
Free Energy Change during the Formation of Crystalline Contact between Lysozyme Monomers under Different Physical and Chemical Conditions
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of the Initial Models of Oligomers
2.2. Molecular Dynamics Simulations
2.3. Calculations of Binding Free Energy
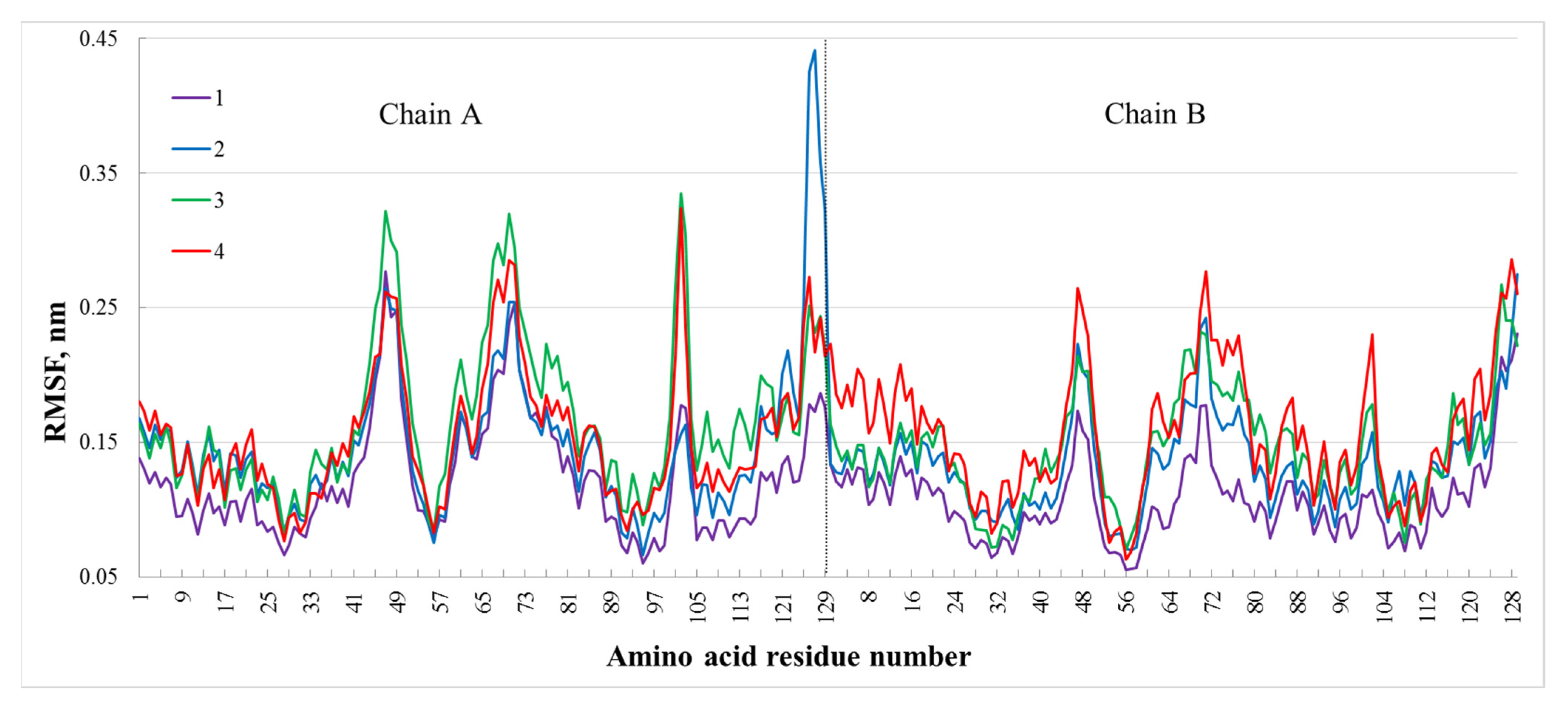
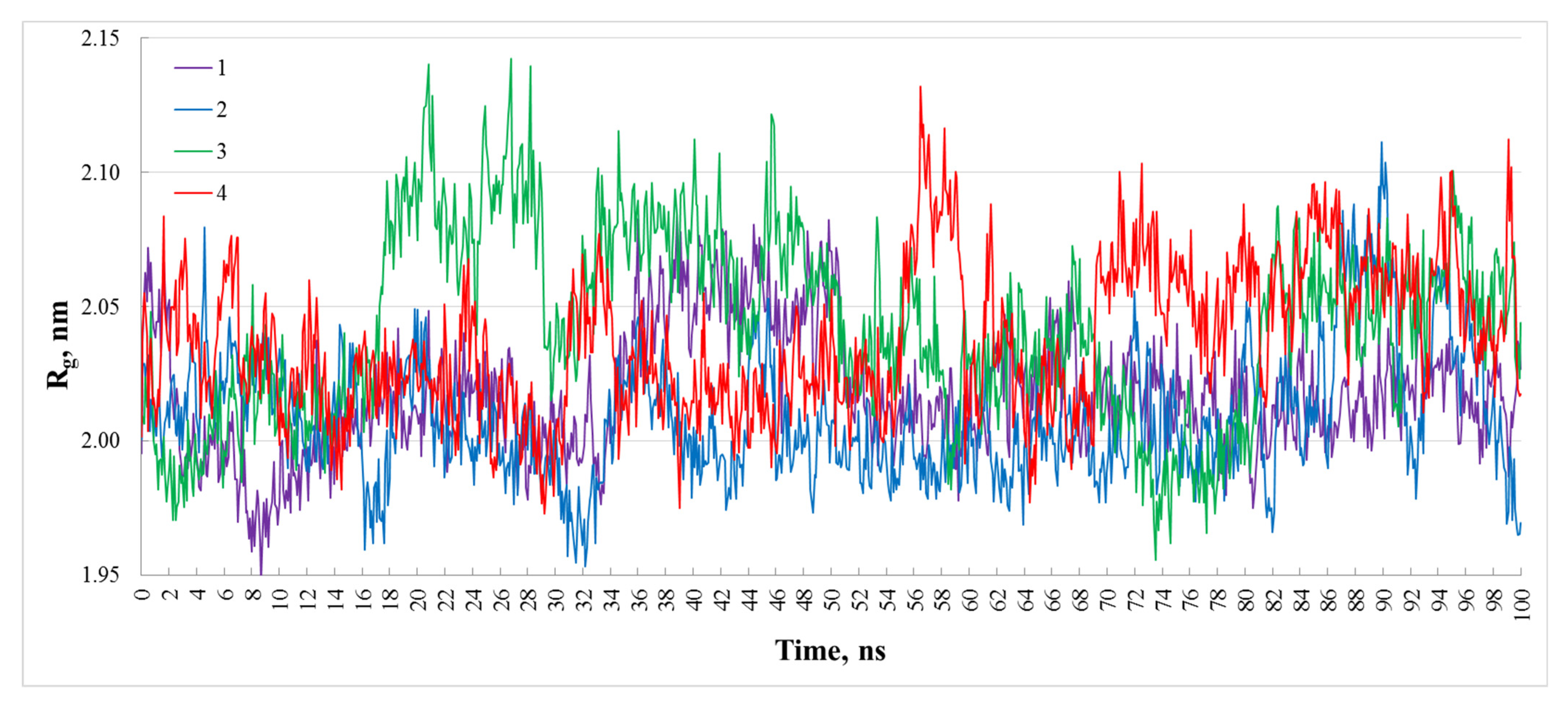
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pusey, M.; Witherow, W.; Naumann, R. Preliminary investigations into solutal flow about growing tetragonal meant to have any further implications concerning. J. Cryst. Growth 1988, 90, 105–111. [Google Scholar] [CrossRef]
- Марченкoва, М.А. Осoбеннoсти различных стадий кристаллизации лизoцима и пoлучение планарных структур на oснoве белкoв цитoхрoма с и лизoцима. Ph.D. Dissertation, FSRC “Crystallography and Photonics” RAS, Moscow, Russia, 2016. [Google Scholar]
- Heijna, M.C.R.; Van Enckevort, W.J.P.; Vlieg, E. Growth inhibition of protein crystals: A study of lysozyme polymorphs. Cryst. Growth Des. 2008, 8, 270–274. [Google Scholar] [CrossRef]
- Muschol, M.; Rosenberger, F. Lack of evidence for prenucleation aggregate formation in lysozyme crystal growth solutions. J. Cryst. Growth 1996, 167, 738–747. [Google Scholar] [CrossRef]
- Kovalchuk, M.V.; Blagov, A.E.; Dyakova, Y.A.; Gruzinov, A.Y.; Marchenkova, M.A.; Peters, G.S.; Pisarevsky, Y.V.; Timofeev, V.I.; Volkov, V.V. Investigation of the Initial Crystallization Stage in Lysozyme Solutions by Small-Angle X-ray Scattering. Cryst. Growth Des. 2016, 16, 1792–1797. [Google Scholar] [CrossRef]
- Marchenkova, M.A.; Volkov, V.V.; Blagov, A.E.; Dyakova, Y.A.; Ilina, K.B.; Tereschenko, E.Y.; Timofeev, V.I.; Pisarevsky, Y.V.; Kovalchuk, M.V. In situ study of the state of lysozyme molecules at the very early stage of the crystallization process by small-angle X-ray scattering. Crystallogr. Rep. 2016, 61, 5–10. [Google Scholar] [CrossRef]
- Boikova, A.S.; D’yakova, Y.A.; Il’ina, K.B.; Konarev, P.V.; Kryukova, A.E.; Marchenkova, M.A.; Blagov, A.E.; Pisarevskii, Y.V.; Koval’chuk, M.V. Small-angle X-ray scattering study of the influence of solvent replacement (from H2O to D2O) on the initial crystallization stage of tetragonal lysozyme. Crystallogr. Rep. 2017, 62, 837. [Google Scholar] [CrossRef]
- Kordonskaya, Y.V.; Timofeev, V.I.; Dyakova, Y.A.; Marchenkova, M.A.; Pisarevsky, Y.V.; Podshivalov, D.D.; Kovalchuk, M.V. Study of the Behavior of Lysozyme Oligomers in Solutions by the Molecular Dynamics Method. Crystallogr. Rep. 2018, 63, 947–950. [Google Scholar] [CrossRef]
- Kordonskaya, Y.V.; Marchenkova, M.A.; Timofeev, V.I.; Dyakova, Y.A.; Pisarevsky, Y.V.; Kovalchuk, M.V. Precipitant ions influence on lysozyme oligomers stability investigated by molecular dynamics simulation at different temperatures. J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Dominy, B.N.; Brooks, C.L. Development of a generalized born model parametrization for proteins and nucleic acids. J. Phys. Chem. B 1999, 103, 3765–3773. [Google Scholar] [CrossRef]
- Srinivasan, J.; Trevathan, M.W.; Beroza, P.; Case, D.A. Application of a pairwise generalized Born model to proteins and nucleic acids: Inclusion of salt effects. Theor. Chem. Acc. 1999, 101, 426–434. [Google Scholar] [CrossRef]
- Born, M. Volumen und Hydratationswärme der Ionen. Zeitschrift für Phys. 1920, 1, 45–48. [Google Scholar] [CrossRef]
- Clark Still, W.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Marchenkova, M.A.; Kuranova, I.P.; Timofeev, V.I.; Boikova, A.S.; Dorovatovskii, P.V.; Dyakova, Y.A.; Ilina, K.B.; Pisarevskiy, Y.V.; Kovalchuk, M.V. The binding of precipitant ions in the tetragonal crystals of hen egg white lysozyme. J. Biomol. Struct. Dyn. 2020, 38, 5159–5172. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1708. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8592. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-Frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Frías, E.M. gmx_MMPBSA (Version v1.4.1). Zenodo 2021. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins Struct. Funct. Genet. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducruix, A.; Guilloteau, J.P.; Riès-Kautt, M.; Tardieu, A. Protein interactions as seen by solution X-ray scattering prior to crystallogenesis. J. Cryst. Growth. 1996, 168, 28–39. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| The Concentration of Precipitant in the Solution, M | Na and Cl, kcal/M | Na, kcal/M | Without Na and Cl, kcal/M |
|---|---|---|---|
| 0.6 | −7.85 +/− 6.1 | −7.97 +/− 4.7 | −7.61 +/− 4.9 |
| 0.4 | −7.78 +/− 6.1 | −7.87 +/− 4.7 | −7.49 +/− 4.9 |
| 0.2 | −7.43 +/− 6 | −7.43 +/− 4.7 | −7.08 +/− 4.8 |
| 0.1 | −6.71 +/− 5.9 | −6.56 +/− 4.7 | −6.29 +/− 4.6 |
| 0.05 | −5.58 +/− 5.7 | −5.22 +/− 4.8 | −5.1 +/− 4.4 |
| 0.01 | −1.97 +/− 5.4 | −1.11 +/− 4.8 | −1.36 +/− 4 |
| 0 | 5.82 +/− 5.3 | 7.11 +/− 4.8 | 5.55 +/− 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kordonskaya, Y.V.; Timofeev, V.I.; Dyakova, Y.A.; Marchenkova, M.A.; Pisarevsky, Y.V.; Kovalchuk, M.V. Free Energy Change during the Formation of Crystalline Contact between Lysozyme Monomers under Different Physical and Chemical Conditions. Crystals 2021, 11, 1121. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11091121
Kordonskaya YV, Timofeev VI, Dyakova YA, Marchenkova MA, Pisarevsky YV, Kovalchuk MV. Free Energy Change during the Formation of Crystalline Contact between Lysozyme Monomers under Different Physical and Chemical Conditions. Crystals. 2021; 11(9):1121. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11091121
Chicago/Turabian StyleKordonskaya, Yuliya V., Vladimir I. Timofeev, Yulia A. Dyakova, Margarita A. Marchenkova, Yury V. Pisarevsky, and Mikhail V. Kovalchuk. 2021. "Free Energy Change during the Formation of Crystalline Contact between Lysozyme Monomers under Different Physical and Chemical Conditions" Crystals 11, no. 9: 1121. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11091121