; Xu, Q.; Lai, C.; Peng, C. Pomegranate-inspired SnS/ZnS@C heterostructural nanocubes towards high-performance sodium ion battery. Appl. Surf. Sci. 2019, 496, 143631. [Google Scholar] [CrossRef]
- Tang, H.; Li, Y.; Ye, H.; Hu, F.; Gao, C.; Tao, L.; Tu, T.; Gou, G.; Chen, X.; Fan, X.; et al. High-performance humidity sensor using Schottky-contacted SnS nanoflakes for noncontact healthcare monitoring. Nanotechnology 2020, 31, 055501. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.W.; Yoon, J.J.; Kim, Y.D.; Woo, D. Study of the Interaction Between Biomolecule Monolayers Using Total Internal Reflection Ellipsometry. J. Korean Phys. Soc. 2020, 58, 1031–1034. [Google Scholar] [CrossRef]
- Lefebvre, P.; Gil, B.; Allegre, J.; Mathieu, H.; Chen, Y. Nonparabolic behavior of GaSb-Alsb quantum wells under hydrostatic pressure. Phys. Rev. B 1987, 35, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Lukes, F.; Humlicek, J.; Schmidt, E. Electroreflectance and thermoreflectance spectra of SnS. Solid State Commun. 1983, 45, 445–448. [Google Scholar] [CrossRef]
- Hegde, S.S.; Murahari, P.; Fernandes, B.J.; Venkatesh, R.; Ramesh, K. Synthesis, thermal stability and structural transition of cubic SnS nanoparticles. J. Alloys Compd. 2020, 820, 153116. [Google Scholar] [CrossRef]
- Li, Z.; Sun, F.H.; Tang, H.; Dong, J.F.; Li, J.F. Enhanced thermoelectric properties of p-type SnS0.2Se0.8 solid solution doped with Ag. J. Alloys Compd. 2018, 745, 172–178. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, X.; Wu, D. Enhanced Thermoelectric Properties of Graphene/Cu2SnSe3 Composites. Crystals 2017, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Tang, C.; Zhang, C.; Li, G.; Zhao, Y.; Li, W.; Chen, G.; Yang, T. CuZnSn(SxSe1-x)4 Solar Cell Prepared by the Sol-Gel Method Following a Modified Three-Step Selenization Process. Crystals 2019, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Butt, F.K.; Haq, B.; Rehman, S.U.; Ahmed, S.R.; Cao, C.; AlFaifi, S. Investigation of thermoelectric properties of novel cubic phase SnSe: A promising material for thermoelectric applications. J. Alloys Compd. 2017, 715, 438–444. [Google Scholar] [CrossRef]
- Chu, F.; Zhang, Q.; Zhou, Z.; Hou, D.; Wang, L.; Jiang, W. Enhanced thermoelectric and mechanical properties of Na-doped polycrystalline SnSe thermoelectric materials via CNTs dispersion. J. Alloys Compd. 2018, 741, 756–764. [Google Scholar] [CrossRef]
- Guan, X.; Lu, P.; Wu, L.; Han, L.; Liu, G.; Song, Y.; Wang, S. Thermoelectric properties of SnSe compound. J. Alloys Compd. 2015, 643, 116–120. [Google Scholar] [CrossRef]
- Arepalli, V.K.; Kim, J. Effect of substrate temperature on the structural and optical properties of radio frequency sputtered tin sulfide thin films for solar cell application. Thin Solid Films 2018, 666, 34–39. [Google Scholar] [CrossRef]
- Cho, J.Y.; Shin, K.; Lee, H.S.; Neerugatti, K.E.; Heo, J. Influence of sodium diffusion from substrates on performance of SnS/CdS thin-film solar cells. J. Mater. Chem. A 2019, 7, 24186–24190. [Google Scholar] [CrossRef]
- Choi, H.; Lee, N.; Park, H.; Choi, Y.; Kim, K.; Choi, Y.; Kim, J.; Song, S.; Yuk, H.; Jeon, H. Development of a SnS film process for energy device applications. Appl. Sci. 2019, 9, 4606. [Google Scholar] [CrossRef] [Green Version]
- Chua, D.; Kim, S.B.; Sinsermsuksakul, P.; Gordon, R. Atomic layer deposition of energy band tunable tin germanium oxide electron transport layer for the SnS-based solar cells with 400 mV open-circuit voltage. Appl. Phys. Lett. 2019, 114, 213901. [Google Scholar] [CrossRef]
- Shown, I.; Samireddi, S.; Chang, Y.C.; Putikam, R.; Chang, P.H.; Sabbah, A.; Fu, F.Y.; Chen, W.F.; Wu, C.I.; Yu, T.Y.; et al. Carbon-doped SnS2 nanostructure as a high-efficiency solar fuel catalyst under visible light. Nat. Commun. 2018, 9, 169. [Google Scholar] [CrossRef] [Green Version]
- Chao, D.; Zhu, C.; Yang, P.; Xia, X.; Liu, J.; Wang, J.; Fan, X.; Savilov, S.V.; Lin, J.; Fan, H.J.; et al. Array of nanosheets render ultrafast and high-capacity Na-ion storage by tunable pseudocapacitance. Nat. Commun. 2016, 7, 12122. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.F.; Wang, W.; Fong, W.K.; Yu, Y.; Surya, C. Tin Compensation for the SnS Based Optoelectronic Devices. Sci. Rep. 2017, 7, 39704. [Google Scholar] [CrossRef] [Green Version]
- Bushell, Z.L.; Broderick, C.A.; Nattermann, L.; Joseph, R.; Keddie, J.L.; Rorison, J.M.; Volz, K.; Sweeney, S.J. Giant bowing of the band gap and spin-orbit splitting energy in GaP 1−xBix dilute bismide alloys. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bedair, T.M.; Cho, Y.; Kim, T.J.; Kim, Y.D.; Park, B.J.; Joung, Y.K.; Han, D.K. Reinforcement of interfacial adhesion of a coated polymer layer on a cobalt-chromium surface for drug-eluting stents. Langmuir 2014, 30, 8020–8028. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, J.; Liu, Y.; Peng, Y.; Maraj, M.; Peng, B.; Wang, Y.; Sun, W. Effects of Thermal Annealing on Optical Properties of Be-Implanted GaN Thin Films by Spectroscopic Ellipsometry. Crystals 2020, 10, 439. [Google Scholar] [CrossRef]
- Zalamai, V.V.; Rusu, E.V.; Syrbu, N.N.; Tiron, A.V. Optical properties and electronic band structure of SnS single crystals. Phys. B Condens. Matter 2019, 575, 411712. [Google Scholar] [CrossRef]
- Rehman, S.U.; Butt, F.K.; Hayat, F.; Haq, B.; Tariq, Z.; Aleem, F.; Li, C. An insight into a novel cubic phase SnSe for prospective applications in optoelectronics and clean energy devices. J. Alloys Compd. 2018, 733, 22–32. [Google Scholar] [CrossRef]
- Cao, M.; Wu, C.; Yao, K.; Jing, J.; Huang, J.; Cao, M.; Zhang, J.; Lai, J.; Ali, O.; Wang, L.; et al. Chemical bath deposition of single crystal SnS nanobelts on glass substrates. Mater. Res. Bull. 2018, 104, 244–249. [Google Scholar] [CrossRef]
- Rana, C.; Saha, S. Structural, optical and electrical characterization of SnS nanomaterials grown at different temperatures. J. Mater. Sci. Mater. Electron. 2019, 30, 21160–21169. [Google Scholar] [CrossRef]
- Mirabella, F.; Ghijsen, J.; Johnson, R.L.; Golacki, Z.; Orlowski, B.A. Photoemission study of Sn1-xMnxSe2. J. Alloys Compd. 2001, 328, 166–170. [Google Scholar] [CrossRef]
- Raadik, T.; Grossberg, M.; Raudoja, J.; Traksmaa, R.; Krustok, J. Temperature-dependent photoreflectance of SnS crystals. J. Phys. Chem. Solids 2019, 74, 1683–1685. [Google Scholar] [CrossRef]
- Losurdo, M.; Bergmair, M.; Bruno, G.; Cattelan, D.; Cobet, C.; De Martino, A.; Fleischer, K.; Dohcevic-Mitrovic, Z.; Esser, N.; Galliet, M.; et al. Spectroscopic ellipsometry and polarimetry for materials and systems analysis at the nanometer scale: State-of-the-art, potential, and perspectives. J. Nanopart. Res. 2009, 11, 1521–1554. [Google Scholar] [CrossRef] [Green Version]
- Aspnes, D.E.; Studna, A.A. Dielectric functions and optical parameters of Si, Ge, GaP, GaAs, GaSb, InP, InAs, and InSb from 1.5 to 6.0 eV. Phys. Rev. B 1983, 27, 985. [Google Scholar] [CrossRef]
- Logothetidis, S.; Via, L.; Cardona, M. Temperature dependence of the dielectric function and the interband critical points of InSb. Phys. Rev. B 1985, 31, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Logothetidis, S.; Polatoglou, H.M. Ellipsometric studies of the dielectric function of SnSe and a simple model of the electronic structure and the bonds of the orthorhombic IV-VI compounds. Phys. Rev. B 1987, 36, 7491. [Google Scholar] [CrossRef] [PubMed]
- Banai, R.E.; Burton, L.A.; Choi, S.G.; Hofherr, F.; Sorgenfrei, T.; Walsh, A.; To, B.; Cröll, A.; Brownson, J.R.S. Ellipsometric characterization and density-functional theory analysis of anisotropic optical properties of single-crystal α-SnS. J. Appl. Phys. 2014, 116, 013511. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Le, V.L.; Nguyen, T.M.H.; Kim, T.J.; Nguyen, X.A.; Kim, B.; Kim, K.; Lee, W.; Cho, S.; Kim, Y.D. Temperature Dependence of the Dielectric Function and Critical Points of α-SnS from 26 to 350 K. Sci. Rep. 2020, 10, 18396. [Google Scholar] [CrossRef] [PubMed]
- Le, V.L.; Cuong, D.D.; Nguyen, X.A.; Nguyen, H.T.; Nguyen, T.M.H.; Cho, S.; Hong, S.C.; Rhim, S.H.; Kim, T.J.; Kim, Y.D. Anisotropic Behavior of Excitons in Single Crystal α-SnS. AIP Adv. 2020, 10, 105003. [Google Scholar] [CrossRef]
- Ly, T.T.; Duvjir, G.; Min, T.; Byun, J.; Kim, T.; Saad, M.M.; Hai, N.T.M.; Cho, S.; Lee, J.; Kim, J. Atomistic study of the alloying behavior of crystalline SnSe1-xSx. Phys. Chem. Chem. Phys. 2017, 19, 21648–21654. [Google Scholar] [CrossRef]
- Nguyen, T.M.H.; Nguyen, Q.V.; Duong, A.T.; Cho, S. Growth and electrical properties of SnS1-xSex (0 ≤ x ≤ 1) single crystals grown using the temperature gradient method. J. Korean Phys. Soc. 2021. [Google Scholar] [CrossRef]
- Aspnes, D.E. Approximate solution of ellipsometric equations for optically biaxial crystals. J. Opt. Soc. Am. 1980, 10, 1275–1277. [Google Scholar] [CrossRef]
- Logothetidis, S.; Via, L.; Cardona, M. Ellipsometric study of interband transitions in orthorhombic GeS. Phys. Rev. B 1985, 31, 2180. [Google Scholar] [CrossRef]
- Schubert, M.; Hofmann, T.; Herzinger, C.M.; Dollase, W. Generalized ellipsometry for orthorhombic, absorbing materials: Dielectric functions, phonon modes and band-to-band transitions of Sb2S3. Thin Solid Film. 2004, 455–456, 619–623. [Google Scholar] [CrossRef]
- Jellison, G.E., Jr.; McGuire, M.A.; Boatner, L.A.; Budai, J.D.; Specht, E.D.; Singh, D.J. Spectroscopic dielectric tensor of monoclinic crystals: CdWO4. Phys. Rev. B 2011, 84, 195439. [Google Scholar] [CrossRef]
- Schmidt, D.; Booso, B.; Hofmann, T.; Schubert, E.; Sarangan, A.; Schubert, M. Generalized ellipsometry for monoclinic absorbing materials: Determination of optical constants of Cr columnar thin films. Opt. Lett. 2009, 34, 992. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.K.S.; Gong, Y.; Yang, P.; Ng, C.M. Characterization of biaxial stressed silicon by spectroscopic ellipsometry and synchrotron x-ray scattering. Semicond. Sci. Technol. 2007, 22, 1232. [Google Scholar] [CrossRef]
- Bruggeman, D.A.G. Berechnung verschiedener physikalischer Konstanten von heterogenen substanzen. Ann. Phys. 1953, 416, 636. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D.; Johnson, E.R. A simple effective potential for exchange. J. Chem. Phys. 2006, 124, 221101. [Google Scholar] [CrossRef]
- Tran, F.; Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 2009, 102, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekhar, H.R.; Humphreys, R.G.; Zwick, U.; Cardona, M. Infrared and Raman spectra of the IV–VI compounds SnS and SnSe. Phys. Rev. B 1977, 15, 2177–2183. [Google Scholar] [CrossRef]
- Luo, W.; Ismail-Beigi, S.; Cohen, M.L.; Louie, S.G. Quasiparticle band structure of ZnS and ZnSe. Phys. Rev. B 2002, 66, 195215. [Google Scholar] [CrossRef]
- Waler, J.P.; Cqhen, M.L. Calculation of the Refiectivity, Modulated Refiectivity, and Band Structure of GaAs, GaP, ZnSe, and ZnS. Phys. Rev. 1969, 183, 763. [Google Scholar] [CrossRef]
- Erbarut, E. Optical response functions of ZnS, ZnSe, ZnTe by the LOM method. Solid State Commun. 2003, 127, 515–519. [Google Scholar] [CrossRef]
- Gomes, L.C.; Trevisanutto, P.E.; Carvalho, A.; Rodin, A.S.; Neto, A.H.C. Strongly bound Mott-Wannier excitons in GeS and GeSe monolayers. Phys. Rev. B 2016, 94, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Eymard, R.; Otto, A. Optical anil electron-energy-loss spectroscopy of GeS, GeSe, SnS, anil SnSe single crystals. Phys. Rev. B 1977, 16, 541–559. [Google Scholar] [CrossRef]
- Cardona, M. Modulation Spectroscopy, 1st ed.; Academic Press: Cambridge, MA, USA, 1969; Volume 11. [Google Scholar]
- Le, V.L.; Kim, T.J.; Kim, Y.D.; Aspnes, D.E. Combined interpolation, scale change, and noise reduction in spectral analysis. J. Vac. Sci. Technol. B 2019, 37, 052903. [Google Scholar] [CrossRef]
- Le, V.L.; Kim, T.J.; Kim, Y.D.; Aspnes, D.E. External removal of endpoint-discontinuity artifacts in the reciprocal-space analysis of spectra. Curr. Appl. Phys. 2020, 20, 232–236. [Google Scholar] [CrossRef]
- Lautenschlager, P.; Garriga, M.; Vina, L.; Cardona, M. Temperature dependence of the dielectric function and interband critical points in silicon. Phys. Rev. B 1987, 36, 4821. [Google Scholar] [CrossRef] [PubMed]
- Lautenschlager, P.; Garriga, M.; Cardona, M. Temperature dependence of the interband critical-point parameters of InP. Phys. Rev. B 1987, 36, 4813. [Google Scholar] [CrossRef]
- Zollner, S.; Garriga, M.; Kircher, J.; Humlicek, J.; Cardona, M.; Neuhold, G. Temperature dependence of the dielectric function and the interband critical-point parameters of GaP. Phys. Rev. B 1993, 48, 7915. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yoon, J.J.; Kim, T.J.; Kim, Y.D.; Lee, E.H.; Bae, M.H.; Song, J.D.; Choi, W.J.; Liang, C.T.; Chang, Y.C. Optical properties of AlAsxSb1-x alloys determined by in situ ellipsometry. Appl. Phys. Lett. 2013, 103, 011901. [Google Scholar] [CrossRef]
- Davydov, Y.V.; Goncharuk, I.N.; Smirnov, A.N.; Nikolaev, A.E.; Lundin, W.V.; Usikov, A.S.; Klochikhin, A.A.; Aderhold, J.; Graul, J.; Semchinova, O.; et al. Composition dependence of optical phonon energies and Raman line broadening in hexagonal (formula presented) alloys. Phys. Rev. B 2002, 65, 125203. [Google Scholar] [CrossRef]
- Lu, X.; Beaton, D.A.; Lewis, R.B.; Tiedje, T.; Zhang, Y. Composition dependence of photoluminescence of GaAs1-xBix alloys. Appl. Phys. Lett. 2009, 95, 129–132. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
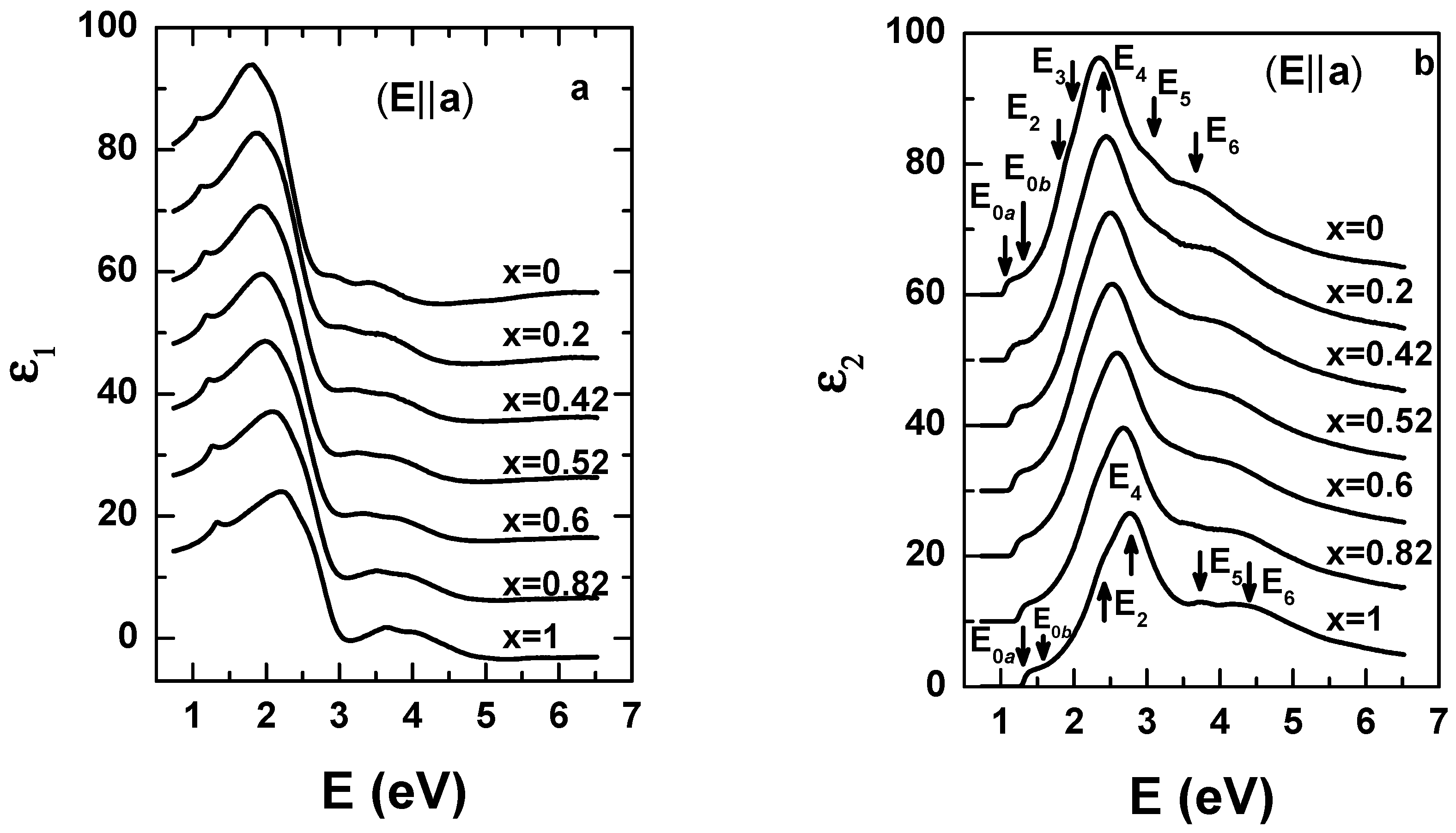{kind=link}
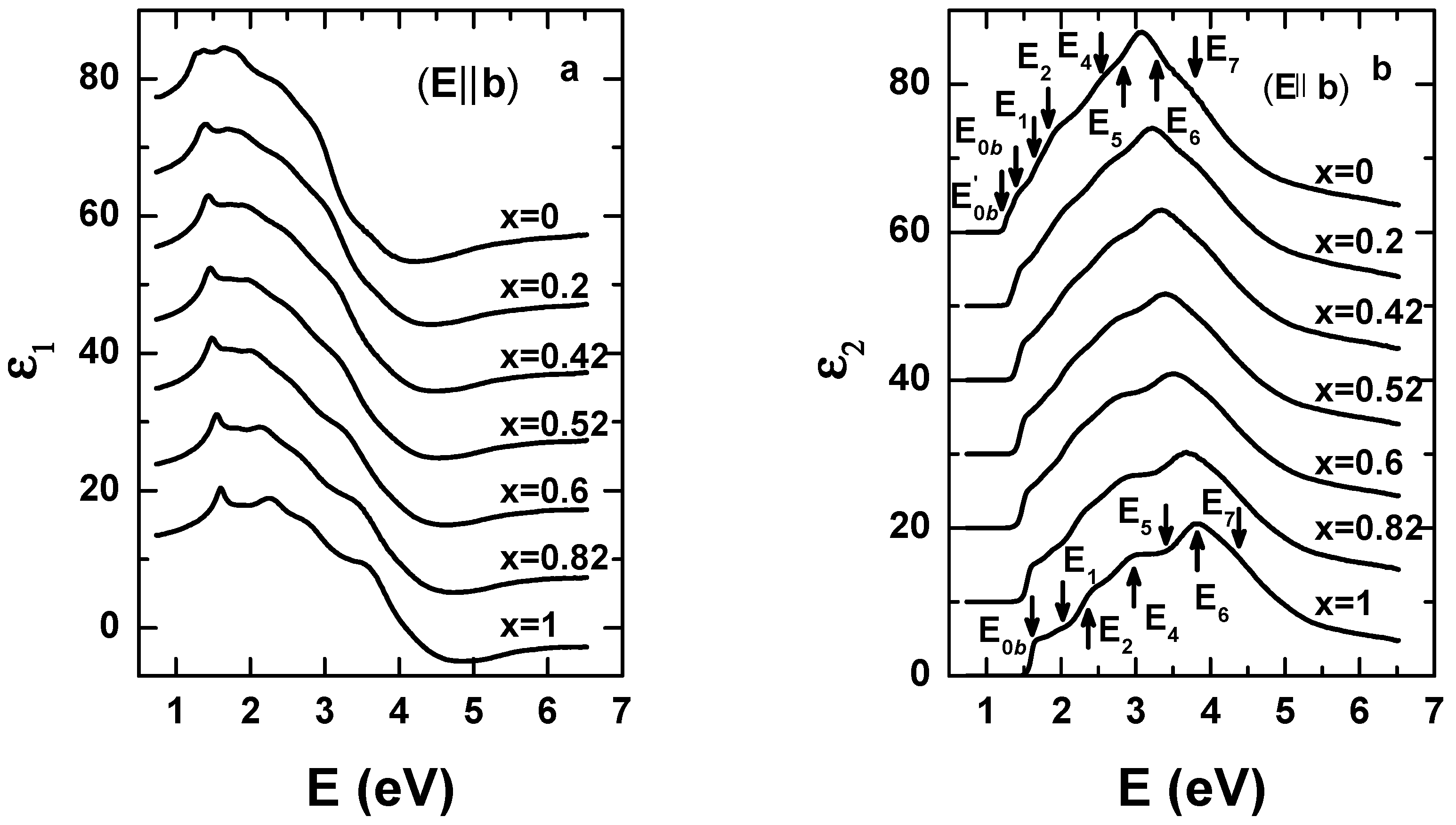{kind=link}
{kind=link}
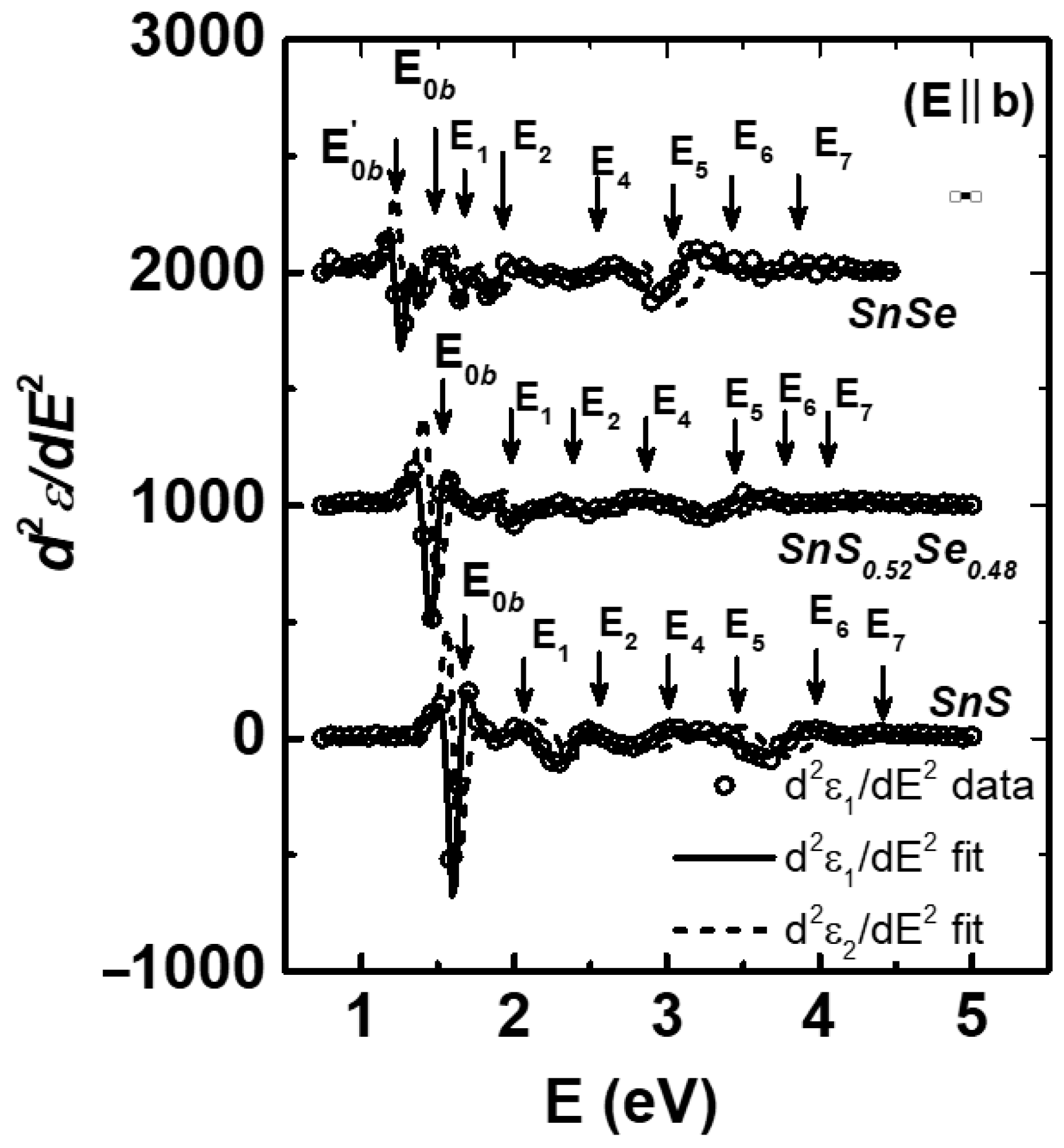{kind=link}
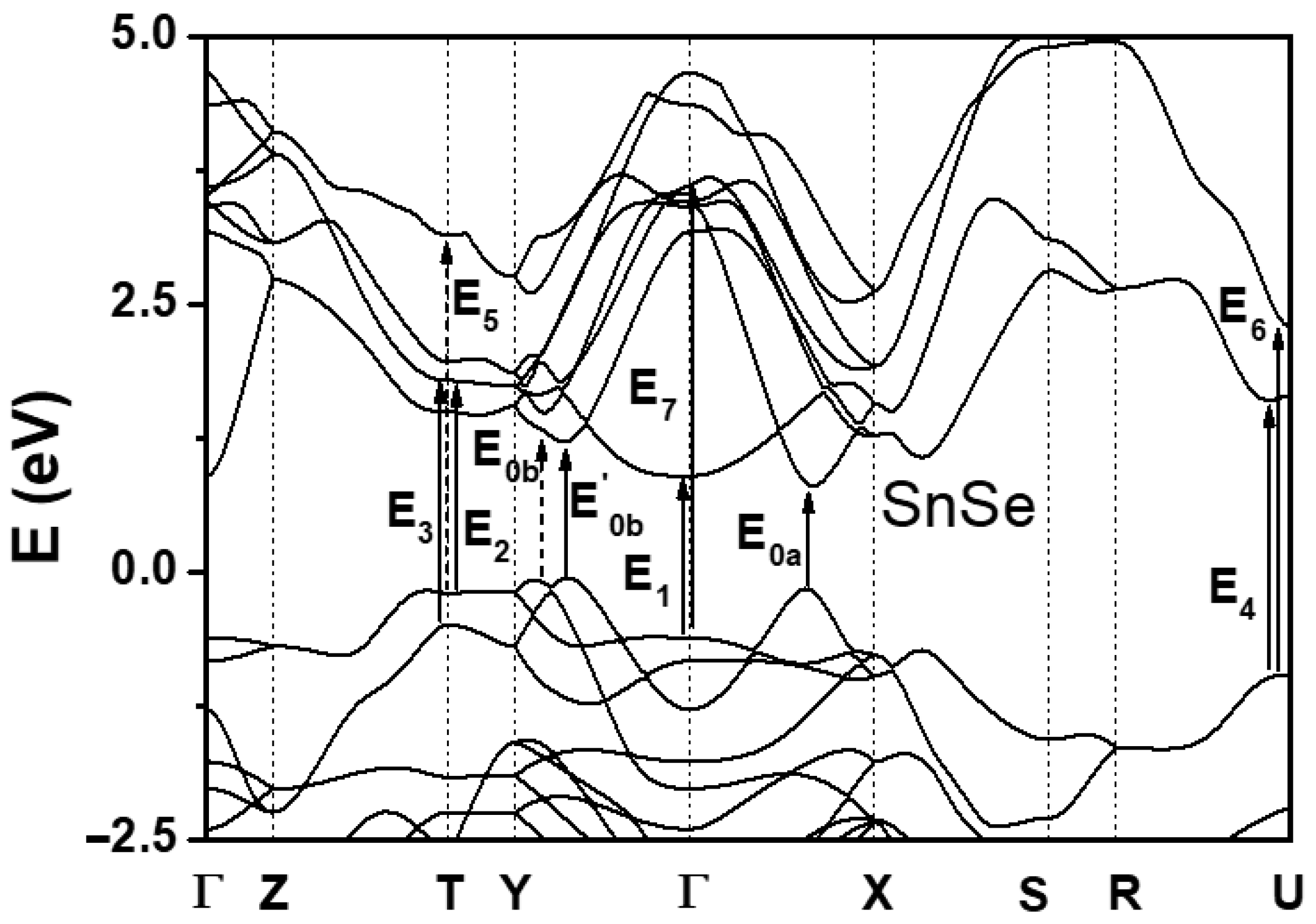{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CP | SnSe | ||
|---|---|---|---|
| Theory | a-Axis | b-Axis | |
| 0.97 a— | 1.06 a | ||
| 1.20 a— | 1.21 a | ||
| 1.37 a— | 1.31 a, 1.26 d | 1.37 a, 1.35 d | |
| 1.49 a— | 1.65 a, 1.64 d | ||
| 1.90 a— | 1.90 a, 1.81 d | 1.85 a, 1.86 d | |
| 2.24 a— | 2.23 a, 2.14 d | ||
| 2.62 a— | 2.33 a, 2.31 d | 2.56 a, 2.43 d | |
| 3.29 a— | 2.99 a, 3.06 d | ||
| 3.32 a— | 3.02 a, 2.91 d | 3.25 a | |
| 4.20 a— | 3.41 a, 3.83 d | 3.68 a, 3.58 d | |
| CP | SnS | ||
|---|---|---|---|
| Theory | a-Axis | b-Axis | |
| 1.33 a— | 1.33 a, 1.31 b | ||
| 1.72 a— | 1.65 a, 1.60 b, 1.59 c | 1.60 a, 1.59 b | |
| 1.87 a— | 1.90 a, 1.98 b, 1.91 c | ||
| 2.40 a— | 2.36 a, 2.34 b, 2.35 c | 2.32 a, 2.28 b, 2.36 c | |
| 3.08 a— | 2.83 a, 2.76 b, 2.80 c | 2.92 a, 2.98 b, 2.82 c | |
| 3.80 a— | 3.44 a, 3.29 b, 3.47 c | ||
| 4.04 a— | 3.64 a, 3.70 b, 3.68 c | 3.88 a, 3.71 b, 3.70 c | |
| 4.55 a— | 3.98 a, 4.06 b | 4.24 a, 4.30 b, 4.41 c | |
| CP | c | b | a |
|---|---|---|---|
| 1.0591 | 0.2208 | 0.0443 | |
| 1.3135 | 0.2404 | 0.0943 | |
| 1.9049 | 0.0641 | 0.3997 | |
| 2.2326 | 0.3654 | 0 | |
| 2.3257 | 0.3055 | 0.1987 | |
| 3.0126 | 0.1776 | 0.4509 | |
| 3.4121 | 0.2852 | 0.2863 |
| CP | c | b | a |
|---|---|---|---|
| 1.234 | 0.2939 | 0 | |
| 1.3716 | 0.1006 | 0.1341 | |
| 1.6577 | 0.0555 | 0.1865 | |
| 1.8535 | 0.2844 | 0.1815 | |
| 2.5555 | 0.1627 | 0.2013 | |
| 2.9756 | 0.7565 | −0.2862 | |
| 3.2598 | 0.3873 | 0.2298 | |
| 3.6728 | 0.3259 | 0.2421 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, X.A.; Nguyen, T.M.H.; Kim, T.J.; Le, L.V.; Nguyen, T.H.; Kim, B.; Kim, K.; Lee, W.; Cho, S.; Kim, Y.D. A Systematic Study of Compositionally Dependent Dielectric Tensors of SnSxSe1-x Alloys by Spectroscopic Ellipsometry. Crystals 2021, 11, 548. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11050548
Nguyen XA, Nguyen TMH, Kim TJ, Le LV, Nguyen TH, Kim B, Kim K, Lee W, Cho S, Kim YD. A Systematic Study of Compositionally Dependent Dielectric Tensors of SnSxSe1-x Alloys by Spectroscopic Ellipsometry. Crystals. 2021; 11(5):548. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11050548
Chicago/Turabian StyleNguyen, Xuan Au, Thi Minh Hai Nguyen, Tae Jung Kim, Long Van Le, Tung Hoang Nguyen, Bogyu Kim, Kyujin Kim, Wonjun Lee, Sunglae Cho, and Young Dong Kim. 2021. "A Systematic Study of Compositionally Dependent Dielectric Tensors of SnSxSe1-x Alloys by Spectroscopic Ellipsometry" Crystals 11, no. 5: 548. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11050548