
Theoretical Model for a Novel Electronic State in a Dirac Electron System Close to Merging: An Imaginary Element between Sulphur and Selenium


Abstract
:1. Introduction
2. Calculation Methods
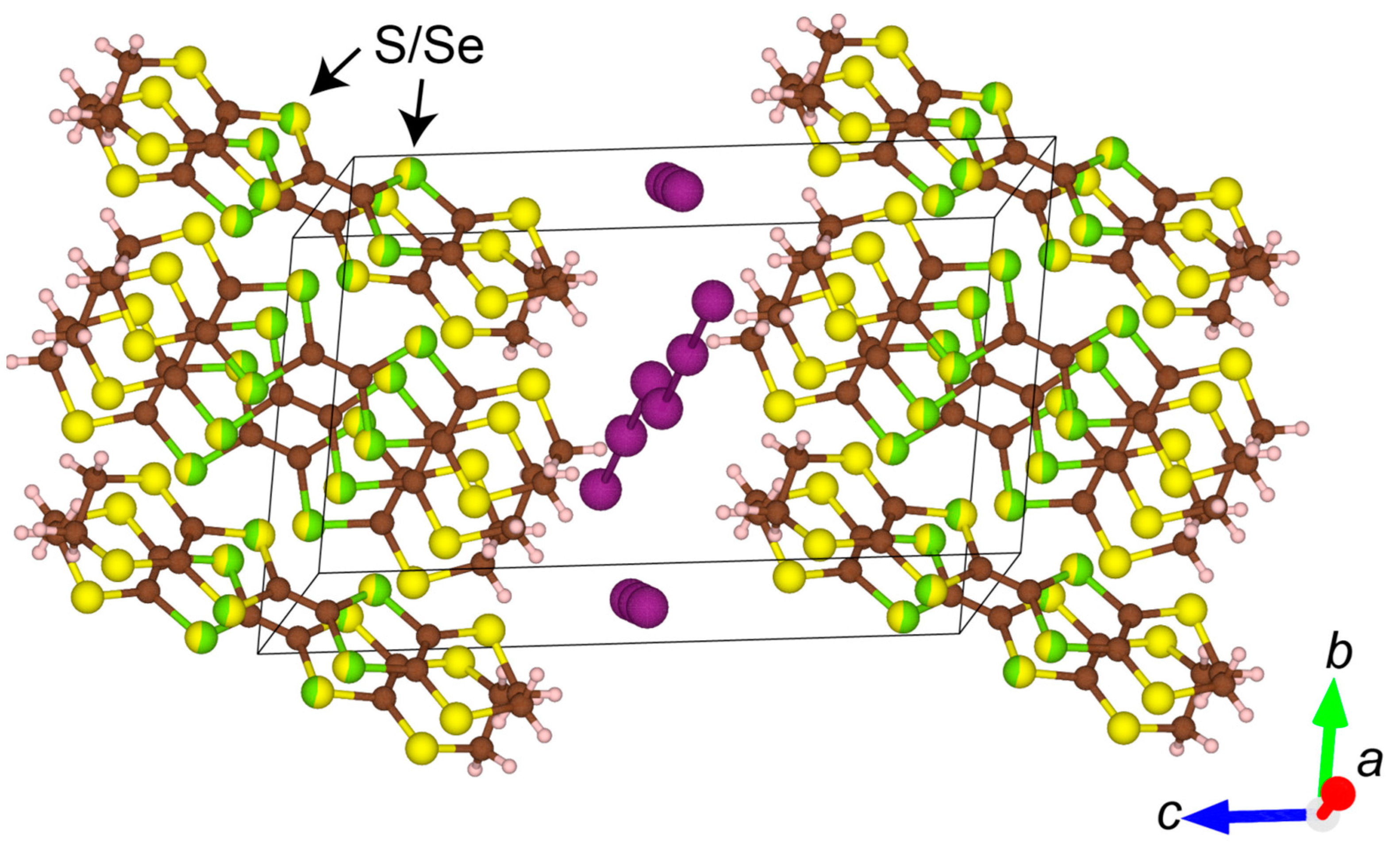
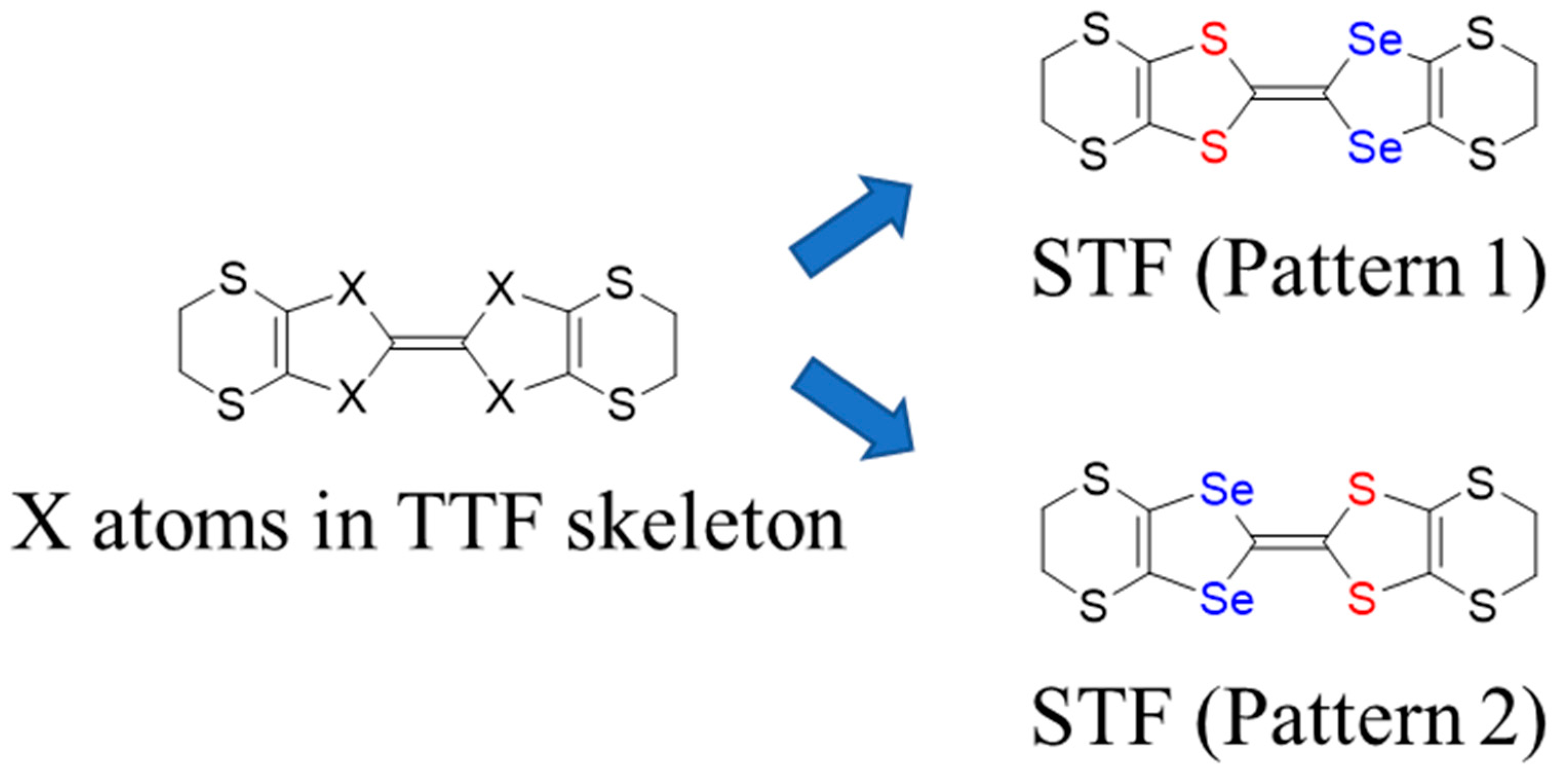
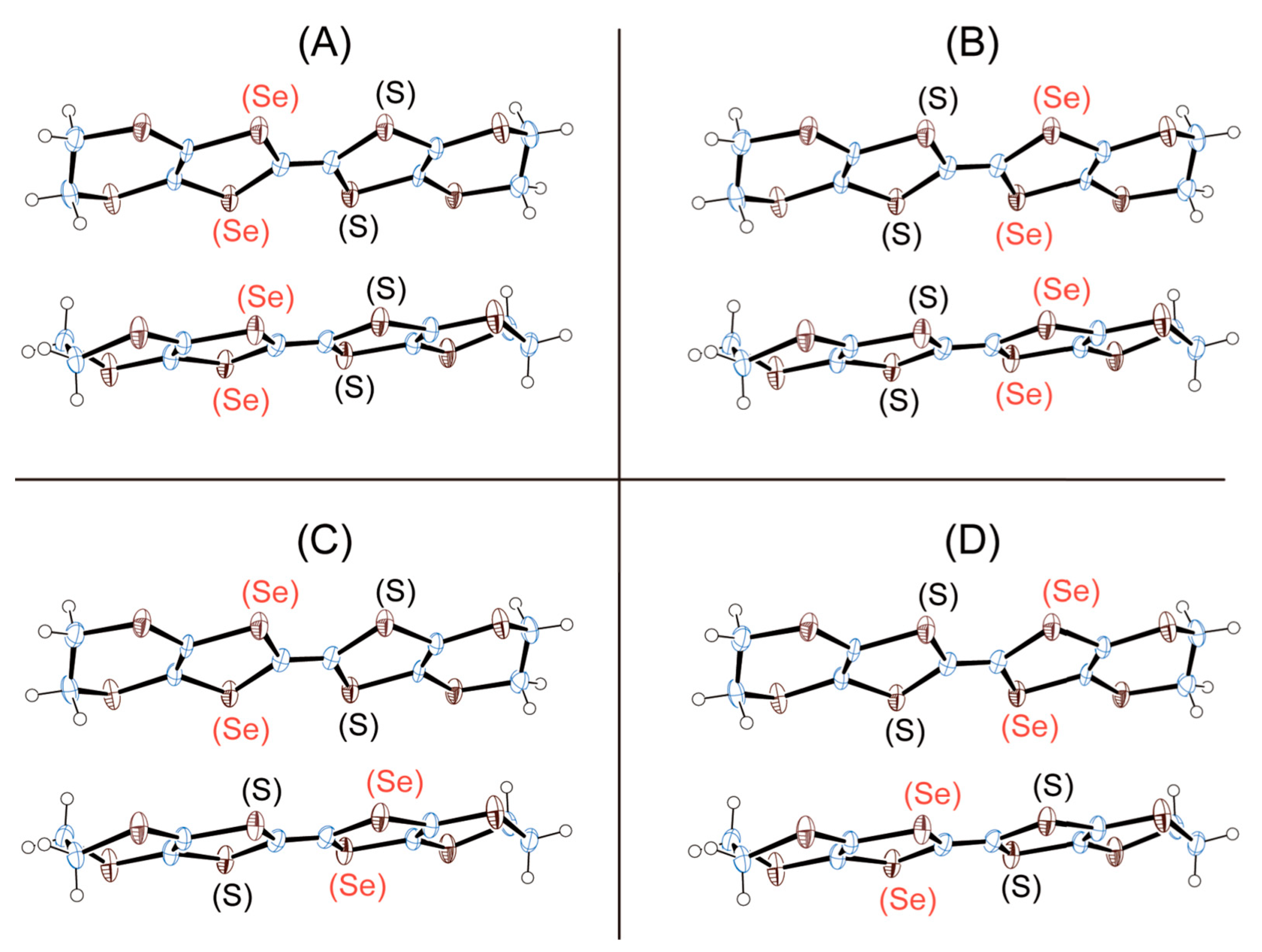
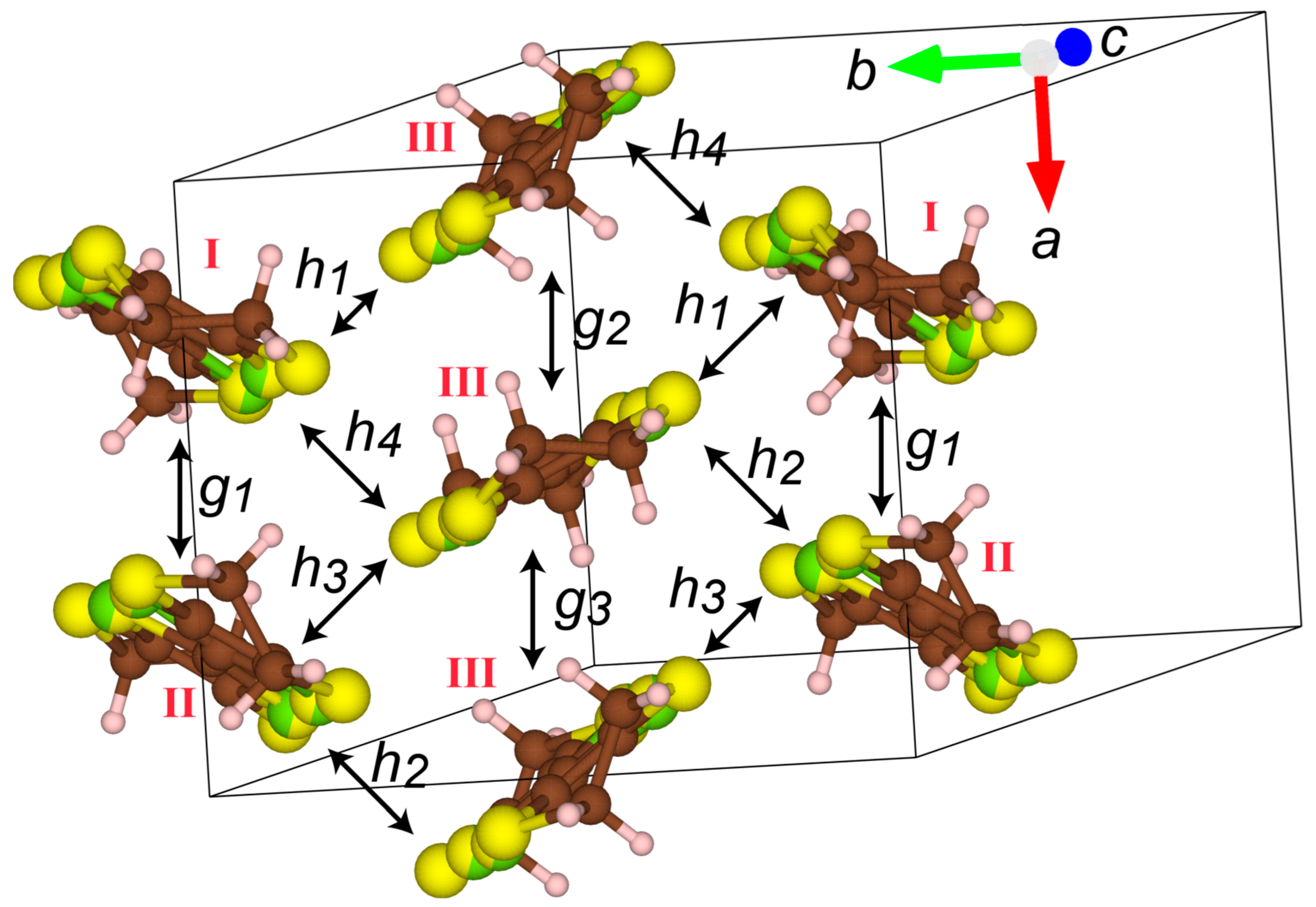
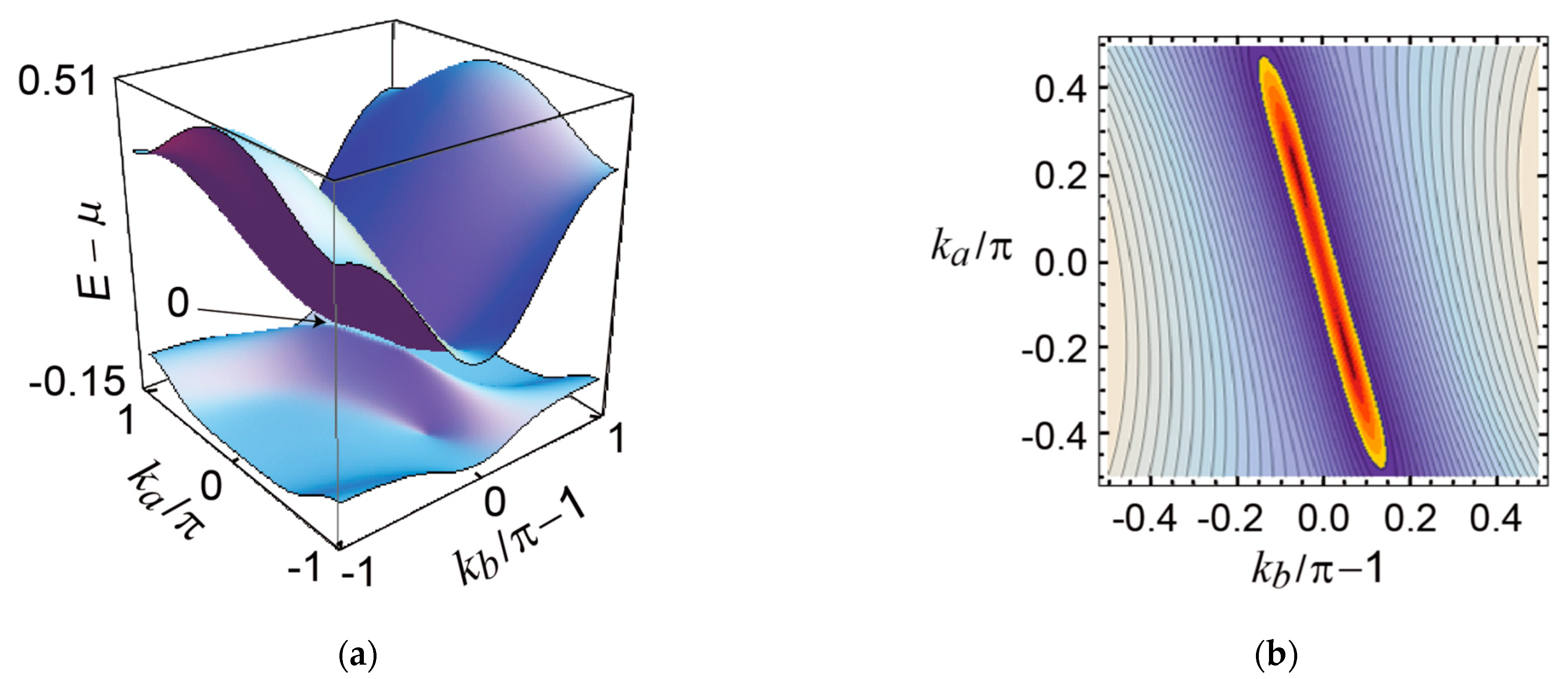
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Details of Our Previous Method of Calculation of Overlap and Transfer Integrals in α-STF2I3
References
- Shwetha, G.; Chandra, S.; Chandra Shekar, N.V.; Kalavathi, S. Existence of spin-polarized Dirac cone in Sc2CrB6: A DFT study. Phys. B Cond. Mat. 2022, 624, 413369. [Google Scholar] [CrossRef]
- Yi, W.; Jiang, X.; Wang, Z.; Yang, T.; Yang, B.; Liu, X. ABX6 monolayers: A new Dirac material family containing high Fermi velocities and topological properties. Appl. Surf. Sci. 2021, 570, 151237. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Wang, G.; Yuan, Z.; Song, W.; Lou, R.; Liu, Z.; Huang, Y.; Liu, Z.; Lei, H.; et al. Dirac cone, flat band and saddle point in kagome magnet YMn6Sn6. Nat. Commun. 2021, 12, 3129. [Google Scholar] [CrossRef]
- Kitayama, K.; Tanaka, Y.; Ogata, M.; Mochizuki, M. Floquet theory of photoinduced topological phase transitions in the organic salt α-(BEDT-TTF)2I3 irradiated with elliptically polarized light. J. Phys. Soc. Jpn. 2021, 90, 104705. [Google Scholar] [CrossRef]
- Li, P.; Guo, Z.-X. The Dirac half-semimetal and quantum anomalous Hall effect in two-dimensional Janus Mn2X3Y3 (X, Y = F, Cl, Br, I). Phys. Chem. Chem. Phys. 2021, 23, 19673–19679. [Google Scholar] [CrossRef] [PubMed]
- Bittner, N.; Golež, D.; Casula, M.; Werner, P. Photoinduced Dirac-cone flattening in BaNiS2. Phys. Rev. B 2021, 104, 115138. [Google Scholar] [CrossRef]
- Herrera, S.A.; Naumis, G.G. Optoelectronic fingerprints of interference between different charge carriers and band flattening in graphene superlattices. Phys. Rev. B 2021, 104, 115424. [Google Scholar] [CrossRef]
- Tanaka, Y.; Mochizuki, M. Real-time dynamics of the photoinduced topological state in the organic conductor α-(BEDT-TTF)2I3 under continuous-wave and pulse excitations. Phys. Rev. B 2021, 104, 085123. [Google Scholar] [CrossRef]
- Jiang, W.; Ni, X.; Liu, F. Exotic topological bands and quantum states in metal-organic and covalent-organic frameworks. Acc. Chem. Res. 2021, 54, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Kino, H.; Miyazaki, T. First-Principles Study of Electronic Structure in α-(BEDT-TTF)2I3 at Ambient Pressure and with Uniaxial Strain. J. Phys. Soc. Jpn. 2006, 75, 034704. [Google Scholar] [CrossRef] [Green Version]
- Tsumuraya, T.; Suzumura, Y. First-principles study of the effective Hamiltonian for Dirac fermions with spin-orbit coupling in two-dimensional molecular conductor α-(BETS)2I3. Eur. Phys. J. B 2021, 94, 17. [Google Scholar] [CrossRef]
- Briganti, M.; Totti, F. Magnetic anisotropy on demand exploiting high-pressure as remote control: An ab initio proof of concept. Dalton Trans. 2021, 50, 10621–10628. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Wehling, T.O.; Black-Schaffer, A.M.; Balatsky, A.V. Dirac materials. Adv. Phys. 2014, 63, 1–76. [Google Scholar] [CrossRef] [Green Version]
- Naito, T. Modern History of Organic Conductors: An Overview. Crystals 2021, 11, 838. [Google Scholar] [CrossRef]
- Kato, R.; Suzumura, Y. Novel Dirac electron in single-component molecular conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate). J. Phys. Soc. Jpn. 2017, 86, 064705. [Google Scholar] [CrossRef]
- Kato, R.; Cui, H.-B.; Tsumuraya, T.; Miyazaki, T.; Suzumura, Y. Emergence of the Dirac electron system in a single-component molecular conductor under high pressure. J. Am. Chem. Soc. 2017, 139, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Tajima, N.; Sugawara, S.; Tamura, M.; Kato, R.; Nishio, Y.; Kajita, K. Transport properties of massless Dirac Fermions in an organic conductor α-(BEDT-TTF)2I3 under pressure. EPL 2007, 80, 47002. [Google Scholar] [CrossRef]
- Kobayashi, A.; Suzumura, Y.; Fukuyama, H. Hall effect and orbital diamagnetism in zerogap state of molecular conductor α-(BEDT-TTF)2I3. J. Phys. Soc. Jpn. 2008, 77, 064718. [Google Scholar] [CrossRef]
- Naito, T.; Doi, R.; Suzumura, Y. Exotic Dirac Cones on the Band Structure of α-STF2I3 at Ambient Temperature and Pressure. J. Phys. Soc. Jpn. 2020, 89, 023701. [Google Scholar] [CrossRef] [Green Version]
- Naito, T.; Doi, R. Band Structure and Physical Properties of α-STF2I3: Dirac Electrons in Disordered Conduction Sheets. Crystals 2020, 10, 270. [Google Scholar] [CrossRef] [Green Version]
- Kobara, R.; Igarashi, S.; Kawasugi, Y.; Doi, R.; Naito, T.; Tamura, M.; Kato, R.; Nishio, Y.; Kajita, K.; Tajima, N. Universal Behavior of Magnetoresistance in Organic Dirac Electron Systems. J. Phys. Soc. Jpn. 2020, 89, 113703. [Google Scholar] [CrossRef]
- Hiraki, K.-I.; Harada, S.; Arai, K.; Takano, Y.; Takahashi, T.; Tajima, N.; Kato, R.; Naito, T. Local Spin Susceptibility of α-D2I3 (D = bis(ethylendithio)tetraselenafulvalene (BETS) and bis(ethylendithio)dithiadiselenafulvalene (BEDT-STF)) Studied by 77Se NMR. J. Phys. Soc. Jpn. 2011, 80, 014715. [Google Scholar] [CrossRef]
- Inokuchi, M.; Tajima, H.; Kobayashi, A.; Ohta, T.; Kuroda, H.; Kato, R.; Naito, T.; Kobayashi, H. Electrical and Optical Properties of α-(BETS)2I3 and α-(BEDT-STF)2I3. Bull. Chem Soc. Jpn. 1995, 68, 547–553. [Google Scholar] [CrossRef]
- Naito, T.; Kobayashi, H.; Kobayashi, A. The Electrical Behavior of Charge-Transfer Salts Based on an Unsymmetrical Donor Bis(ethylenedithio)diselenadithiafulvalene (STF): Disorder Effect on the Transport Properties. Bull. Chem. Soc. Jpn. 1997, 70, 107–114. [Google Scholar] [CrossRef]
- Zhao, P.-L.; Wang, A.-M. Interplay between tilt, disorder, and Coulomb interaction in type-I Dirac fermions. Phys. Rev. B 2019, 100, 125138. [Google Scholar] [CrossRef] [Green Version]
- Papaj, M.; Isobe, H.; Fu, L. Nodal arc of disordered Dirac fermions and non-Hermitian band theory. Phys. Rev. B 2019, 99, 201107. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Velarte, M.C.; Kretz, B.; Moro-Lagares, M.; Aguirre, M.H.; Riedemann, T.M.; Lograsso, T.A.; Morellón, L.; Ibarra, M.R.; Garcia-Lekue, A.; Serrate, D. Chemical disorder in topological insulators: A route to magnetism tolerant topological surface states. Nano Lett. 2017, 17, 4047–4054. [Google Scholar] [CrossRef] [Green Version]
- Suzumura, Y.; Proskurin, I.; Ogata, M. Effect of tilting on the in-plane conductivity of Dirac electrons in organic conductor. J. Phys. Soc. Jpn. 2014, 83, 023701. [Google Scholar] [CrossRef]
- Montambaux, G.; Piéchon, F.; Fuchs, J.-N.; Goerbig, M.O. Merging of Dirac points in a two-dimensional crystal. Phys. Rev. B 2009, 80, 153412. [Google Scholar] [CrossRef]
- Pellegrino, F.M.D.; Angilella, G.G.N.; Pucci, R. Strain effect on the optical conductivity of graphene. Phys. Rev. B 2010, 81, 035411. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Suzumura, Y.; Piéchon, F.; Montambaux, G. Emergence of Dirac electron pair in the charge-ordered state of the organic conductor α-(BEDT-TTF)2I3. Phys. Rev. B 2011, 84, 075450. [Google Scholar] [CrossRef] [Green Version]
- Samal, S.S.; Nandy, S.; Saha, K. Nonlinear transport without spin-orbit coupling or warping in two-dimensional Dirac semimetals. Phys. Rev. B 2021, 103, L201202. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pairs (a) | X = Se | X = S | X = Se or S (50%) |
|---|---|---|---|
| g1 | +9.0 | −19.7 | −5.35 |
| g2 | −8.50 | −17.9 | −13.2 |
| g3 | +2.2 | −11.7 | −4.75 |
| h1 | +31.8 | −25.9 | +2.95 |
| h2 | −33.7 | −25.3 | −29.5 |
| h3 | −15.0 | −13.3 | −14.15 |
| h4 | +6.8 | −8.6 | −0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naito, T.; Suzumura, Y. Theoretical Model for a Novel Electronic State in a Dirac Electron System Close to Merging: An Imaginary Element between Sulphur and Selenium. Crystals 2022, 12, 346. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030346
Naito T, Suzumura Y. Theoretical Model for a Novel Electronic State in a Dirac Electron System Close to Merging: An Imaginary Element between Sulphur and Selenium. Crystals. 2022; 12(3):346. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030346
Chicago/Turabian StyleNaito, Toshio, and Yoshikazu Suzumura. 2022. "Theoretical Model for a Novel Electronic State in a Dirac Electron System Close to Merging: An Imaginary Element between Sulphur and Selenium" Crystals 12, no. 3: 346. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030346