
H2XP:OH2 Complexes: Hydrogen vs. Pnicogen Bonds



Abstract
:1. Introduction

2. Methods
2.1. Cambridge Structural Database Search
2.2. Ab Initio Calculations
3. Results and Discussion
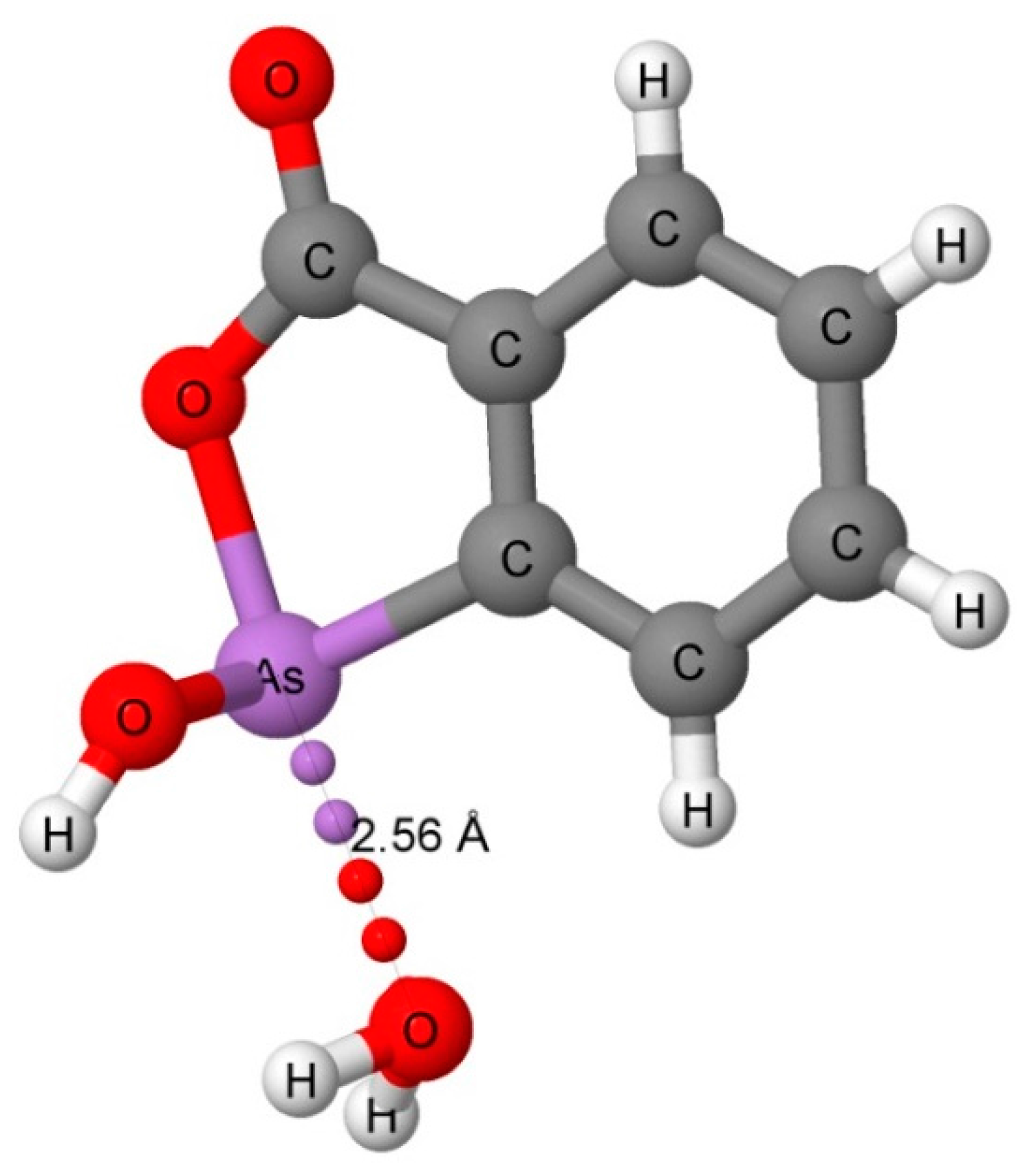
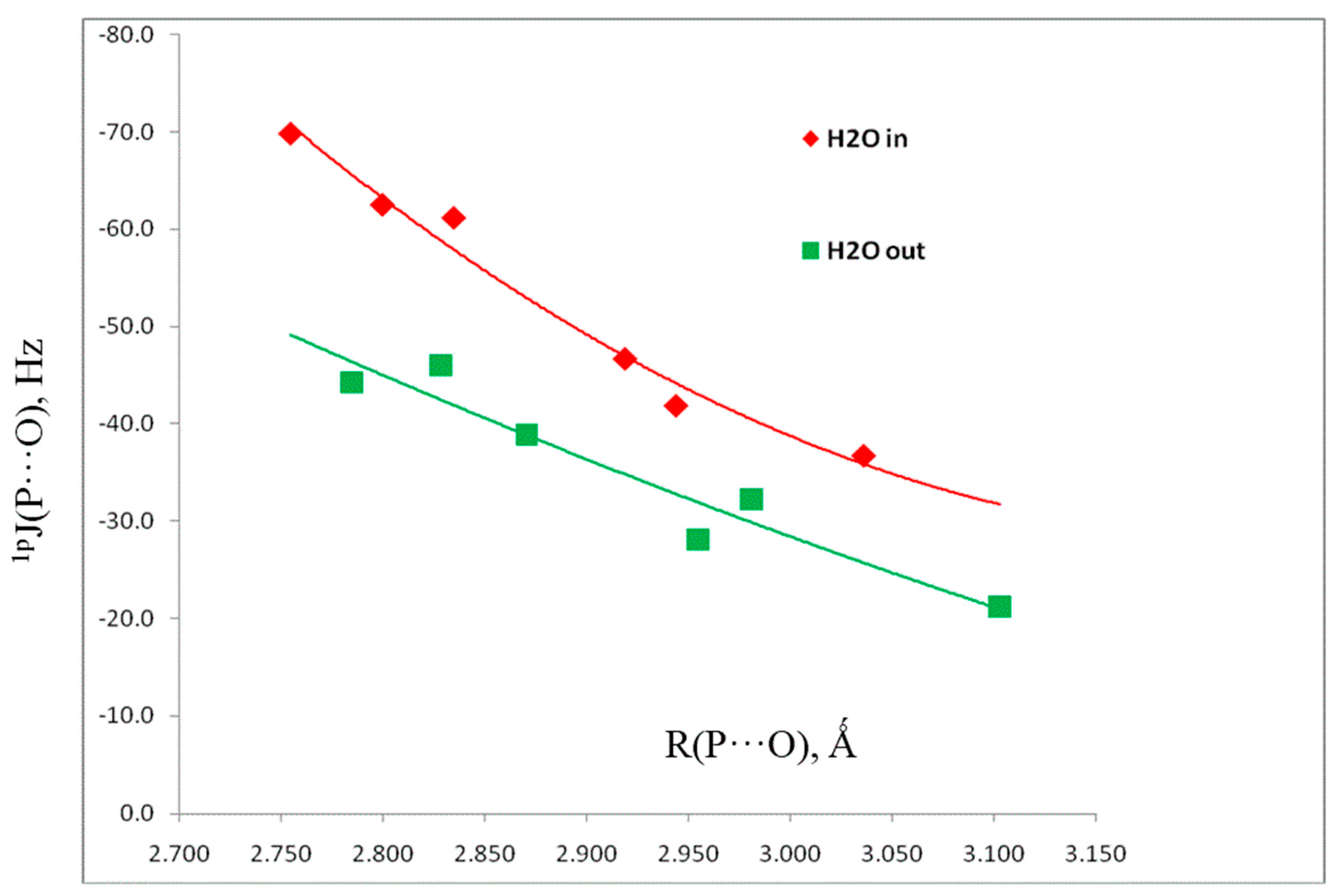
3.1. CSD Search

3.2. Computational Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2XP, X = | Binding Energies (ΔE, kJ·mol−1) | |
|---|---|---|
| ZB | HB | |
| NC | –21.1 | |
| F | –19.5 | |
| Cl | –17.3 | –9.4 |
| CN | –16.9 | |
| OH | –12.8 | |
| CCH | –11.3 | –10.8 |
| H | –11.1 | |
| CH3 | –15.4 | |
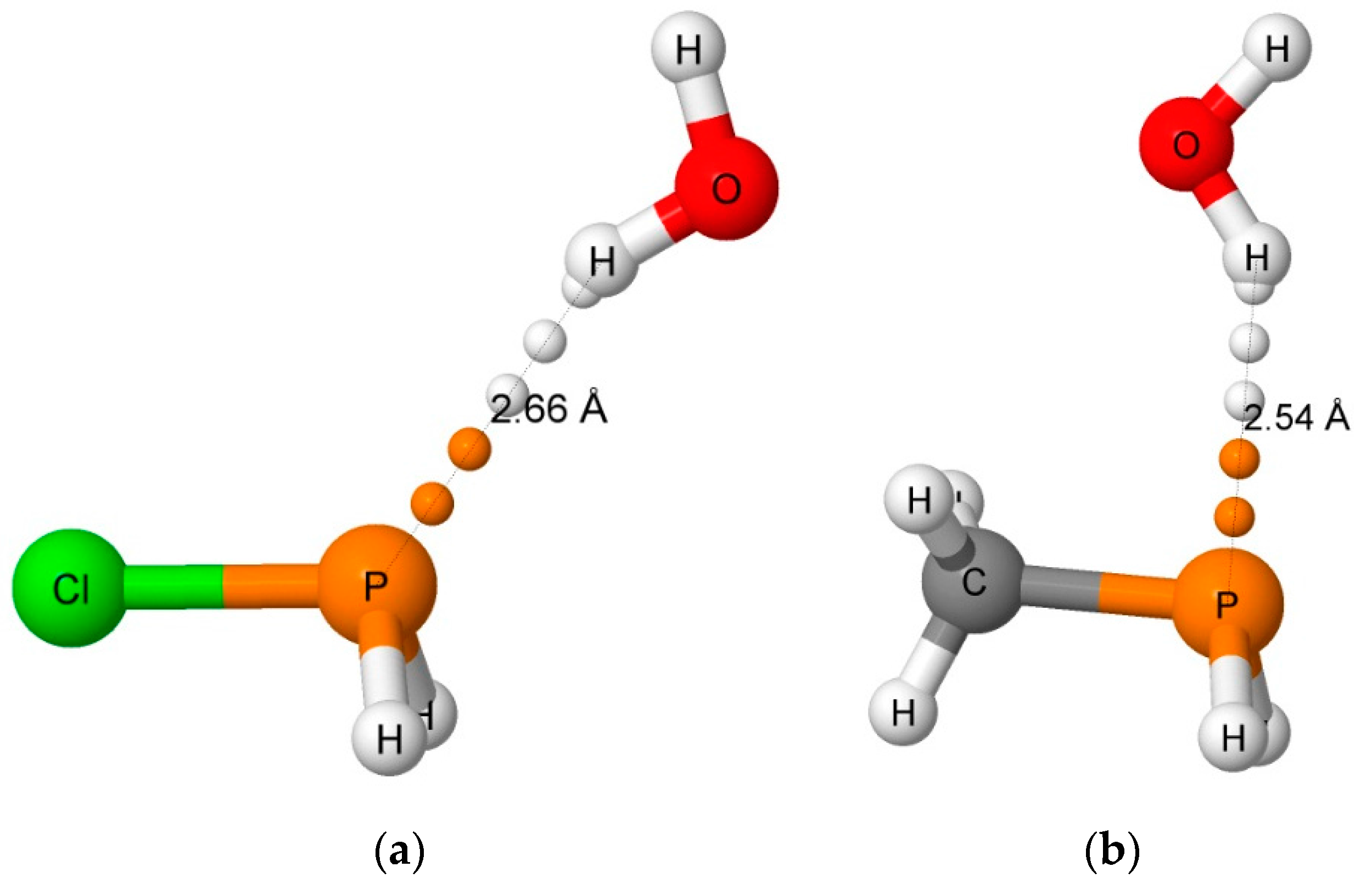
3.2.1. Hydrogen-Bonded Complexes
| H2XP, X = | Distance (R, Å) | Angles (<, °) | ||
|---|---|---|---|---|
| R(O···P) | R(H···P) | R(O-H) a | <H-O-P | |
| Cl | 3.553 | 2.656 | 0.965 | 19 |
| CCH | 3.585 | 2.629 | 0.966 | 7 |
| H | 3.575 | 2.620 | 0.966 | 7 |
| CH3 | 3.360 | 2.536 | 0.967 | 27 |

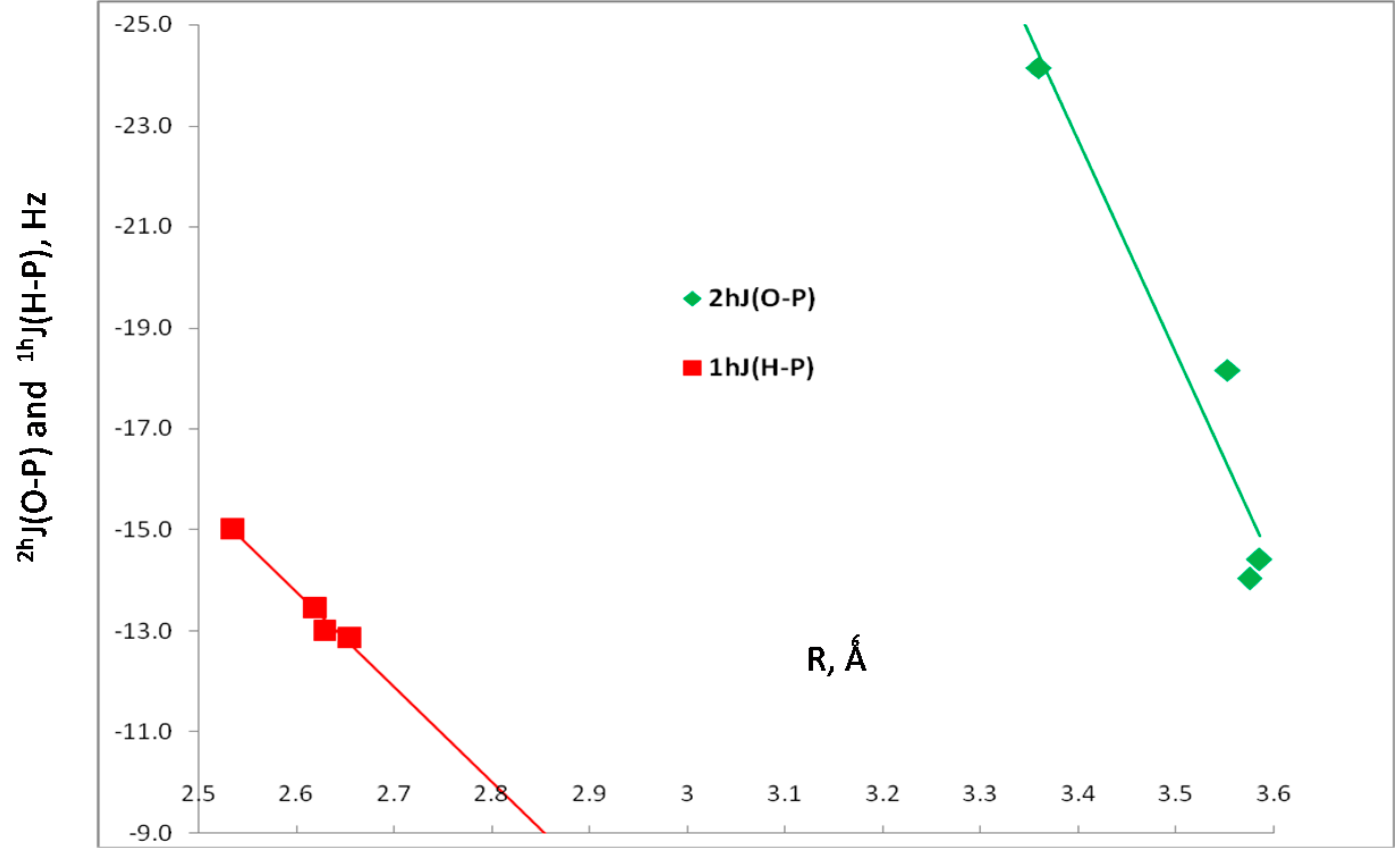
| H2XP, X = | Charge-Transfer Energies (kJ·mol−1) | Coupling Constants (Hz) | ||
|---|---|---|---|---|
| Plp→σ*H-O a | 2hJ(O-P) | 1hJ(H-P) | 1J(O-H) b | |
| Cl | 9.0 | –18.2 | –12.9 | –78.0 |
| CCH | 13.8 | –14.4 | –13.0 | –77.9 |
| H | 14.0 | –14.0 | –13.5 | –78.1 |
| CH3 | 15.0 | –24.1 | –15.0 | –78.4 |

3.2.2. Pnicogen-Bonded Complexes
| H2XP, X = | Equilibrium Symmetry | ΔE (kJ·mol−1) for Equilibrium Structures | ΔE (kJ·mol−1) for Cs Structures with H2O in-Plane | ΔE (kJ·mol−1) for Cs Structures with H2O out-of-Plane |
|---|---|---|---|---|
| NC | Cs | –21.1 | –21.1 | –19.9b |
| F | C1 | –19.5 | –19.2a | –18.5b |
| Cl | C1 | –17.3 | –17.2a | –16.3b |
| CN | Cs | –16.9 | –16.9 | –15.7b |
| OH | C1 | –12.8 | –12.4a | –12.1b |
| CCH | Cs | –11.3 | –11.3 | –10.1b |

| H2XP, X = | Distance (R, Å) | Angles (<, °) | Charge-Pransfer Energies (kJ·mol−1) | Coupling Constants (Hz) |
|---|---|---|---|---|
| R(P···O) | <O-P-Aa | Olp→σ*P-A | 1pJ(P-O) | |
| NCb | 2.800 | 165 | 20.3 | –62.5 |
| F | 2.755 | 167 | 19.5 | –69.8 |
| Cl | 2.835 | 166 | 17.4 | –61.2 |
| CNb | 2.944 | 161 | 12.3 | –41.9 |
| OH | 2.919 | 166 | 11.6 | –46.7 |
| CCH | 3.036 | 167 | 8.0 | –36.7 |

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allen, F.H.; Wood, P.A.; Galek, P.T.A. Role of chloroform and dichloromethane solvent molecules in crystal packing: An interaction propensity study. Acta Cryst. 2013, B69, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Van de Streek, J. All series of multiple solvates (including hydrates) from the Cambridge Structural Database. CrystEngComm 2007, 9, 350–352. [Google Scholar] [CrossRef]
- Infantes, L.; Chisholm, J.; Motherwell, S. Extended motifs from water and chemical functional groups in organic molecular crystals. CrystEngComm 2003, 5, 480–486. [Google Scholar] [CrossRef]
- Infantes, L.; Motherwell, S. Water clusters in organic molecular crystals. CrystEngComm 2002, 4, 454–461. [Google Scholar] [CrossRef]
- Gillon, A.L.; Feeder, N.; Davey, R.J.; Storey, R. Hydration in Molecular Crystals-A Cambridge Structural Database Analysis. Cryst. Growth Des. 2003, 3, 663–673. [Google Scholar] [CrossRef]
- Infantes, L.; Fabian, L.; Motherwell, S. Organic crystal hydrates: What are the important factors for formation. CrystEngComm 2007, 9, 65–71. [Google Scholar] [CrossRef]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Arunan, E.; Desiraju Gautam, R.; Klein Roger, A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary David, C.; Crabtree Robert, H.; Dannenberg Joseph, J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju Gautam, R.; Klein, R.-A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1,2-dicarba-closo-dodecaboranes with short P···P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Bauer, S.; Tschirschwitz, S.; Lönnecke, P.; Frank, R.; Kirchner, B.; Clarke, M.L.; Hey-Hawkins, E. Enantiomerically pure bis(phosphanyl)carbaborane(12) compounds. Eur. J. Inorg. Chem. 2009, 2009, 2776–2788. [Google Scholar] [CrossRef]
- Tschirschwitz, S.; Lonnecke, P.; Hey-Hawkins, E. Aminoalkylferrocenyldichlorophosphanes: Facile synthesis of versatile chiral starting materials. Dalton Trans. 2007, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.; Janjić, G.; Zarić, S. σ-Hole interactions of covalently-bonded nitrogen, phosphorus and arsenic: A survey of crystal structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. Intramolecular pnicogen interactions in PHF-(CH2)n-PHF (n = 2–6) systems. ChemPhysChem 2013, 14, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intramolecular pnicogen interactions in phosphorus and arsenic analogues of proton sponges. Phys. Chem. Chem. Phys. 2014, 16, 15900–15909. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The pnicogen bond in review: Structures, binding energies, bonding properties, and spin-spin coupling constants of complexes stabilized by pnicogen bonds. In Noncovalent Forces; Challenges and Advances in Computational Chemistry and Physics Series; Scheiner, S., Ed.; Springer: Gewerbestrasse, Switzerland, 2015; Volume 19, pp. 191–263. [Google Scholar]
- Allen, F. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Pople, J.A.; Binkley, J.S.; Seeger, R. Theoretical models incorporating electron correlation. Int. J. Quantum Chem. Quantum Chem. Symp. 1976, 10, 1–19. [Google Scholar] [CrossRef]
- Krishnan, R.; Pople, J.A. Approximate fourth-order perturbation theory of the electron correlation energy. Int. J. Quantum Chem. 1978, 14, 91–100. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Silver, D.M. Many-body perturbation theory applied to electron pair correlation energies. I. Closed-shell first-row diatomic hydrides. J. Chem. Phys. 1975, 62, 3258–3268. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Del Bene, J.E. Proton affinities of ammonia, water, and hydrogen fluoride and their anions: A quest for the basis-set limit using the dunning augmented correlation-consistent basis sets. J. Phys. Chem. 1993, 97, 107–110. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron Through Neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian–09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keith, T.A. AIMALL, Version 15.09.27; TK Gristmill Software: Overland Park, KS, USA, 2011. Available online: http://aim.tkgristmill.com (accessed on 31 December 2015).
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a Natural Bond Orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785789. [Google Scholar] [CrossRef]
- Perera, S.A.; Nooijen, M.; Bartlett, R.J. Electron correlation effects on the theoretical calculation of Nuclear Magnetic Resonance spin–spin coupling constants. J. Chem. Phys. 1996, 104, 3290–3305. [Google Scholar] [CrossRef]
- Perera, S.A.; Sekino, H.; Bartlett, R.J. Coupled–cluster calculations of indirect nuclear coupling constants: The importance of non–Fermi-contact contributions. J. Chem. Phys. 1994, 101, 2186–2196. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Watts, J.D.; Nooijen, M.; Oliphant, N.; Perera, S.A.; Szalay, P.S.; Lauderdale, W.J.; Gwaltney, S.R.; Beck, S.; et al. ACES II; University of Florida: Gainesville, FL, USA, 2014. [Google Scholar]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Characterizing Complexes with Pnicogen Bonds Involving sp2 Hybridized Phosphorus Atoms: (H2C=PX)2 with X = F, Cl, OH, CN, NC,CCH, H, CH3, and BH2. J. Phys. Chem. A 2013, 117, 6893–6903. [Google Scholar]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. 31P-31P Spin-spin coupling constants for Pnicogen Homodimers. Chem. Phys. Lett. 2011, 512, 184–187. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, Energies, Bonding, and NMR Properties of Pnicogen Complexes H2XP:NXH2 (X = H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Del Bene, J.E.; Elguero, J. H2XP:OH2 Complexes: Hydrogen vs. Pnicogen Bonds. Crystals 2016, 6, 19. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6020019
Alkorta I, Del Bene JE, Elguero J. H2XP:OH2 Complexes: Hydrogen vs. Pnicogen Bonds. Crystals. 2016; 6(2):19. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6020019
Chicago/Turabian StyleAlkorta, Ibon, Janet E. Del Bene, and Jose Elguero. 2016. "H2XP:OH2 Complexes: Hydrogen vs. Pnicogen Bonds" Crystals 6, no. 2: 19. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6020019