
4-Phenylnaphtho[2,3-c]furan-1(3H)-one, 9-Phenylnaphtho[2,3-c]furan-1(3H)-one and 3a,4-Dihydro-9-phenylnaphtho[2,3-c]furan-1(3H)-one Crystal Structures


Abstract
:1. Introduction
2. Materials and Instrumentation
2.1. Synthesis
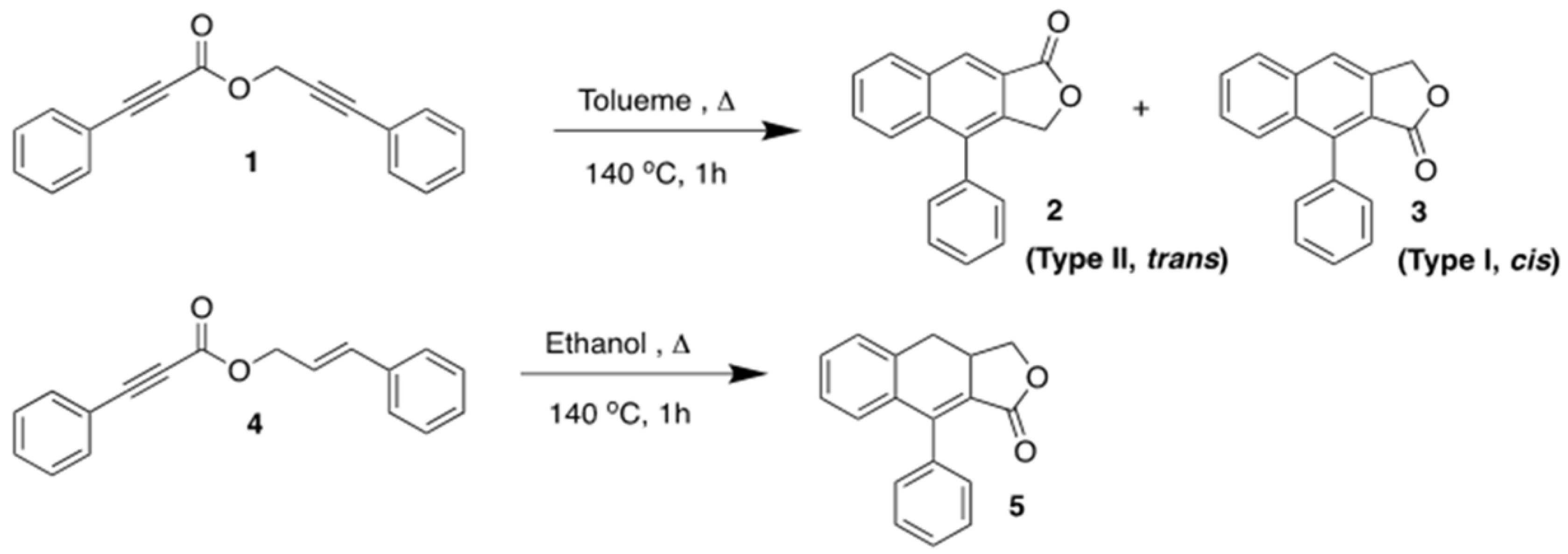
2.1.1. Synthesis of 3-Phenylprop-2-yn-1-yl 3-phenylpropiolate (1) and Trans-cinnamyl 3-phenylpropiolate (4)
2.1.2. Synthesis of 4-Phenylnaphtho [2,3-c]furan-1(3H)-one (2) and 9-Phenylnaphtho [2,3-c]furan-1(3H)-one (3)
2.1.3. Synthesis of 3a,4-Dihydro-9-phenylnaphtho[2,3-c]furan-1(3H)-one (5)
2.2. X-ray Analysis
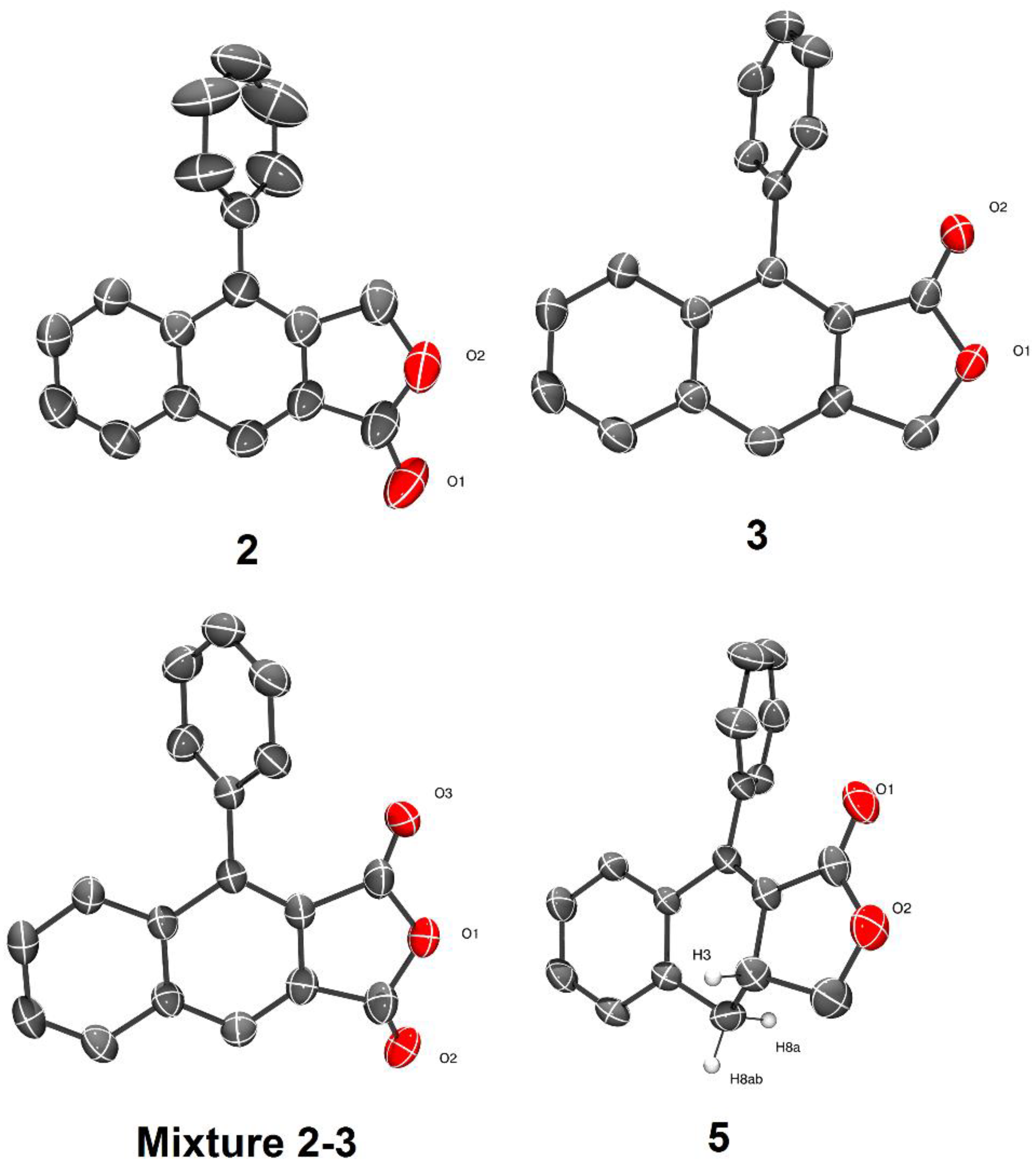
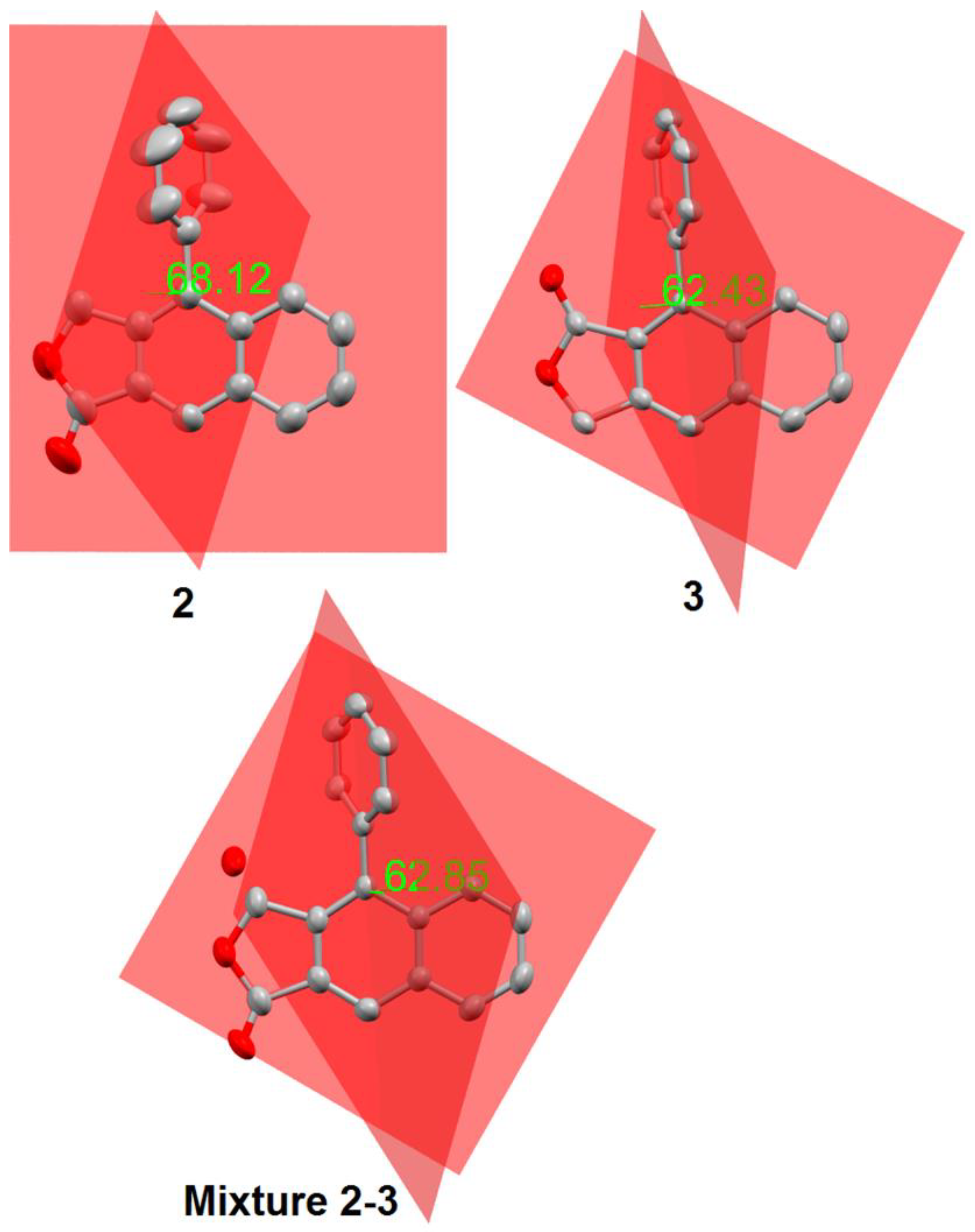
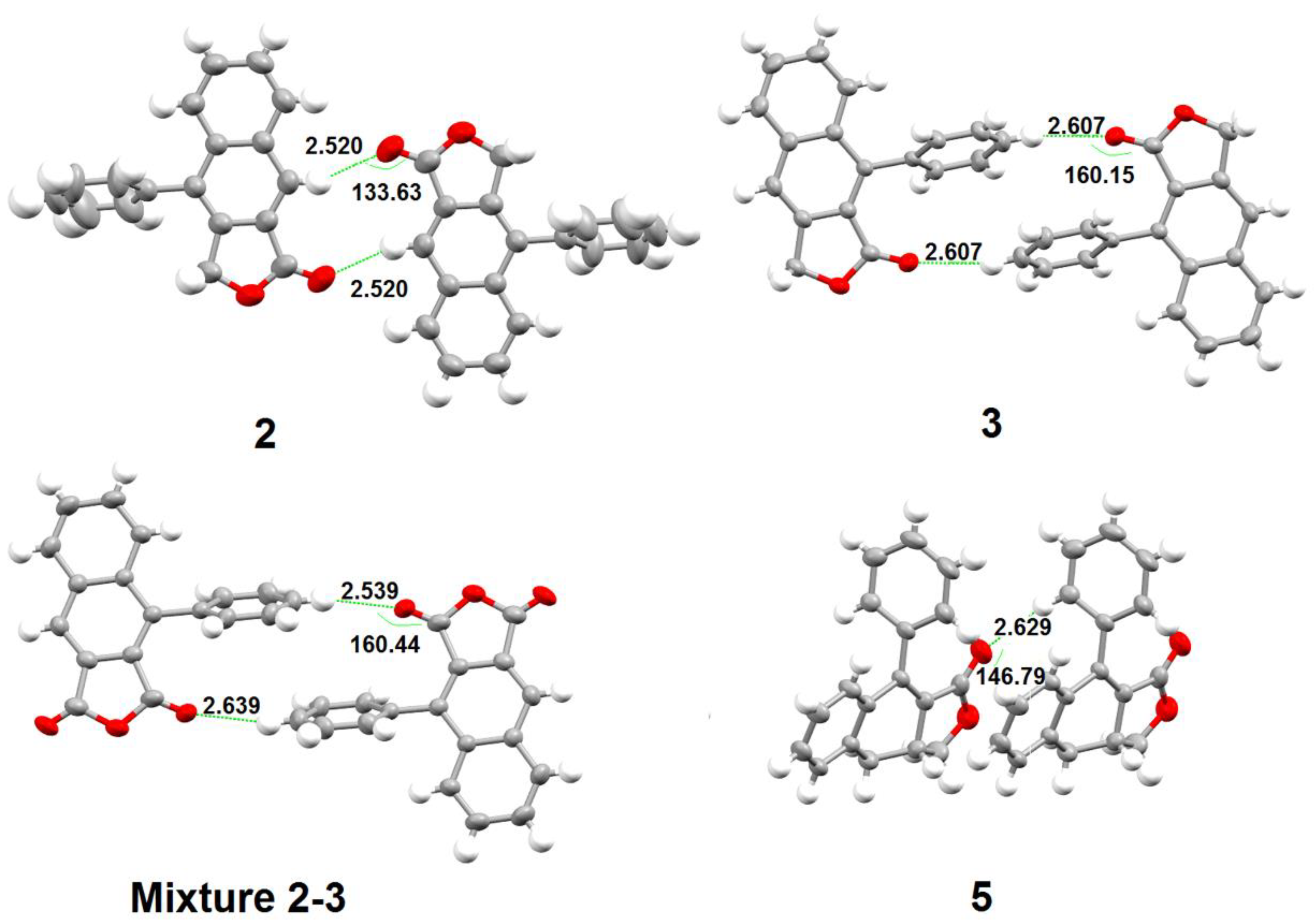
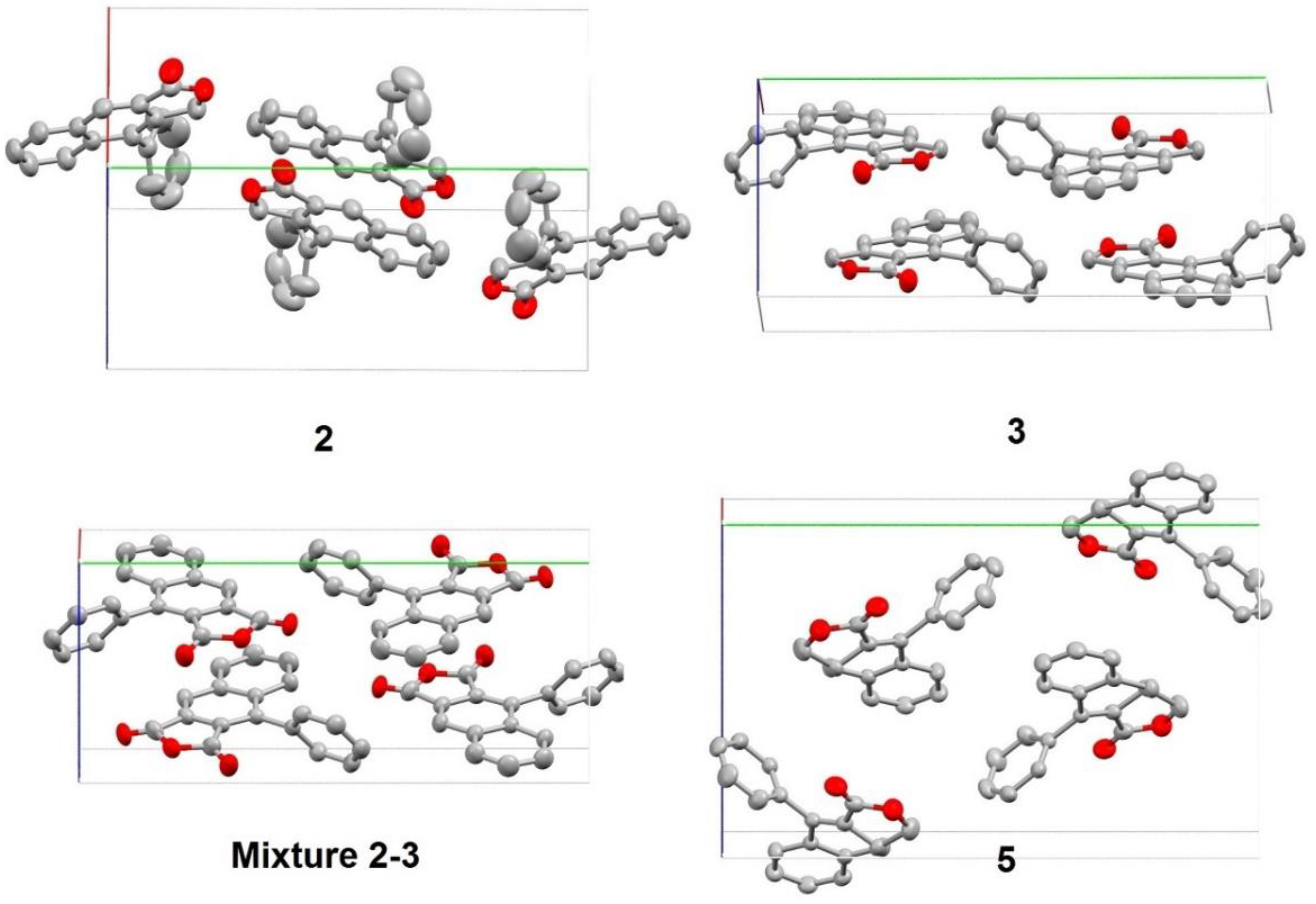
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calvo-Flores, F.G.; Dobado, J.A.; Isac-García, J.; Martín-MartíNez, F.J. (Eds.) Biological Properties of Lignans. Lignin and Lignans as Renewable Raw Materials; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 369–454. [Google Scholar]
- Vasilev, N.P.; Ionkova, I. Cytotoxic activity of extracts from Linum cell cultures. Fitoterapia 2005, 76, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Asano, J.; Chiba, K.; Tada, M.; Yoshii, T. Antiviral activity of lignans and their glycosides from Justicia procumbens. Phytochemistry 1996, 42, 713–717. [Google Scholar] [CrossRef]
- Cow, C.; Leung, C.; Charlton, J.L. Antiviral activity of arylnaphthalene and aryldihydronaphthalene lignans. Can. J. Chem. 2000, 78, 553–561. [Google Scholar] [CrossRef]
- Navarro, E.; Alonso, S.J.; Trujillo, J.; Jorge, E.; Pérez, C. Central nervous activity of elenoside. Phytomedicine 2004, 11, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Rakesh, K.P.; Mumtaz, S.; Moku, B.; Asiri, A.M.; Marwani, H.M.; Manukumar, H.M.; Qin, H.-L. Arylnaphthalene lactone analogues: Synthesis and development as excellent biological candidates for future drug discovery. RSC Adv. 2018, 8, 9487–9502. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, R.; Weber, J.V. Improved Methods of Synthesis of Lignan Arylnaphthalene Lactones via Arylpropargyl Arylpropiolate Esters. J. Nat. Prod. 1989, 52, 367–375. [Google Scholar] [CrossRef]
- Li, J.-H.; Tang, J.-S.; Xie, Y.-X.; Wang, Z.-Q.; Deng, C.-L. Phosphazene Base-Catalyzed Intramolecular Cascade Reactions of Aryl-Substituted Enynes. Synthesis 2010, 18, 3204–3210. [Google Scholar] [CrossRef]
- Foley, P.; Eghbali, N.; Anastas, P.T. Advances in the methodology of a multicomponent synthesis of arylnaphthalene lactones. Green Chem. 2010, 12, 888–892. [Google Scholar] [CrossRef]
- Park, J.-E.; Lee, J.; Seo, S.-Y.; Shin, D. Regioselective route for arylnaphthalene lactones: Convenient synthesis of taiwanin C, justicidin E, and daurinol. Tetrahedron Lett. 2014, 55, 818–820. [Google Scholar] [CrossRef]
- Kao, T.T.; Lin, C.C.; Shia, K.S. The Total Synthesis of Retrojusticidin B, Justicidin E, and Helioxanthin. J. Org. Chem. 2015, 80, 6708–6714. [Google Scholar] [CrossRef] [PubMed]
- Eghbali, N.; Eddy, J.; Anastas, P.T. Silver-Catalyzed One-Pot Synthesis of Arylnaphthalene Lactones. J. Org. Chem. 2008, 73, 6932–6935. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, J.-H.; Kim, S.-H.; Shin, D. Transition Metal-Mediated Annulation Approaches for Synthesis of Arylnaphthalene Lignan Lactones. Front. Chem. 2020, 8, 628. [Google Scholar] [CrossRef] [PubMed]
- Gudla, V.; Balamurugan, R. Synthesis of Arylnaphthalene Lignan Scaffold by Gold-Catalyzed Intramolecular Sequential Electrophilic Addition and Benzannulation. J. Org. Chem. 2011, 76, 9919–9933. [Google Scholar] [CrossRef] [PubMed]
- Naresh, G.; Kant, R.; Narender, T. Silver(I)-Catalyzed Regioselective Construction of Highly Substituted α-Naphthols and Its Application toward Expeditious Synthesis of Lignan Natural Products. Org. Lett. 2015, 17, 3446–3449. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, L.S.; Brummond, K.M. Intramolecular dehydro-Diels-Alder reaction affords selective entry to arylnaphthalene or aryldihydronaphthalene lignans. Org. Lett. 2014, 16, 4158–4161. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, U.C.; Losovyj, Y.; Chen, C.-H.; Zaleski, J.M. Designing Synergistic Nanocatalysts for Multiple Substrate Activation: Interlattice Ag–Fe3O4 Hybrid Materials for CO2-Inserted Lactones. ACS Catal. 2020, 10, 3349–3359. [Google Scholar] [CrossRef]
- SAINT, Version 7.46A; Bruker Analytical X-ray System: Madison, WI, USA, 1997–2007.
- SADABS, V2.10, Bruker Nonius Area Detector Scaling and Absorption Correction; Bruker AXS Inc.: Madison, WI, USA, 2003.
- TWINABS, V2008/2, Bruker Nonius Scaling and Absorption for Twinned Crystals; Bruker AXS Inc.: Madison WI, USA, 2008.
- TWINABS, V1.05, Bruker Nonius Scaling and Absorption for Twinned Crystals; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Altomare, A.; Burla, M.C.; Cammalli, G.; Cascarano, M.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, A. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.J. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- SHELXTL, Version 5.1; Bruker AXS Inc.: Madison, WI, USA, 1997.
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Amer, A.; Ho, D.; Rumpel, K.; Schenkel, R.I.; Zimmer, H. Substituted gamma-butyrolactones. Part 37: Reactions of (arylmethylene)furandiones with nucleophiles. A novel approach to the cyclolignan lactone skeleton. J. Org. Chem. 1991, 56, 5210–5213. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | 2 | 3 | Mixture 2–3 | 5 |
|---|---|---|---|---|
| Empirical formula | C18H12O2 | C18H12O2 | C18H14O2 | C18H14O2 |
| Formula weight | 260.28 | 260.28 | 262.29 | 262.29 |
| Temperature/K | 296(2) | 296(2) | 296(2) | 296(2) |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21/c | P21/c | P21/c | P21/n |
| a/Å | 10.0988(5) | 9.1899(10) | 9.2069(17) | 6.0820(2) |
| b/Å | 16.7625(8) | 17.7239(16) | 17.797(3) | 18.7457(8) |
| c/Å | 8.0793(4) | 7.7263(8) | 7.7530(17) | 11.6861(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 94.699(2) | 91.711(4) | 91.170(8) | 98.759(2) |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å3 | 1363.08(12) | 1257.9(2) | 1270.1(4) | 1316.81(9) |
| Z | 4 | 4 | 4 | 4 |
| ρcalcg/cm3 | 1.268 | 1.374 | 1.372 | 1.323 |
| μ/mm−1 | 0.082 | 0.089 | 0.088 | 0.085 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 4.72 to 60.094 | 4.434 to 58.288 | 4.424 to 58.366 | 4.142 to 61.038 |
| Index ranges | −14 ≤ h ≤ 14, −23 ≤ k ≤ 23, −11 ≤ l ≤ 11 | −12 ≤ h ≤ 12, −24 ≤ k ≤ 22, −8 ≤ l ≤ 10 | −12 ≤ h ≤ 12, −24 ≤ k ≤ 24, −10 ≤ l ≤ 10 | −8 ≤ h ≤ 7, −26 ≤ k ≤ 26, −16 ≤ l ≤ 16 |
| Reflections collected | 32,430 | 15,890 | 22,604 | 28,192 |
| Independent reflections | 3988 [Rint = 0.0239, Rsigma = 0.0151] | 3393 [Rint = 0.0534, Rsigma = 0.0414] | 3436 [Rint = 0.0467, Rsigma = 0.0308] | 4018 [Rint = 0.0337, Rsigma = 0.0232] |
| Data/restraints/parameters | 3988/0/181 | 3393/0/181 | 3436/2/207 | 4018/0/181 |
| Goodness-of-fit on F2 | 1.105 | 1.075 | 1.047 | 1.137 |
| Final R indexes [I > = 2σ (I)] | R1 = 0.0842, wR2 = 0.2262 | R1 = 0.0458, wR2 = 0.1242 | R1 = 0.0495, wR2 = 0.1388 | R1 = 0.0634, wR2 = 0.1530 |
| Final R indexes [all data] | R1 = 0.1191, wR2 = 0.2543 | R1 = 0.0739, wR2 = 0.1470 | R1 = 0.0653, wR2 = 0.1484 | R1 = 0.0896, wR2 = 0.1769 |
| Largest diff. peak/hole/e Å−3 | 1.04/−1.04 | 0.49/−0.34 | 0.25/−0.18 | 0.96/−1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merbouh, N.; Cassegrain, S.; Zhou, W. 4-Phenylnaphtho[2,3-c]furan-1(3H)-one, 9-Phenylnaphtho[2,3-c]furan-1(3H)-one and 3a,4-Dihydro-9-phenylnaphtho[2,3-c]furan-1(3H)-one Crystal Structures. Crystals 2021, 11, 857. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080857
Merbouh N, Cassegrain S, Zhou W. 4-Phenylnaphtho[2,3-c]furan-1(3H)-one, 9-Phenylnaphtho[2,3-c]furan-1(3H)-one and 3a,4-Dihydro-9-phenylnaphtho[2,3-c]furan-1(3H)-one Crystal Structures. Crystals. 2021; 11(8):857. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080857
Chicago/Turabian StyleMerbouh, Nabyl, Simon Cassegrain, and Wen Zhou. 2021. "4-Phenylnaphtho[2,3-c]furan-1(3H)-one, 9-Phenylnaphtho[2,3-c]furan-1(3H)-one and 3a,4-Dihydro-9-phenylnaphtho[2,3-c]furan-1(3H)-one Crystal Structures" Crystals 11, no. 8: 857. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080857