
Processing of Multicrystal Diffraction Patterns in Macromolecular Crystallography Using Serial Crystallography Programs


1
Department of Life Science, Pohang University of Science and Technology, Pohang 37673, Korea
2
POSTECH Biotech Center, Pohang University of Science and Technology, Pohang 37673, Korea
Crystals 2022, 12(1), 103; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12010103
Submission received: 16 December 2021
/
Revised: 7 January 2022
/
Accepted: 11 January 2022
/
Published: 13 January 2022
(This article belongs to the Special Issue Serial X-ray Crystallography)
Abstract
:Cryocrystallography is a widely used method for determining the crystal structure of macromolecules. This technique uses a cryoenvironment, which significantly reduces the radiation damage to the crystals and has the advantage of requiring only one crystal for structural determination. In standard cryocrystallography, a single crystal is used for collecting diffraction data, which include single-crystal diffraction patterns. However, the X-ray data recorded often may contain diffraction patterns from several crystals. The indexing of multicrystal diffraction patterns in cryocrystallography requires more precise data processing techniques and is therefore time consuming. Here, an approach for processing multicrystal diffraction data using a serial crystallography program is introduced that allows for the integration of multicrystal diffraction patterns from a single image. Multicrystal diffraction data were collected from lysozyme crystals and processed using the serial crystallography program CrystFEL. From 360 images containing multicrystal diffraction patterns, 1138 and 691 crystal lattices could be obtained using the XGANDALF and MOSFLM indexing algorithms, respectively. Using this indexed multi-lattice information, the crystal structure of the lysozyme could be determined successfully at a resolution of 1.9 Å. Therefore, the proposed approach, which is based on serial crystallography, is suitable for processing multicrystal diffraction data in cryocrystallography.
1. Introduction
Cryocrystallography is a widely used X-ray crystallographic technique for determining the structure of macromolecules and has the advantage of significantly reducing the radiation damage experienced by the crystals during data collection [1,2]. Typically, a single crystal is mounted under cryogenic conditions (e.g., temperature of 100 K), resulting in higher-quality diffraction data compared with those collected at room temperature using a single crystal [3].
During a typical cryocrystallographic data collection process, the preferred approach is to mount a single crystal in the path of the X-ray beam and obtain the single-crystal diffraction patterns to determine the structure of the crystal [4]. When recording the X-ray images, the Bragg peaks are detected in the diffraction patterns and used to determine the crystal system and dimensions of the unit cell as well as the crystal orientation in the X-ray beam [5]. Based on the unit cell and orientation information of the crystal, the indexing of the diffraction pattern is performed, wherein each Bragg peak in the diffraction pattern is assigned an index, which is in the form of three integers, namely h, k, and l [6]. Based on the indexing results, the additional Bragg peaks in the diffraction patterns are integrated and scaled. This yields the structure factor for determining the crystal structure [7]. Therefore, the accurate indexing of the Bragg peaks in the diffraction patterns is critical for the success of the initial data processing step. To prevent the misindexing of the diffraction patterns, single-crystal diffraction patterns are preferred during typical X-ray data collection.
However, experimentally, crystal samples often grow in multiple layers or in the form of clusters [8,9,10]. When these crystals are exposed to X-rays, they result in multicrystal diffraction patterns. In addition, during the crystal-mounting process, several crystals could be mounted in the crystal-mounting tool (e.g., CryoLoop or MicroLoop). In such instances, when the crystals are exposed to X-rays, the diffraction images recorded could contain diffraction patterns from multiple crystals. From these multicrystal diffraction patterns, a single-crystal lattice must be distinguished and indexed to determine the structure factor.
In such cases, it is generally possible to index the crystal lattice by selecting the highest-intensity Bragg peak detected in the multicrystal diffraction pattern [7]. However, this results in only one diffraction pattern being indexed and integrated for one image, and the unselected Bragg peaks are not used. Furthermore, if the crystal lattice is not clearly distinguishable from the multicrystal diffraction patterns, the indexing program may provide incorrect crystal lattice information [10]. When data indexing based on multicrystal diffraction patterns fails, the collected data cannot be used for structural determination. Therefore, when collecting multicrystal diffraction patterns during cryocrystallography, it is important to use a program that can index the crystal lattice efficiently.
Serial crystallography (SX) is an emerging X-ray crystallography technique that minimizes radiation damage and allows for the crystal structure to be determined at room temperature [11,12,13,14,15,16,17,18]. During SX measurements, a large number of crystals are exposed to the incident X-ray beam using a sample delivery system, such as an injector or syringe [19,20,21,22,23,24,25,26], fixed target [27,28,29], or microfluidic system [30,31], and the diffraction data are collected in a serial fashion. Since each crystal is not accurately delivered uniformly to the X-ray beam every time, the collected images included single-crystal diffraction data, multicrystal diffraction data, and diffraction-free data [32]. During SX data processing, not only single-crystal diffraction patterns but also multicrystal diffraction patterns can be used [33]. Several SX software programs, such as CrystFEL [33,34], cctbx.xfel [35], cppxfel [36], IOTA [37], and Psocake [38], have been developed to index, integrate, and scale diffraction data.
CrystFEL is one of the most commonly employed SX data processing programs [33,34] and is used with various indexing algorithms, such as MOSFLM [39], XDS [40], DirAx [41], TakeTwo [42], FELIX [43], and XGANDALF [44]. CrystFEL can index and integrate multicrystal lattices from images containing multicrystal diffraction patterns [33,34]. Accordingly, it is expected that using an SX program to process multicrystal diffraction images obtained from cryocrystallography would simplify the data processing step and yield a higher number of diffraction patterns compared to the conventional programs. However, to the best of my knowledge, this has not been demonstrated experimentally.
Here, a new approach for processing multicrystal diffraction data obtained by cryocrystallography is presented using an SX program. Several lysozyme crystals were exposed to an X-ray beam to produce multicrystal diffraction patterns. These diffraction patterns were then successfully processed using the CrystFEL program and the XGANDALF and MOSFLM indexing algorithms, and the crystal structure of the lysozyme could be determined successfully at a resolution of 1.9 Å. The results obtained confirmed that SX programs can be employed for the data processing of multicrystal diffraction patterns obtained from conventional cryocrystallography.
2. Materials and Methods
2.1. Sample Preparation
Lysozyme powder made from hen egg white was purchased from Sigma-Aldrich (L6876; St. Louis, MO, USA). The lysozyme powder was crystallized by the batch method as previously reported [31]. Briefly, the lysozyme powder was dissolved in a buffer containing 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl. The lysozyme solution (25 mg/mL) and a crystallization solution containing 0.1 M sodium acetate (pH 4.5), 2 M NaCl, and 8% (w/v) PEG 8000 were transferred to a 1.5 mL microcentrifuge tube and vortexed immediately at 3000 rpm for 30 s. The mixture was then incubated at 20 °C overnight. The size of the crystals formed was approximately 40–100 μm.
2.2. Data Collection
The diffraction data were collected at beamline 11 C of Pohang Light Source II (Republic of Korea) [45]. The lysozyme crystal suspension was transferred to a siliconized cover glass using a pipette. The lysozyme crystals were picked using a CryoLoop device and soaked in a cryoprotectant solution consisting of 0.1 M sodium acetate (pH 4.5), 2 M NaCl, 8% (w/v) Polyethylene glycol 8000, and 20% (v/v) ethylene glycol for 10 s. The CryoLoop device with the crystals was mounted on the goniometer of the beamline in a nitrogen stream at 100 K. Multiple crystals were present in the CryoLoop, and the diffraction data were collected for an X-ray exposure time of 1 s in steps of 1° while rotating the crystals by 360°. During data collection, 3–6 crystals were exposed to X-rays depending on the rotation angle of the CryoLoop. The recorded multicrystal diffraction data were processed using CrystFEL [33]. To record single-crystal diffraction data, a lysozyme crystal was immersed in the cryoprotectant solution and then mounted on the goniometer; the data were collected using the same parameters as those used for multicrystal diffraction data collection. The single-crystal diffraction data were processed using HKL2000 [7].
2.3. Structural Determination
The electron density maps were obtained based on molecular replacement using Molrep [46]. The room-temperature structure of the lysozyme (PDB code 7DTB) [29] was used as the search model. The model was built using Coot [47]. Structural refinement was performed using the phenix.refine program of the Phenix package [48]. The final structure was validated with MolProbity [49]. The structural figures were generated using PyMOL (https://pymol.org (accessed on 12 January 2022)).
3. Results
In the case of the multicrystal diffraction pattern images generated from crystals of different sizes or through partial multicrystal diffraction, crystal lattice indexing can be performed successfully by selecting only the diffraction patterns containing high-intensity Bragg peaks. However, it is relatively difficult to index multiple diffraction patterns with similar intensities generated from crystals of similar sizes. In this study, to process multicrystal diffraction data, which is even more challenging, multicrystal diffraction patterns were obtained by exposing crystals of similar sizes to X-rays such that the individual crystal diffraction patterns could not be distinguished with ease.
Similar-sized lysozyme crystals were obtained using the batch method, which is used widely in SX [29,31]. After being soaked in the cryoprotectant solution, multiple crystals were mounted on a goniometer maintained at 100 K in a nitrogen stream (Figure 1). The CryoLoop containing the lysozyme crystals was aligned such that multiple crystals were exposed to the X-ray beam as the CryoLoop was rotated by 360° during the data collection process (Figure 1).
As a result, all the collected images contained diffraction patterns corresponding to several crystals (Figure 2). In almost all the images, it was difficult to intuitively distinguish the typical diffraction pattern of the lysozyme containing Bragg peaks at regular intervals.
As the crystals were distributed throughout in the CryoLoop, when the CryoLoop was rotated by 360° and exposed to the incident X-ray beam, the number and position of the crystals exposed to the X-rays varied with the rotation angle of the CryoLoop. Accordingly, when X-ray penetrated the two nylon points of the CryoLoop, the image contained maximum multicrystal diffraction patterns. Furthermore, when X-rays passed along the vertical direction of the CryoLoop plane, the images contained a small number of multicrystal diffraction patterns that could be distinguished partially represented the section.
All collected images contained multiple crystal diffraction patterns (Figure 2). The multicrystal diffraction patterns were initially processed using the HKL2000 program, which is employed widely in cryocrystallography, but this program provided the misindexing information with wrong incorrect space group and unit cell dimension for lysozyme crystal for the diffraction pattern collected in this experiment. Next, the multicrystal diffraction patterns were indexed using the CrystFEL program [33]. As the data processing algorithms of different indexing programs would be different, the indexing rate and data statistics would also differ based on the program used. In this study, both single and multiple lattices were indexed from the multicrystal diffraction images using CrystFEL and various indexing algorithms, such as XGANDALF, MOSFLM, XDS, and DirAx. When processing the multicrystal diffraction patterns using the different indexing algorithms, the important indexing parameters, such as the signal-to-noise ratio (SNR), tolerance, and integration radius, were kept constant (set to the default values). The unit cell information of the lysozyme was the input, and the acceptable indexing tolerances for the dimensions and angle of the crystal lattice were set at 5% and 1.5%, respectively; the indexed crystal lattices exceeding these values were excluded from the data. In Braggs peaks indexing, single-lattice refers to obtaining only one crystal lattice from one image, and multiple-lattice refers to obtaining multiple crystal lattice by referring to “subtract and retry” method from one image.
For single-lattice indexing, XGANDALF, MOSFLM, XDS, and DirAx indexed 80, 78, 72, and 1 images, respectively, from 360 images (Table 1). All the indexing algorithms used with the multicrystal diffraction patterns showed single-lattice indexing rates of less than 23%.
For multi-lattice indexing, XGANDALF, MOSFLM, XDS, and DirAx indexed 360, 326, 213, and 1 images, respectively (Table 1). XGANDALF was able to index all 360 images (100%), while MOSFLM and XDS were able to index 90.55% and 59.16% of the images, respectively. In contrast, DirAx could recognize the diffraction pattern in only one image. In the multi-lattice indexing mode, XGANDALF, MOSFLM, and XDS could extract 1288, 385, and 308 single-crystal lattices, respectively (Table 1). Both XGANDAL and MOSFLM were able to extract more single-crystal lattices instead of the complete images. Specifically, XGANDALF yielded 3.53 times more crystal lattices than complete images. A representative example of multi-lattice indexing from a diffraction image using XGANDALF is shown in Figure 3. Therefore, when combined, the multicrystal diffraction patterns obtained using cryocrystallography could be successfully indexed using an SX program, with the indexing results depending on the indexing program used.
Meanwhile, for the single-crystal diffraction data, 360 single-crystal lattices could be obtained from the 360 images using HKL2000 (Table 1).
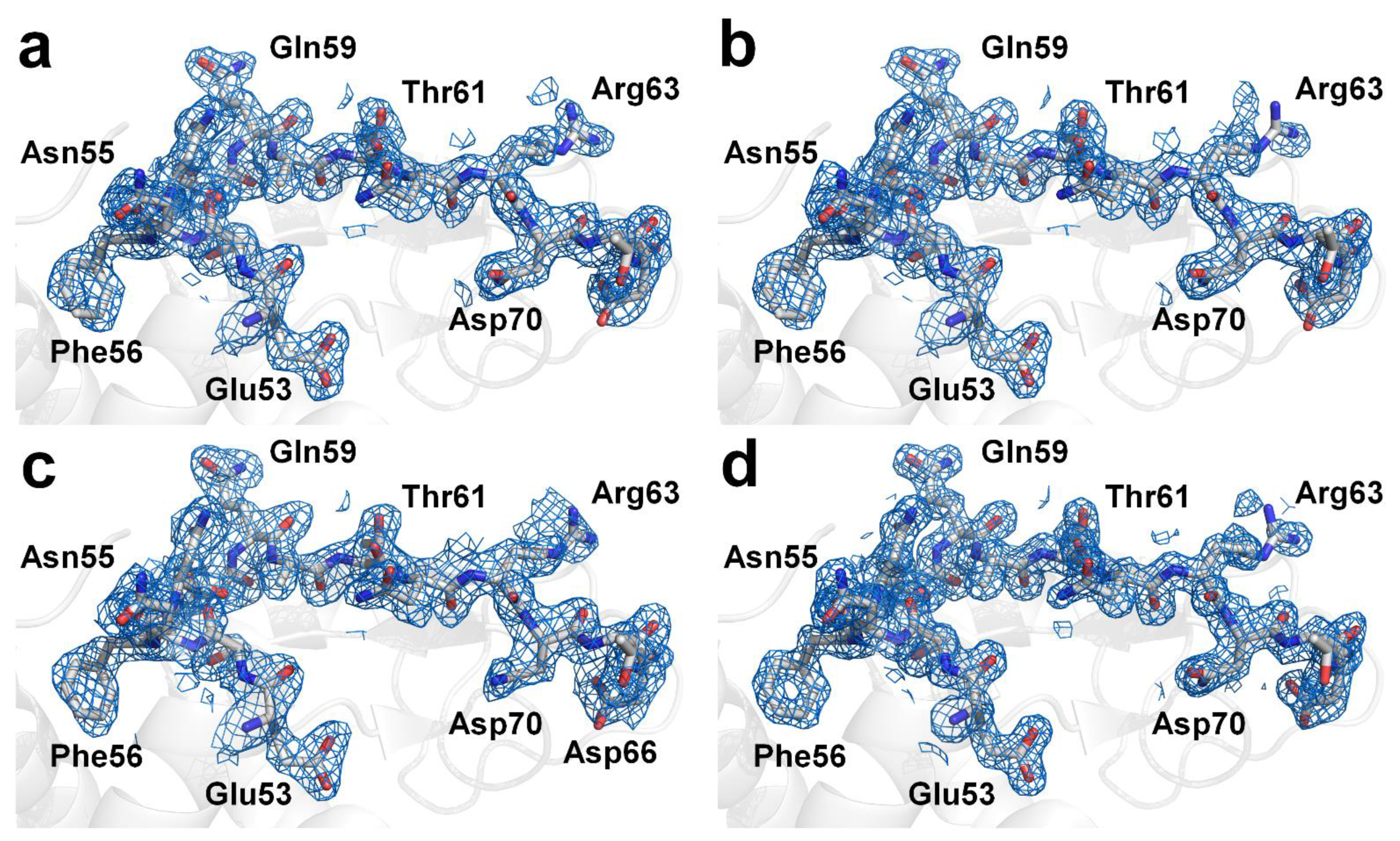
Next, the crystal structure of the lysozyme was determined from the multi- and single-crystal diffraction data. For the multicrystal diffraction data, the Rwork/Rfree values of the final 1.9 Å lysozyme structure indexed using XGANDALF and MOSFLM were 0.207/0.264 and 0.199/0.246, respectively. The overall electron density maps of the lysozyme obtained using XGANDALF and MOSFM were sufficiently clear for interpreting all the amino acids (Figure 4a,b). Concurrently, the refinement statistics for the model structure were different for XGANDALF and MOSFLM (Table 2). Meanwhile, although the data statistics for the multicrystal diffraction patterns processed using the XDS method were poor, these data were also used for structural determination. As expected, the Rwork and Rfree values of the 2.1 Å lysozyme structure in the case of XDS were 0.266 and 0.341, respectively, indicating that the model structure obtained using this method was not suitable (Figure 4c). For the single-crystal diffraction data, the Rwork/Rfree values of the final 1.6 Å lysozyme structure were 0.171 and 0.209, respectively. This dataset also exhibited clear electron density maps (Figure 4d), and almost all the amino acids were well defined.
4. Discussion
In cryocrystallography, diffraction data obtained from a single crystal are preferred, as this facilitates the indexing of the periodic Bragg peaks produced by the crystal, therefore yielding reliable and accurate structural information [7]. However, in the case of multicrystal diffraction patterns, the indexing process not only results in false Bravais lattice information in many cases but also affects the SNR and structure factor, depending on the degree of overlapping of the Bragg peaks.
In this study, both multi- and single-crystal lysozyme diffraction data were collected. After data processing, the single-crystal diffraction data showed higher resolution and SNR and CC values compared with those of the multicrystal diffraction data (Table 1). In addition, with respect to structural refinement, the single-crystal diffraction data yielded a more reliable refinement model in terms of the Rfree value (Table 2). Therefore, it can be surmised that high-quality diffraction data obtained from a single crystal are preferable in cryocrystallography. However, often, the diffraction patterns recorded are from multiple crystals, owing to the nature of the crystal samples or multiple crystal fishing. In such cases, if the multiple crystal diffraction patterns are not indexed, typically, a diffraction experiment is conducted again using another fresh crystal sample. However, in the cases where single-crystal diffraction data collection is difficult or there are no additional crystal samples available, the structural information must be extracted from multicrystal diffraction patterns, even though these may be of relatively low quality.
Multicrystal diffraction patterns can be resolved using popular X-ray programs, such as HKL2000, which are used widely in X-ray crystallography. The following approaches are possible: (i) indexing crystal lattices from images in which single-crystal diffraction patterns are clearly distinguishable from multicrystal ones; (ii) indexing the high-intensity Bragg peaks by increasing the σ-cutoff level; (iii) indexing based on the partial crystal lattice pattern in a specific area, which may have low or high resolution; (iv) indexing the crystal lattice after manually increasing or decreasing the Bragg peaks by peak search mode.
These general approaches are useful for indexing multicrystal diffraction patterns. However, they can be time consuming and tedious if the multicrystal diffraction patterns are complex or of poor quality. Moreover, these approaches can usually extract only one diffraction pattern from multicrystal diffraction patterns. If the possibility of obtaining multi-lattice information from images is realized, the values of parameters, such as the completeness, redundancy, and resolution, would improve.
Here, I attempted to perform the data processing of multicrystal diffraction patterns obtained by cryocrystallography using the CrystFEL program, which is a widely used program for SX. The obtained multicrystal diffraction patterns could be indexed successfully and were used to determine the crystal structure of the lysozyme at a high resolution. The proposed approach has the following advantages: (i) it is not necessary to check the image for indexing multicrystal diffraction patterns; (ii) multi-lattice information can be extracted from a single image; (iii) the indexing efficiency can be improved using various indexing algorithms either individually or in combination.
In this study, multicrystal diffraction patterns were processed using CrystFEL and indexing programs, such as XGANDALF, MOSFLM, XDS, and DirAx. Each indexing method resulted in a different indexing rate, number of multicrystal lattices, and data statistics (Table 1). Overall, XGANDALF and MOSFLM showed higher indexing efficiencies compared with those of XDS and DirAx. However, the results of multicrystal diffraction pattern analysis depend on the actual multicrystal diffraction patterns recorded as well as the data processing parameters used. Hence, the choice of the indexing method will depend on the quality of the multicrystal diffraction patterns. When indexing the multicrystal diffraction patterns obtained during cryocrystallography using an SX program, it is necessary to use an indexing program that maximizes the indexing efficiency; this can be ensured by evaluating several indexing programs. Moreover, it is necessary to optimize the indexing parameters, such as the integration radius and detector geometry, as well.
In this study, the XGANDAF, MOSFLM, and XDS indexing algorithms were used successfully to separately index multiple lattices. Among them, XGANDALF and MOSFLM exhibited the best indexing performances and yielded the highest-quality electron density maps with suitable Rfree values. In contrast, the XDS indexing method showed a poor performance and resulted in low CC and SNR and high Rfree values despite the use of 308 crystal lattices. It is likely that this result is attributable not to the XDS indexing algorithm but to the fact that the multicrystal diffraction images used in this study were not suitable for processing using XDS. Meanwhile, these results indicate that even if a sufficiently high number of images are obtained while considering the space group for the indexing images, the success of the data processing step can be confirmed only by analyzing both the final data statistics and the refinement results.
In this study, four different indexing algorithms were used to index multicrystal diffraction patterns, and the results were compared. However, to obtain better results, it may be appropriate to combine several indexing algorithms, as this would allow for data processing with improved data statistics. To understand the influence of the indexing algorithm combination on the data obtained in this experiment, further data processing was performed using the following three combinations of algorithms: (1) XGANDALF/MOSFLM, (2) XGANDALF/XDS, and (3) MOSFLM/XDS. XGANDAF/MOSFLM and XGANDAN/XDS showed identical results as 360 indexed images with 1288 crystal diffraction patterns. Since this number was the same as when only the XGANDALF algorithm was used for indexing, there was no significant advantage in the indexing algorithm combination. This indicates that data processing by XGANDALF showed the maximum indexing efficiency for the collected data in this experiment. Meanwhile, as a result of combining the MOSFLM/XDS algorithms, 331 indexed images and 617 crystal diffraction patterns were obtained. This is the result of obtaining 232 and 309 more indexed patterns, respectively, compared to the results of indexing MOSFLM and XDS alone.
5. Conclusions
In this study, I showed that the multicrystal diffraction patterns collected during cryocrystallography can be readily and efficiently processed using an SX program. This approach should be suitable for indexing the cryocrystallographic multicrystal diffraction patterns of both macromolecule crystals and those of small molecules.
Funding
This work was funded by the National Research Foundation of Korea (NRF-2017R1D1A1B03033087, NRF-2017M3A9F6029736 and NRF-2021R1I1A1A01050838) and Korea Initiative for Fostering University of Research and Innovation (KIURI) Program of the NRF (NRF-2020M3H1A1075314).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The final coordinates and structure factors were deposited in the Protein Data Bank under accession code 7WBD (Multicrystal diffraction indexed by CrystFEL with XGANDALF), 7WBE (Multicrystal diffraction indexed by CrystFEL with MOSFLM) and 7WBF (Single-crystal diffraction indexed by HKL2000). The images for multicrystal diffraction pattern of lysozyme were deposited at ZENODO (https://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.5783044).
Acknowledgments
I thank the beamline staff at the MX beamlines at PLS-II at Pohang Accelerator Laboratory for assistance in data collection.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Pflugrath, J.W. Practical macromolecular cryocrystallography. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 622–642. [Google Scholar] [CrossRef]
- Garman, E.F.; Schneider, T.R. Macromolecular cryocrystallography. J. Appl. Crystallogr. 1997, 30, 211–237. [Google Scholar] [CrossRef] [Green Version]
- Li, S.J.; Suzuki, M.; Nakagawa, A. Using cryoloops for X-ray data collection from protein crystals at room temperature: A simple applicable method. J. Cryst. Growth 2005, 281, 592–595. [Google Scholar] [CrossRef]
- Ilari, A.; Savino, C. Protein Structure Determination by X-Ray Crystallography. In Bioinformatics; Methods in Molecular Biology™; Humana Press: Totowa, NJ, USA, 2008; pp. 63–87. [Google Scholar] [CrossRef]
- Smyth, M.S.; Martin, J.H. x ray crystallography. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef]
- Kabsch, W. Automatic-Indexing of Rotation Diffraction Patterns. J. Appl. Crystallogr. 1988, 21, 67–71. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Schieferstein, J.M.; Pawate, A.S.; Varel, M.J.; Guha, S.; Astrauskaite, I.; Gennis, R.B.; Kenis, P.J.A. X-ray transparent microfluidic platforms for membrane protein crystallization with microseeds. Lab Chip 2018, 18, 944–954. [Google Scholar] [CrossRef]
- Maruyama, Y.; Ebihara, T.; Nishiyama, H.; Konyuba, Y.; Senda, M.; Numaga-Tomita, T.; Senda, T.; Suga, M.; Sato, C. Direct observation of protein microcrystals in crystallization buffer by atmospheric scanning electron microscopy. Int. J. Mol. Sci. 2012, 13, 10553–10567. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, J.R. Macromolecular crystal twinning, lattice disorders and multiple crystals1. Crystallogr. Rev. 2008, 14, 189–250. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, H.N.; Caleman, C.; Timneanu, N. Diffraction before destruction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [Green Version]
- Stellato, F.; Oberthur, D.; Liang, M.; Bean, R.; Gati, C.; Yefanov, O.; Barty, A.; Burkhardt, A.; Fischer, P.; Galli, L.; et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 2014, 1, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Standfuss, J.; Spence, J. Serial crystallography at synchrotrons and X-ray lasers. IUCrJ 2017, 4, 100–101. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.C.; Stauch, B.; Ishchenko, A.; Cherezov, V. A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem. Sci. 2017, 42, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Sample Delivery Media for Serial Crystallography. Int. J. Mol. Sci. 2019, 20, 1094. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H. Room-Temperature Structure of Xylitol-Bound Glucose Isomerase by Serial Crystallography: Xylitol Binding in the M1 Site Induces Release of Metal Bound in the M2 Site. Int. J. Mol. Sci. 2021, 22, 3892. [Google Scholar] [CrossRef]
- DePonte, D.P.; Weierstall, U.; Schmidt, K.; Warner, J.; Starodub, D.; Spence, J.C.H.; Doak, R.B. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D Appl. Phys. 2008, 41, 195505. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Mizohata, E.; Nango, E.; Suzuki, M.; Tanaka, T.; Masudala, T.; Tanaka, R.; Shimamura, T.; Tanaka, Y.; Suno, C.; et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 2015, 12, 61–63. [Google Scholar] [CrossRef] [Green Version]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Park, S.; Kim, J.; Park, G.; Cho, Y.; Nam, K.H. Polyacrylamide injection matrix for serial femtosecond crystallography. Sci. Rep. 2019, 9, 2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.H. Shortening injection matrix for serial crystallography. Sci. Rep. 2020, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Nam, K.H. Sample delivery using viscous media, a syringe and a syringe pump for serial crystallography. J. Synchrotron Radiat. 2019, 26, 1815–1819. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H. Polysaccharide-Based Injection Matrix for Serial Crystallography. Int. J. Mol. Sci. 2020, 21, 3332. [Google Scholar] [CrossRef]
- Nam, K.H. Lard Injection Matrix for Serial Crystallography. Int. J. Mol. Sci. 2020, 21, 5977. [Google Scholar] [CrossRef]
- Lee, D.; Baek, S.; Park, J.; Lee, K.; Kim, J.; Lee, S.J.; Chung, W.K.; Lee, J.L.; Cho, Y.; Nam, K.H. Nylon mesh-based sample holder for fixed-target serial femtosecond crystallography. Sci. Rep. 2019, 9, 6971. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Park, S.; Lee, K.; Kim, J.; Park, G.; Nam, K.H.; Baek, S.; Chung, W.K.; Lee, J.L.; Cho, Y.; et al. Application of a high-throughput microcrystal delivery system to serial femtosecond crystallography. J. Appl. Crystallogr. 2020, 53, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H.; Kim, J.; Cho, Y. Polyimide mesh-based sample holder with irregular crystal mounting holes for fixed-target serial crystallography. Sci. Rep. 2021, 11, 13115. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Stable sample delivery in viscous media via a capillary for serial crystallography. J. Appl. Crystallogr. 2020, 53, 45–50. [Google Scholar] [CrossRef]
- Nam, K.H.; Cho, Y. Stable sample delivery in a viscous medium via a polyimide-based single-channel microfluidic chip for serial crystallography. J. Appl. Crystallogr. 2021, 54, 1081–1087. [Google Scholar] [CrossRef]
- Barty, A.; Kirian, R.A.; Maia, F.R.; Hantke, M.; Yoon, C.H.; White, T.A.; Chapman, H. Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014, 47, 1118–1131. [Google Scholar] [CrossRef] [Green Version]
- White, T.A. Processing serial crystallography data with CrystFEL: A step-by-step guide. Acta Crystallogr. D Struct. Biol. 2019, 75, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.A.; Mariani, V.; Brehm, W.; Yefanov, O.; Barty, A.; Beyerlein, K.R.; Chervinskii, F.; Galli, L.; Gati, C.; Nakane, T.; et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, N.K.; Hattne, J.; Grosse-Kunstleve, R.W.; Echols, N. New Python-based methods for data processing. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginn, H.M.; Evans, G.; Sauter, N.K.; Stuart, D.I. On the release of cppxfel for processing X-ray free-electron laser images. J. Appl. Crystallogr. 2016, 49, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Lyubimov, A.Y.; Uervirojnangkoorn, M.; Zeldin, O.B.; Brewster, A.S.; Murray, T.D.; Sauter, N.K.; Berger, J.M.; Weis, W.I.; Brunger, A.T. IOTA: Integration optimization, triage and analysis tool for the processing of XFEL diffraction images. J. Appl. Crystallogr. 2016, 49, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Thayer, J.; Damiani, D.; Ford, C.; Dubrovin, M.; Gaponenko, I.; O’Grady, C.P.; Kroeger, W.; Pines, J.; Lane, T.J.; Salnikov, A.; et al. Data systems for the Linac coherent light source. Adv. Struct. Chem. Imaging 2017, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duisenberg, A.J.M. Indexing in Single-Crystal Diffractometry with an Obstinate List of Reflections. J. Appl. Crystallogr. 1992, 25, 92–96. [Google Scholar] [CrossRef]
- Ginn, H.M.; Roedig, P.; Kuo, A.; Evans, G.; Sauter, N.K.; Ernst, O.P.; Meents, A.; Mueller-Werkmeister, H.; Miller, R.J.; Stuart, D.I. TakeTwo: An indexing algorithm suited to still images with known crystal parameters. Acta Crystallogr. D Struct. Biol. 2016, 72, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, K.R.; White, T.A.; Yefanov, O.; Gati, C.; Kazantsev, I.G.; Nielsen, N.F.; Larsen, P.M.; Chapman, H.N.; Schmidt, S. FELIX: An algorithm for indexing multiple crystallites in X-ray free-electron laser snapshot diffraction images. J. Appl. Crystallogr. 2017, 50, 1075–1083. [Google Scholar] [CrossRef]
- Gevorkov, Y.; Yefanov, O.; Barty, A.; White, T.A.; Mariani, V.; Brehm, W.; Tolstikova, A.; Grigat, R.R.; Chapman, H.N. XGANDALF—Extended gradient descent algorithm for lattice finding. Acta. Crystallogr. A Found Adv. 2019, 75, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.H.; Eo, C.; Hwangbo, S.A.; Ha, S.C.; Kim, J.H.; Kim, H.; Lee, C.S.; Seo, I.D.; Yun, Y.D.; Lee, W.; et al. BL-11C Micro-MX: A high-flux microfocus macromolecular-crystallography beamline for micrometre-sized protein crystals at Pohang Light Source II. J. Synchrotron Radiat. 2021, 28, 1210–1215. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
Figure 1.
Photograph of multiple crystals mounted in CryoLoop. Data collection point is indicated by yellow dotted circle.
Figure 1.
Photograph of multiple crystals mounted in CryoLoop. Data collection point is indicated by yellow dotted circle.

Figure 2.
Magnified views of multicrystal diffraction patterns recorded at oscillation angles of 60°, 120°, 180°, 240°, 300°, and 360°.
Figure 2.
Magnified views of multicrystal diffraction patterns recorded at oscillation angles of 60°, 120°, 180°, 240°, 300°, and 360°.

Figure 3.
Multi-lattice indexing from image of multicrystal diffraction pattern using XGANDALF indexing method. (a–d) show identical images; identical standard positions are indicated by red circles. (b–d) show three different indexed multicrystal lattices obtained from image in (a). Circles indicate peak positions predicted by CrystFEL.
Figure 3.
Multi-lattice indexing from image of multicrystal diffraction pattern using XGANDALF indexing method. (a–d) show identical images; identical standard positions are indicated by red circles. (b–d) show three different indexed multicrystal lattices obtained from image in (a). Circles indicate peak positions predicted by CrystFEL.

Figure 4.
Electron density maps (2mFo-DFc, marine mesh, 1.0 σ) of lysozyme structure obtained from multicrystal diffraction images processed with CrystFEL using (a) XGANDALF, (b) MOSFLM, and (c) XDS methods. (d) Electron density maps (2mFo-DFc, marine mesh, 1.0 σ) of lysozyme structure obtained from single-crystal diffraction images processed using HKL2000.
Figure 4.
Electron density maps (2mFo-DFc, marine mesh, 1.0 σ) of lysozyme structure obtained from multicrystal diffraction images processed with CrystFEL using (a) XGANDALF, (b) MOSFLM, and (c) XDS methods. (d) Electron density maps (2mFo-DFc, marine mesh, 1.0 σ) of lysozyme structure obtained from single-crystal diffraction images processed using HKL2000.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Results of image indexing of multiple and single lysozyme crystals.
| Data Collection | Multicrystal Diffraction a | Single-Crystal Diffraction | ||||
|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.9796 | 0.9796 | ||||
| Temperature (K) | 100 | 100 | ||||
| Rotation range per image (degree) | 1 | 1 | ||||
| Total rotation range (degree) | 360 | 360 | ||||
| Exposure time per image (s) | 1 | 1 | ||||
| Space group | P43212 | P43212 | ||||
| Unit cell dimension (Å) a = b, c | 79.51, 37.62 | 79.14, 37.02 | ||||
| Indexing method | XGANDAF | MOSFLM | XDS | DirAx | HKL2000 | |
| Single-lattice | 80 | 78 | 72 | 1 | 360 | |
| Multi-lattice | Image | 360 | 326 | 213 | 1 | |
| Crystals | 1288 | 385 | 308 | 1 | ||
a Multicrystal diffraction patterns were indexed using CrystFEL.
Table 2.
Refinement of multicrystal lysozyme patterns by CrystFEL using multicrystal lattice indexing.
Table 2.
Refinement of multicrystal lysozyme patterns by CrystFEL using multicrystal lattice indexing.
| Diffraction | Multiple Crystals a | Single Crystal | ||
|---|---|---|---|---|
| Data processing method | XGANDAF | MOSFLM | XDS | HKL2000 |
| No. of indexed images | 360 | 326 | 213 | 360 |
| No. of indexed patterns | 1288 | 385 | 308 | 360 |
| Resolution range (Å) | 80.00–1.90 (1.97–1.90) | 80.00–1.90 (1.97–1.90) | 80.00–2.10 (2.17–2.10) | 50.00–1.60 (1.60–1.63) |
| Unique reflections | 10,003 (966) | 10,003 (966) | 7473 (710) | 16,048 (767) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) |
| Redundancy | 235.6 (168.0) | 273.5 (192.7) | 85.9 (61.3) | 25.3 (23.9) |
| Mean I/σ(I) | 4.79 (2.58) | 4.73 (1.86) | 1.98 (1.13) | 36.24 (4.15) |
| CC* | 0.9867 (0.8013) | 0.9917 (0.8758) | 0.9347 (0.7437) | 0.999 (0.984) |
| Rsplit | 19.97 (47.24) | 16.63 (60.95) | 45.28 (93.05) | |
| Average B factor (Å2) | 16.43 | 25.44 | 23.53 | 12.38 |
| Refinement | ||||
| Resolution range (Å) | 56.22–1.90 (2.05–1.90) | 56.22–1.90 (2.05–1.90) | 56.22–2.10 (2.31–2.10) | 39.57–1.60 (1.65–1.60) |
| σ cutoff | F > 1.35σ(F) | F > 1.34σ(F) | F > 1.34σ(F) | F > 1.37σ(F) |
| No. of reflections (working) | 9318 (1816) | 9320 (1817) | 6924 (1674) | 14,403 (1258) |
| No. of reflections (test) | 647 (127) | 642 (124) | 512 (124) | 1601 (140) |
| Final Rcryst | 0.207 (0.2663) | 0.199 (0.2423) | 0.266 (0.2692) | 0.172 (0.1945) |
| Final Rfree | 0.264 (0.3360) | 0.246 (0.3179) | 0.341 (0.3543) | 0.210 (0.2625) |
| No. of non-H atoms | ||||
| Protein | 1957 | 1957 | 1957 | 1957 |
| Ion | 2 | 2 | 2 | 2 |
| Water | 97 | 97 | 76 | 122 |
| Total | 2056 | 2056 | 2035 | 2081 |
| RMS deviations | ||||
| Bonds (Å) | 0.014 | 0.013 | 0.013 | 0.010 |
| Angles (degree) | 1.418 | 1.386 | 1.556 | 1.251 |
| Average B factor (Å2) | ||||
| Protein | 23.13 | 30.91 | 26.34 | 17.81 |
| Ion | 18.56 | 25.46 | 25.37 | 15.03 |
| Water | 24.58 | 34.48 | 26.07 | 26.61 |
| Ramachandran plot | ||||
| Most favored (%) | 96.1 | 97.6 | 92.9 | 98.4 |
| Allowed (%) | 3.9 | 2.4 | 7.1 | 1.6 |
a Multicrystal diffraction patterns were indexed using CrystFEL.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nam, K.H. Processing of Multicrystal Diffraction Patterns in Macromolecular Crystallography Using Serial Crystallography Programs. Crystals 2022, 12, 103. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12010103
AMA Style
Nam KH. Processing of Multicrystal Diffraction Patterns in Macromolecular Crystallography Using Serial Crystallography Programs. Crystals. 2022; 12(1):103. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12010103
Chicago/Turabian StyleNam, Ki Hyun. 2022. "Processing of Multicrystal Diffraction Patterns in Macromolecular Crystallography Using Serial Crystallography Programs" Crystals 12, no. 1: 103. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12010103
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.