1. Introduction
In 1972, Fujishima and Honda first established that titania (TiO
2) could lead to splitting of water when exposed to visible light, thereby producing gaseous oxygen and hydrogen [
1]. Since then, there has been considerable scrutiny of the properties of aqueous solutions in contact with titania surfaces. More generally, beyond titania, there are a great deal of renewable-energy applications potentially of interest involving photo-electrochemical splitting of water in dye-sensitised solar cells. Naturally, though, the non-toxic, inexpensive and abundant nature of titania render it especially attractive in this regard. Potential photo-active materials may involve support-metal-support-interaction (SMSI) changes to photo-catalytic properties [
2]. In any event, given that titania is one of the most scrutinised oxides, there is somewhat of a paucity in our understanding of the characteristics of interfacial water molecules, together with their potential reactivity at surfaces. This is in spite of progress recently [
3,
4], including via molecular-simulation approaches and theoretical techniques [
5], although the outlook in this regard is surely improved in recent years [
4].
Titania-water interfaces allow for a detailed study of confined water molecules’ dynamical properties. This is very important for the case where hydrogen-bonded molecules play an part in stabilising solutes by solvent interactions, and also forming thereby “cages” [
3,
4]. For instance, Inelastic Neutron Scattering (INS) measurements have led to vibrational spectra of adsorbed water on anatase powder and rutile rods [
6,
7]; this led to insight that confined adsorbed water molecules have dynamical and vibrational behaviour more redolent of less mobile ice vis-à-vis to a liquid [
6,
7]. This adsorbed-water vibrational behaviour has been studied in detail via molecular dynamics (MD) at interfaces of anatase-(101) and rutile-(110) with water [
8,
9]. Further,
ab initio MD (AIMD) has led to important results in interesting studies recently [
9,
10] on librational and higher-frequency adsorbed-water modes. The rutile-(110)-water interface has been particularly scrutinised by MD [
11,
12,
13,
14,
15,
16]. Ion adsorption has been studied by Zhang
et al. [
11]. Importantly, Predota
et al. have investigated structure of the electric double layer [
12,
13,
14], establishing the underlying structural nature of water layers [
12], adsorption of ions [
13], and diffusivity and viscosity properties of water layers [
14]. Crucially, Predota
et al. concluded that diffusivity increases farther away from the interface, increasing to values synonymous with the bulk state [
14]. The effects of protonation on surface properties and characteristics have been considered by Machesky
et al. [
15].
We have performed classical MD to characterise strain within adsorbed water molecules at a variety of titania-water interfaces, as well as considering orientations of their dipoles relative to the normal to the surface [
16]. For a wide range of titania surfaces, we have also considered hydrogen-bonding kinetics between bridging oxygen atoms and water molecules adsorbed physically [
17]. For confined water molecules, we have reproduced well vibrational-spectra data with respect to INS spectra [
18]. For these rather confined layers, a particularly important feature in [
18] was computing of self-diffusivity within these layers. We concluded recently that spatial distribution functions (SDF), in three dimensions, of adsorbed water show there is a more open-like topography for anatase-101 relative to rutile-(110) [
19], facilitating easier contact of adsorbed water with that beyond the first layer. This boosts the degree of hydrogen bonding with water molecules outside the adsorbed layer. We concluded tentatively that this may rationalise different values for mobility in water layers [
19],
i.e., lower mobility for rutile-110 interfaces, and rather higher for anatase-(101), providing extremes in the self-diffusivity of the adsorbed monolayer [
18]. In any event, an empirical-potential model treatment of titania-water interfaces may well fail to capture the subtleties of their physico-chemical characteristics, together with hydroxylation properties [
19]. To clarify this singularly important question, we performed AIMD of rutile-(110) surfaces with partial hydroxylation to determine vibrational properties of hydrogen bonds between bridging oxygen atoms and water, and of orientations of water molecules’ dipoles with respect to the surface normal, for surfaces featuring oxygen-atom vacancies, as well as pristine ones [
20].
Quite apart from the interest in rutile- and anatase- water interfaces’ physico-chemical characteristics
per se, there is the pertinent question of the link between these ground-state characteristics and photo-activity. Titania’s most photo-active polymorph tends to be anatase, possessing greater stability vis-à-vis rutile for nanoparticles [
21]. Pan
et al. have measured and assessed a wide variety of facets of titania crystals, determining that clean anatase-(101) surfaces are more photo-active than their (001) analogues, contrary to many previous findings [
22]; it was also determined that anatase-(101) surfaces afford greater photo-activity than rutile-(110). Naturally, in the context of the previous discussion of DFT-based and classical- MD modelling of titania-water interfaces, this raises the tantalising question of how greater photo-activity in anatase-(101) may be rationalised with respect to rutile-(110) from the perspective of structure, hydrogen-bonding arrangements and kinetics, and also, intriguingly, in terms of ease of access of water beyond the adsorbed layer to the titania surface. Given the tentative evidence of [
19] from SDF considerations of anatase-(101)’s more accessible and open architecture facilitating hydrogen bonding with water beyond the adsorbed layer, this suggests that greater levels of mobility, or self-diffusivity, in the adsorbed layer of water at the anatase-(101) surface, observed from classical MD in [
18], allow for greater scope of more “promiscuous” water contact with the surface. Naturally, this would increase photo-activity and water-splitting rates [
22]. However, the contention of inter-layer water “swopping”, or exchange, to allow for penetration of water molecules into the adsorbed layer in a dynamic equilibrium between layers, has not been explored in the literature to any extent, although Predota
et al. have indeed studied self-diffusivity (via MD) increasing towards bulk-like values further away from the rutile-(110) surfaces [
14]. In the present work, motivated by tackling these open questions of water self-diffusivity in layers for “extremes” of adsorbed-layer diffusional behaviour in anatase-(101) (higher) and rutile-(110) (low) [
18], we study this in various layers, as well as exchange “events” into the adsorbed layer and outer ones, allowing conjecture as to the influence of these water-mobility characteristics, together with architecture of surfaces, on experimentally observed trends in titania-facet photo-activity.
2. Simulation Methodology
We carried out a 1 ns NVT MD simulation [
23] for rutile-(110) and anatase-(101) TiO
2 surfaces, using in-house code, using classical dynamics under equilibrium conditions. We applied three-dimensional Ewald treatment for non-bonded interactions with a relative precision of 10
−5 (in terms of variation of the number of reciprocal-space wavevectors and the real-space contributions [
23]). A Nosé-Hoover NVT ensemble was employed at 300 K, in conjunction with a Velocity-Verlet integration with a 0.33 fs timestep. Bulk liquid water was relaxed for around 200 ps using the Anderson-Hoover NPT ensemble (300 K and 1 bar pressure), prior to running for 1 ns under NVT at 300 K with mild thermostat coupling (period of 0.5 ps). The necessary number of water molecules were added to realise an appropriate bulk-like density of circa 1 g/cm
3 between the titania surface and its periodically-imaged couterpart. The initial MD relaxation was performed for liquid water under NPT conditions, so as to achieve a box density corresponding to 1 bar pressure. The Matsui-Akaogi (MA) [
24] model was applied for titania, whilst a flexible-SPC (FwSPC) [
25] potential was used for water. Ti-Ow parameters were established using the Buckingham potential and O-Ow LJ potential (
cf. Table 1) [
16]. The application of these potentials has led to good accord between computed and experimental vibrational density of states, and also hydrogen-bonding characteristics on the presently-considered titania-water interfaces [
18]. The MA model involves Buckingham-type interactions (see
Table 1). All slabs used were free to move. The details of system size and box dimensions are specified in
Table 2.
Table 1.
Force-field parameters. Taken from [
24] (titania) and [
25] (water), with titania-water interactions as described in [
16].
Table 1.
Force-field parameters. Taken from [24] (titania) and [25] (water), with titania-water interactions as described in [16].
| Buckingham Potential for TiO2 and Water Oxygen: Aij × exp(-rij/ρij) − Cij/rij6 |
| i–j | Aij (kcal·mol−1) | ρij (Å) | Cij (kcal·mol−1 Å6) |
| Ti–O | 391049.1 | 0.194 | 290.331 |
| Ti–Ti | 717647.4 | 0.154 | 121.067 |
| O–O | 271716.3 | 0.234 | 696.888 |
| Ti–Ow | 28593.0 | 0.265 | 148.000 |
| Lennard-Jones potential for water: εij[(σij/rij)1−σij/rij)6] |
| i–j | εij (kcal·mol−1) | σij (Å) |
| Ow–Ow | 0.1554 | 3.165492 |
| Harmonic potential for water: k/2 × (rij − r0)2 |
| i–j | kij (kcal·mol−1 Å−2) | R0ij (Å) |
| Ow–Hw | 1059.162 | 1.012 |
| Harmonic angle bending potential for water: k/2 × (θ – θ0) |
| i–j–k | θ0 deg | k (kcal·mol−1 rad−2) |
| H–O–H | 113.24 | 75.900 |
| Atomic charges: q(Ti) = 2.196 e, q(O) = −1.098 e, q(Ow) = −0.82 e, q(Hw) = 0.41 e; Ow, Hw = water oxygen and hydrogen atoms |
Table 2.
Simulation-box dimensions and number of particles.
Table 2.
Simulation-box dimensions and number of particles.
| Phase (surface) X, Y, Z (Å) | System Size |
|---|
| Rutile (110) 26.26, 45.47, 69.490 | (TiO2)630 (H2O)2000 |
| Anatase (101) 71.46, 26.43, 72.680 | (TiO2)1176 (H2O)3162 |
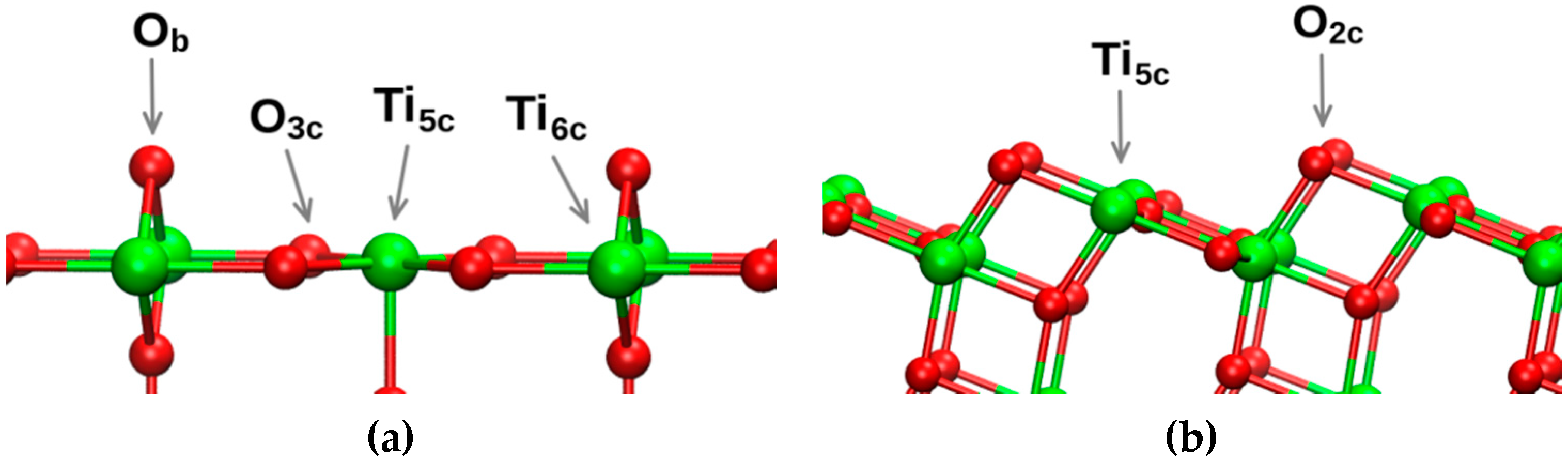
Surfaces (
cf. Figure 1) were prepared from bulk rutile featuring lattice vectors a
0 = b
0 = 4.593 Å, c
0 = 2.959 Å (
P42/MNM) and bulk anatase [a
0 = b
0 = 3.776 Å and c
0 = 9.486 Å (
I41/AMD)]. Details of topography and construction of the surfaces are detailed elsewhere more completely [
16]. The normal to the surface coincided with the
z-axis. We estimated water self-diffusivity in the
x-
y plane (parallel to the surfaces) and
z-direction (perpendicular thereto) via the mean squared displacement (MSD) over 1 ns, sampled in regions of increasing distance from the interface in 0.5 Å “bins”, taking care with length of MSD and statistical sampling level to ensure establishment of the Fickian regime in each bin [
23], whilst also monitoring swopping/exchange events between layers (
vide infra).
Figure 1.
Structure of surfaces; laboratory z-direction is vertical. Ob stands for bridging oxygen, O3c a three-coordinated surface oxygen, Ti5c a penta-coordinated surface Ti atom, and Ti6c denotes a hexa-coordinated Ti atom. (a) rutile-110, and (b) anatase-101.
Figure 1.
Structure of surfaces; laboratory z-direction is vertical. Ob stands for bridging oxygen, O3c a three-coordinated surface oxygen, Ti5c a penta-coordinated surface Ti atom, and Ti6c denotes a hexa-coordinated Ti atom. (a) rutile-110, and (b) anatase-101.
3. Results and Discussion
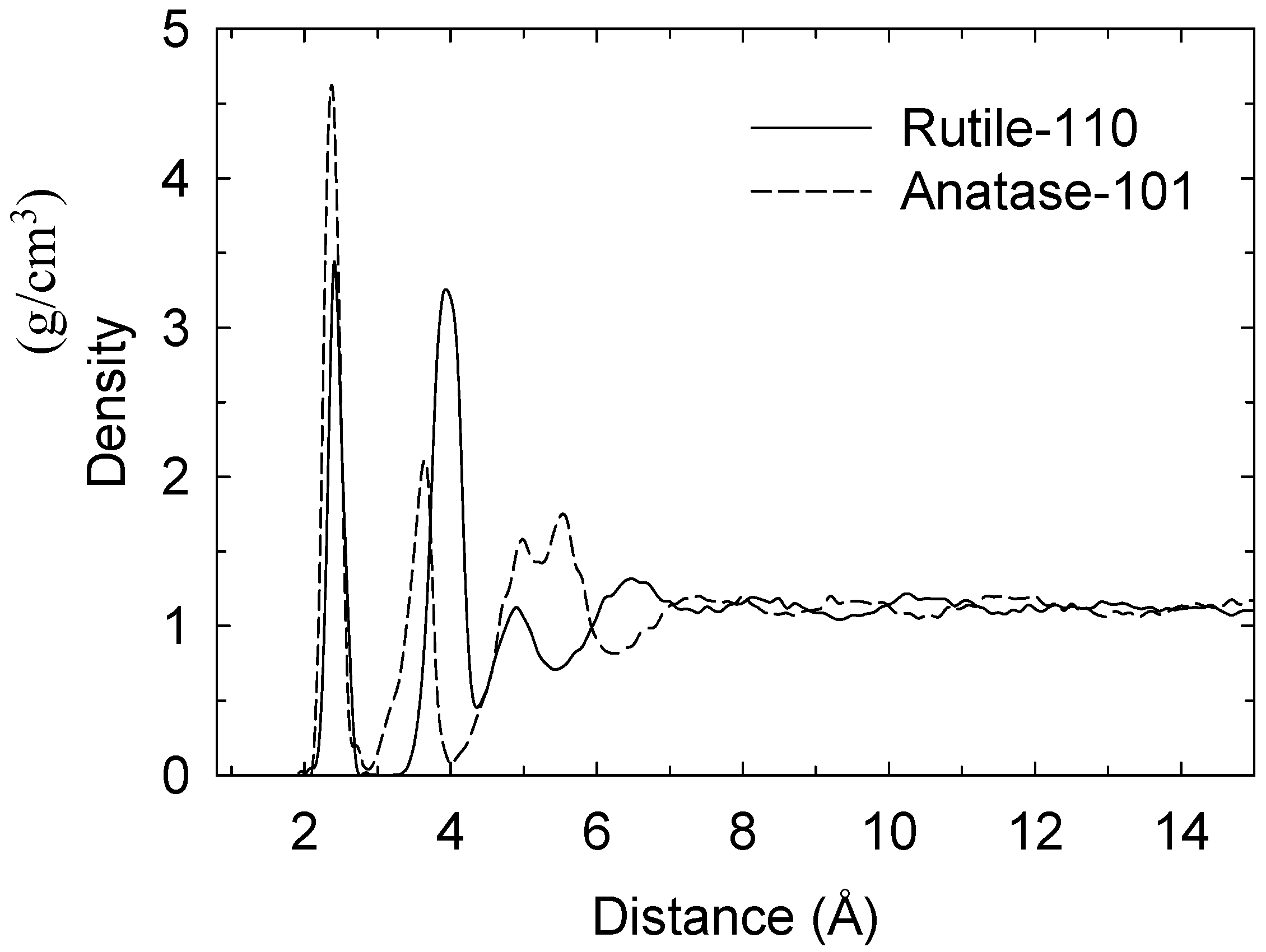
The water-density “profiles”, in terms of
z-axis displacement from interfaces are very similar to those reported previously in [
16] (
cf. Figure 2). In this case, the first minimum is at ~2.9 Å distance from the plane of “uppermost” titanium atoms in the case of both surfaces (
cf. Figure 2). This minimum is the
de facto “border” between the adsorbed monolayer (referred to hereinafter as “ML”) and layers further out from the surfaces, and, in both cases, uniform density is achieved within ~9 Å of the surfaces. There is a second, less distinct adsorbed layer (termed “2L”) between ~2.9 and 4.5 Å distance which is not in any direct contact with the surface at any instant, and beyond this distance, the density layers become less clear-cut as more bulk-like behaviour is achieved, with a third density layer (dubbed “3L”) partly evident between 4.5 and 6.3 Å.
Figure 2.
Density profile along z-axis of water molecules from topmost layers of titanium atoms.
Figure 2.
Density profile along z-axis of water molecules from topmost layers of titanium atoms.
The self-diffusivities of each of the three labelled density layers are specified in
Table 3. The self-diffusivity of bulk water was found to be ~2.8 × 10
−9 m
2·s
−1 (or ~0.93 × 10
−9 m
2·s
−1 in each
x-,
y-,
z-direction given its isotropic nature), whilst the experimental bulk-water value is 2.3 × 10
−9 m
2·s
−1 [
26]. The diffusivity values in the ML exhibit a certain anisotropy due to preferential motion along the local “ridged” architecture (
cf. Figure 1), and are consistent with those in [
18] (determined from a Green-Kubo integral of centre-of-mass velocity autocorrelation functions [
23]). In any event, they tend towards bulk-like values with increasing distance from the surfaces (
cf. Table 3). Those evaluated in 0.5 Å bins along the
z-direction from the surfaces also show a gradual increase to recover bulk values within ~9–10 Å, consistent with recovering a bulk-like density profile (
cf. Figure 3 of [
16]). Predota
et al. observed this in MD simulations for water in contact with rutile-110 [
14], while Mamontov
et al. have studied multi-layer water absorption via neutron scattering and MD, reporting also that the self-diffusivity of water increases away from the surface [
27]. The markedly low value for ML water at rutile-(110) results from the atomistic architecture of the surface (
cf. Figure 1a): adsorbed molecules are confined relatively rigidly in the region between O
b atoms, in contrast to the more “terraced” anatase-101 (
cf. Figure 1b). The latter surface is more accessible to water molecules and allows for greater hydrogen-bonding interactions of somewhat localised “bound” water molecules with those beyond this layer. In [
19], it was found that there are essentially two hydrogen bonds with molecules outside the adsorbed layer in anatase-(101), whilst there is around one such bond per adsorbed water molecule in rutile-(110). This disparity originates from the anatase-(101) surface’s more accessible architecture. The present work confirms the tentative conclusion of [
19] that this open surface structure and greater hydrogen-bond interaction and “communication” of anatase-(101) 1L water molecules with those in the second “2L” layer facilitates a greater self-diffusivity in the ML for anatase-(101), in stark contrast with the much less mobile, “ice-like” and “trapped” ML water molecules at rutile–(110). Indeed, [
18] has determined these more ice-like vibrational features for ML water molecules at rutile-(110) via MD from velocity autocorrelation functions (to obtain the ML’s vibrational density of states) with inelastic neutron scattering spectra.
Table 3.
Self-diffusivities [×10
−9 m
2·s
−1] (x,y,z) in adsorbed layer (ML) and second and third layers (2L & 3L, respectively) from each surface (
cf. density profiles in Figure 3 of [
16]. Note that the sum of the different laboratory directions gives the total self-diffusivity. That of bulk water is ~2.8 × 10
−9 m
2·s
−1 (or ~0.93 × 10
−9 m
2·s
−1 in x,y,z), whilst the experimental bulk-water value is 2.3 × 10
−9 m
2·s
−1.
Table 3.
Self-diffusivities [×10−9 m2·s−1] (x,y,z) in adsorbed layer (ML) and second and third layers (2L & 3L, respectively) from each surface (cf. density profiles in Figure 3 of [16]. Note that the sum of the different laboratory directions gives the total self-diffusivity. That of bulk water is ~2.8 × 10−9 m2·s−1 (or ~0.93 × 10−9 m2·s−1 in x,y,z), whilst the experimental bulk-water value is 2.3 × 10−9 m2·s−1.
| Surface | ML | 1L | 2L |
|---|
| Rutile-(110) | 0.011 ± 0.002, 0.063 ± 0.007, 0.021 ± 0.004 | (0.31,0.37,0.34) ± 0.03 | (0.58,0.60,0.66) ± 0.06 |
| Anatase-(101) | 0.70 ± 0.05, 0.75 ± 0.06, 0.67 ± 0.05 | (0.80, 0.81, 0.76) ± 0.07 | (0.83,0.85,0.80) ± 0.08 |
We now turn to the key point of inter-layer mobility, with special focus on exchange events between the ML and 2L density layers. It was found over 1 ns runs that the probability of an exchange event was only ~0.3% from the ML molecules to transition to the 2L layer at the rutile-(110) surface, but was markedly higher at ~4.2% at anatase-(101), with a similar number of reverse exchanges. Given the greater level of hydrogen-bond interactions (essentially double) for ML with 2L water molecules at anatase-(101), this “communication”, coupled with a substantially more mobile ML layer (
cf. Table 3), affords a much greater likelihood of such exchanges. It was found that there were a great deal more such events between 2L and 3L, approaching levels seen in bulk water, with less disparity between the two surfaces, as one would expect for a transition towards bulk-like diffusive behaviour (
cf. Table 3).



{kind=link}
{kind=link}

