
Theoretical Studies on Hydrogen Bonds in Anions Encapsulated by an Azamacrocyclic Receptor

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussions
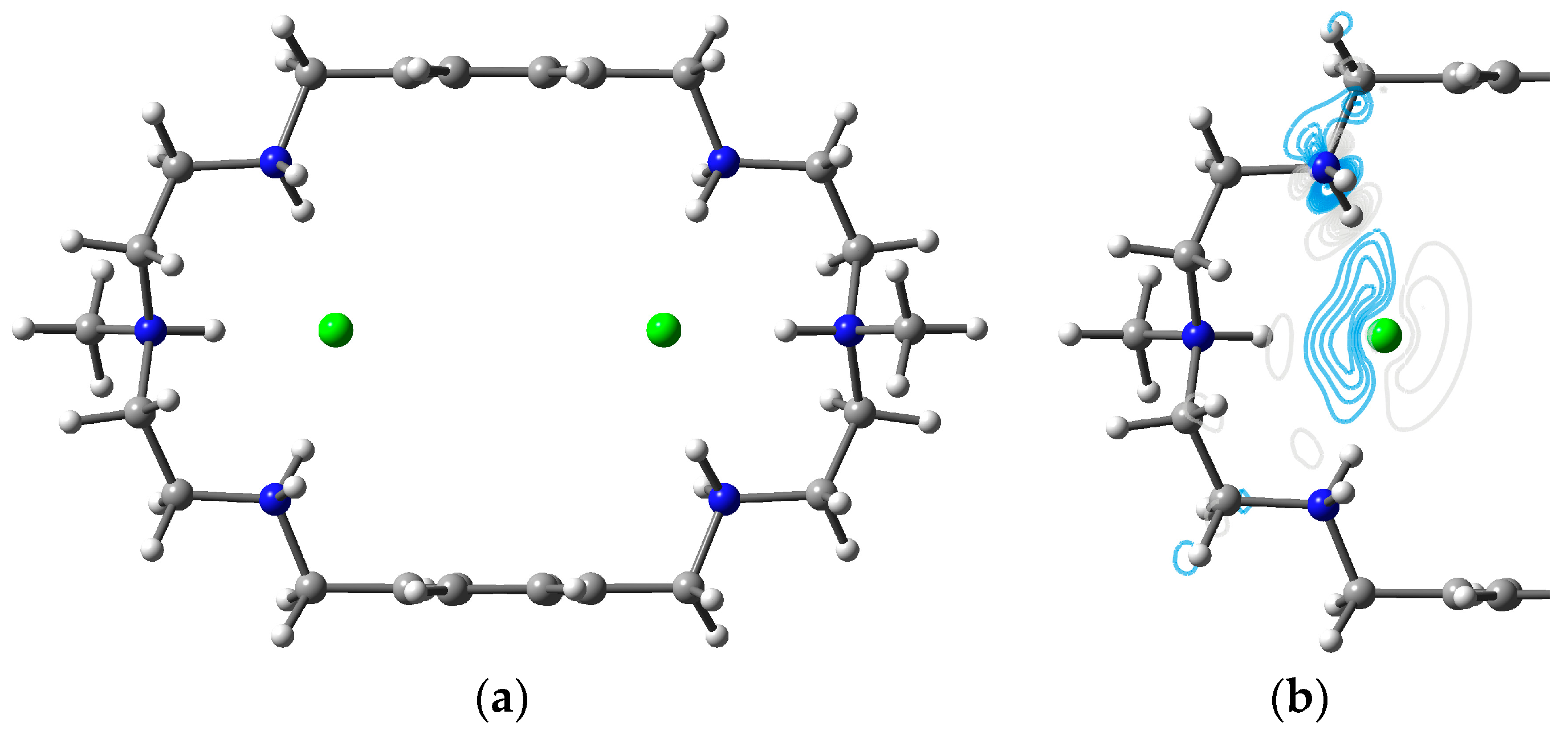
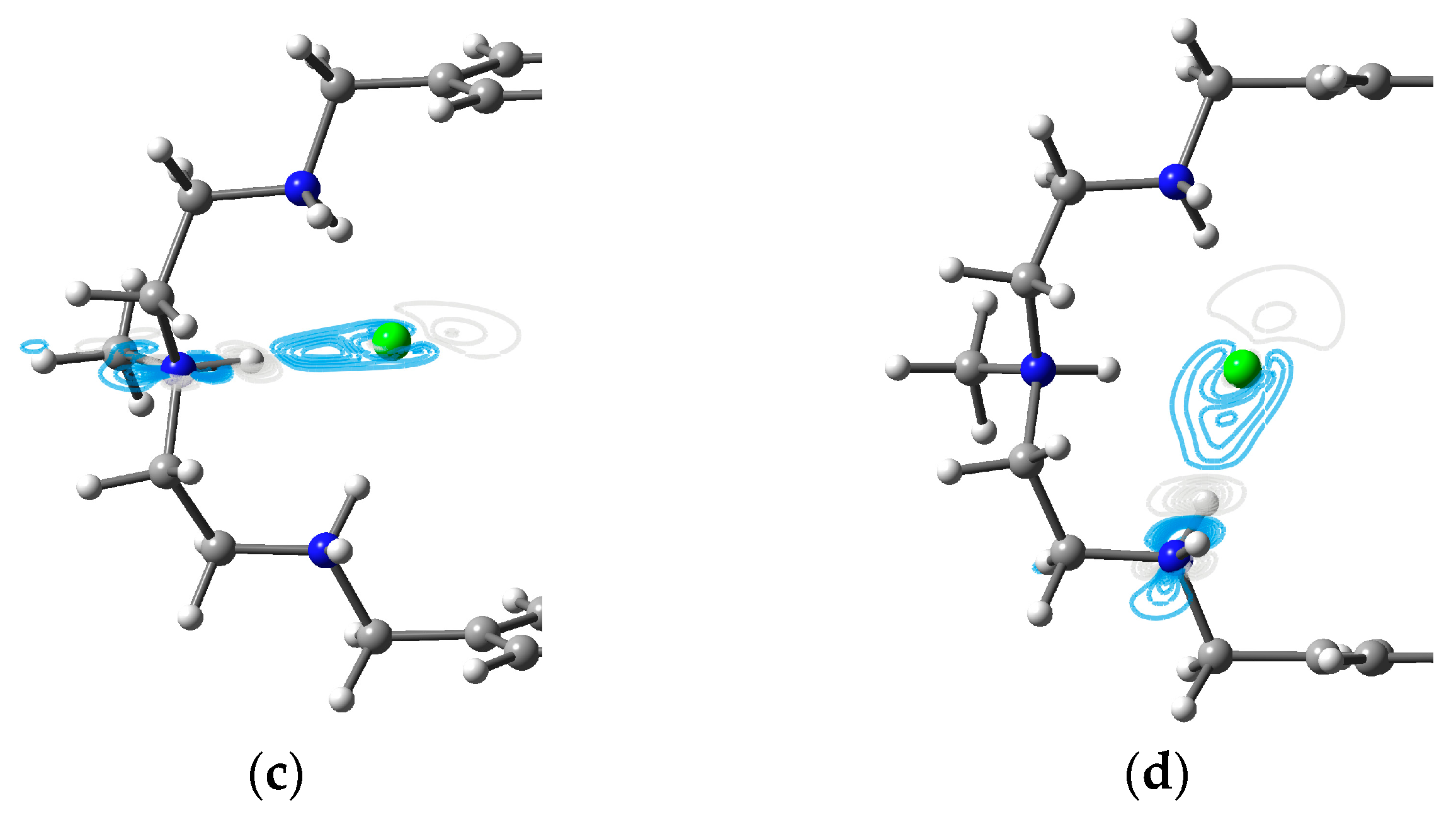
3.1. Two Chlorides Encapsulated in [H6L]6+
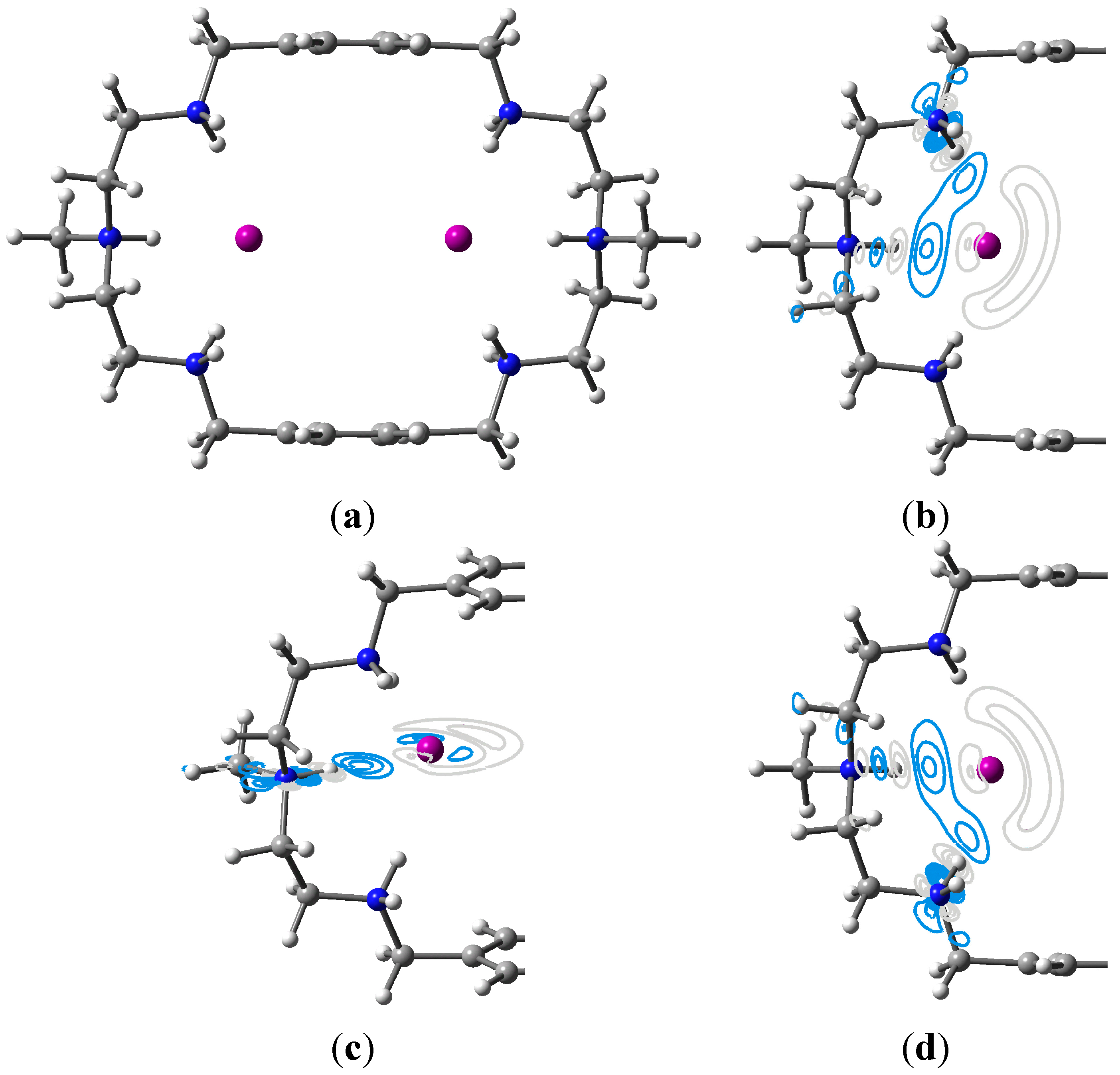
3.2. Two Bromides Encapsulated in [H6L]6
3.3. Two Iodides Encapsulated in [H6L]6
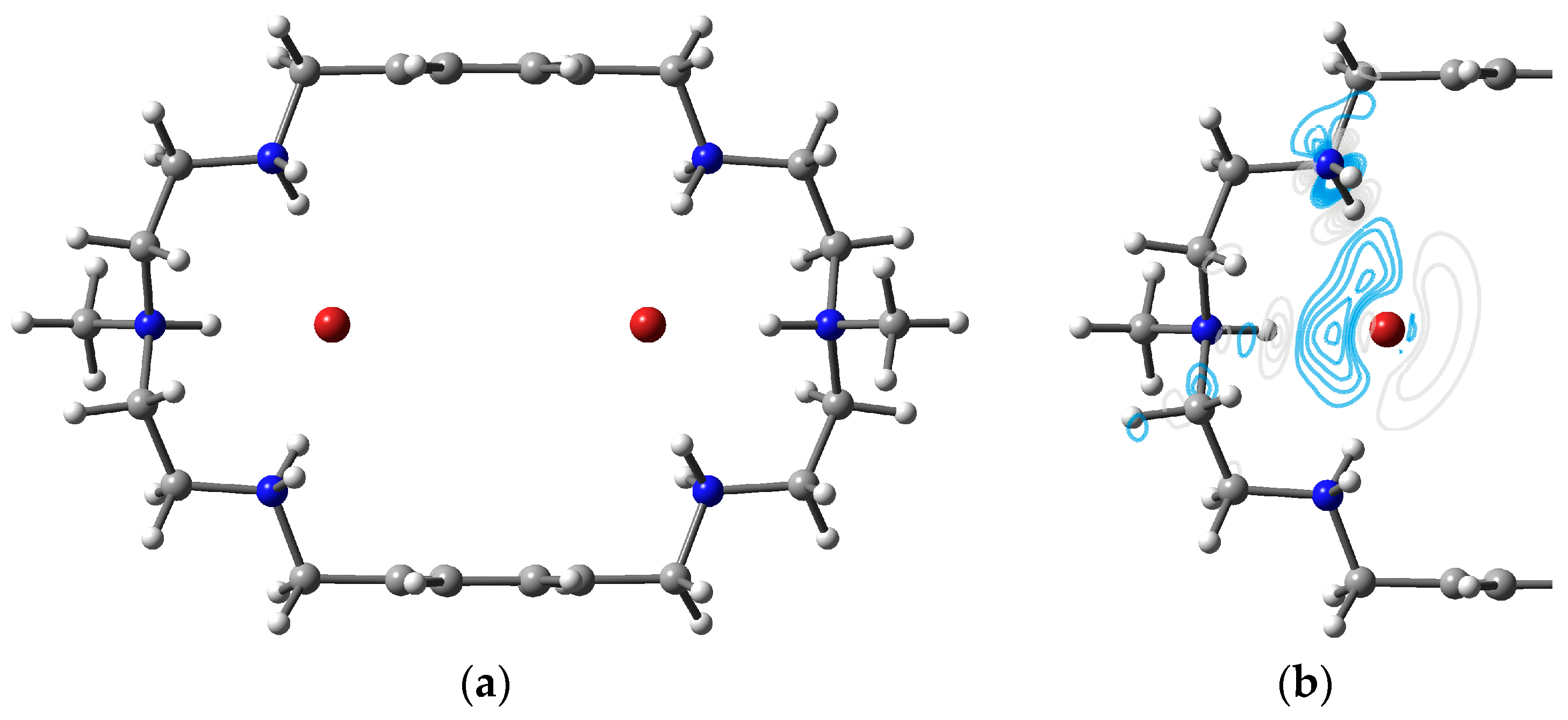
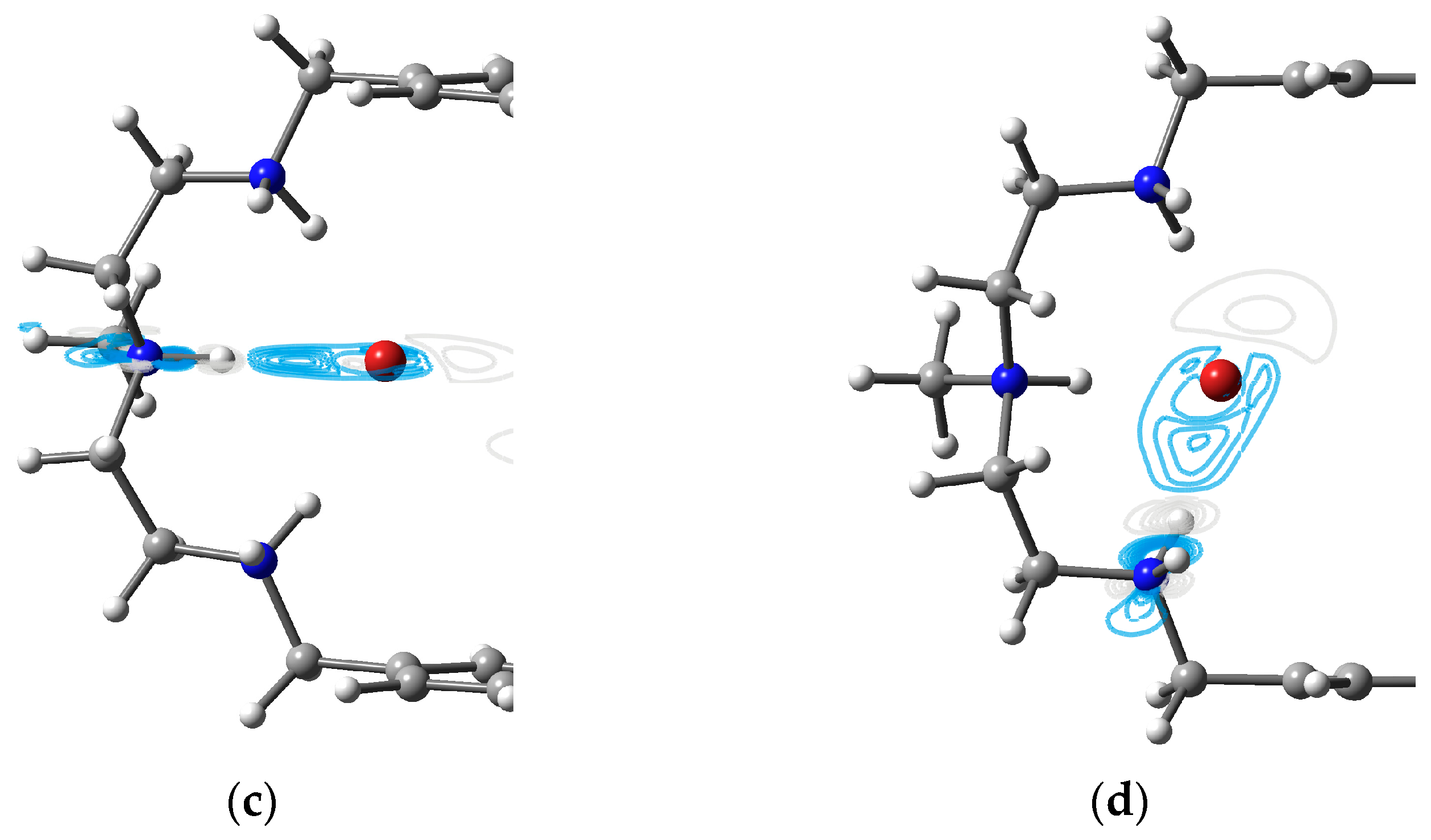
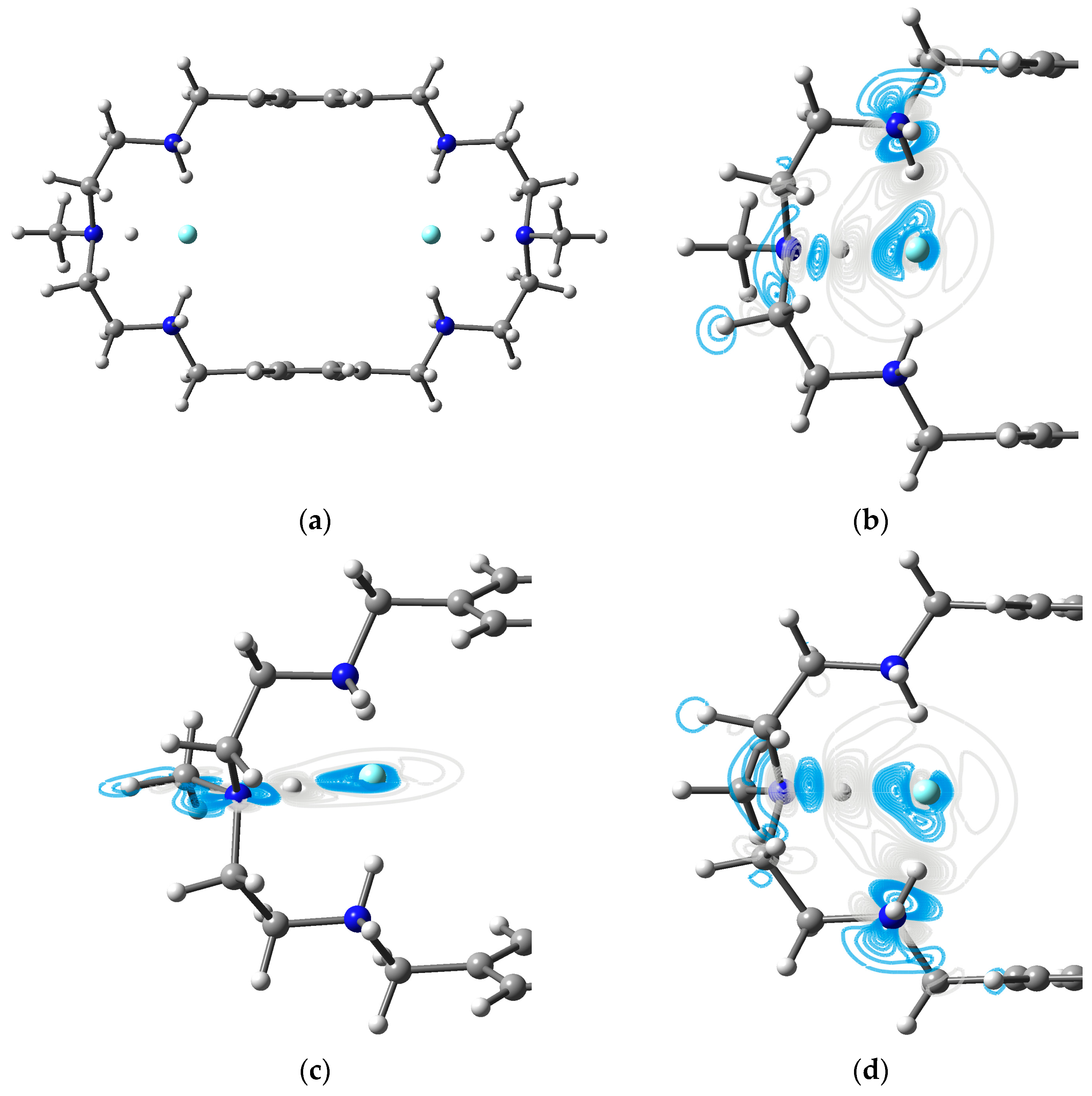
3.4. Two Fluorides Encapsulated in [H6L]6
4. Conclusions
Supplementary Materials
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Wenzel, M.; Hiscock, J.R.; Gale, P.A. Anion receptor chemistry: Highlights from 2010. Chem. Soc. Rev. 2012, 41, 480–520. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Bowman-James, K.; Garcia-Espana, E. Supramolecular Chemistry of Anions; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Amendola, V.; Esteban-Gomez, D.; Fabbrizzi, L.; Licchelli, M. What anions do to N–H-Containing Receptors. Acc. Chem. Res. 2006, 39, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, K.; Antonioli, B.; Sohnel, T.; Wenzel, M.; Gloe, K.; Gloe, K.; Price, J.R.; Lindoy, L.F.; Blake, A.J.; Schroder, M. Pylymine-based anion receptors: Extraction and structural studies. Coord. Chem. Rev. 2006, 250, 2987–3003. [Google Scholar] [CrossRef]
- Hossain, M.A. Inclusion complexes of halide anions with macrocyclic receptors. Curr. Org. Chem. 2008, 12, 1231–1256. [Google Scholar] [CrossRef]
- Révész, Á.; Schröder, D.; Svec, J.; Wimmerová, M.; Sindelar, V. Anion binding by bambus[6]uril probed in the gas phase and in solution. J. Phys. Chem. A 2011, 115, 11378–11386. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, A.; Powell, D.R.; Wong, B.M.; Hossain, M.A. Spectroscopic, structural, and theoretical studies of halide complexes with a urea-based tripodal receptor. Inorg. Chem. 2012, 51, 4274–4284. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.R.; Paterson, M.J.; Steed, J.W. A conformationally flexible, urea-based tripodal anion receptor: solid-state, solution, and theoretical studies. J. Org. Chem. 2006, 71, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Theoretical study of interactions between halogen-substituted s-triazine and halide anions. J. Phys. Chem. A 2013, 117, 8081–8090. [Google Scholar] [CrossRef] [PubMed]
- Chaumont, A.; Wipff, G. Macrotricyclic quaternary tetraammonium receptors: Halide anion recognition and interfacial activity at an aqueous interface. A molecular dynamics investigation. J. Comput. Chem. 2002, 23, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.X.; Zheng, Q.Y.; Wang, Q.Q.; Wang, M.X. Halide recognition by tetroxacalix[2]arene[2]triazine receptors: Concurrent nocovalent halide-pi and lone-pair-pi interactions in host-halide-water ternary complexes. Angew. Chem. Int. Ed. Engl. 2008, 47, 7485–7488. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Z.; Yuan, K.; Lv, L.-L.; Zhu, Y.-C.; Yuan, Z. Designation and exploration of halide-anion recognition based on cooperative noncovalent interactions including hydrogen bonds and anion-π. J. Phys. Chem. A 2015, 119, 5842–5852. [Google Scholar] [CrossRef] [PubMed]
- Rhaman, M.M.; Ahmed, L.; Wang, J.; Powell, D.R.; Leszczynski, J.; Hossain, M.A. Encapsulation and selectivity of sulfate with a furan-based hexaazamacrocyclic receptor in water. Organ. Biomol. Chem. 2014, 12, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Valencia, L.; Bastida, R.; García-España, E.; de Julián-Ortiz, J.V.; Llinares, J.M.; Macías, A.; Pérez Lourido, P. Nitrate encapsulation within the cavity of polyazapyridinophane. Considerations on nitrate-pyridine interactions. Cryst. Growth Des. 2010, 10, 3418–3423. [Google Scholar] [CrossRef]
- Chauhan, S.M.S.; Garg, B.; Bisht, T. Synthesis and anion binding of 2-Arylalazo-meso-octamethylcalix[4] pyrroles. Supramol. Chem. 2009, 21, 394–400. [Google Scholar] [CrossRef]
- Juwarker, H.; Lenhardt, J.M.; Castillo, J.C.; Zhao, E.; Krishnamurthy, S.; Jamiolkowski, R.M.; Kim, K.-H.; Graig, S.L. Anion binding of short, flexible aryl triazole oligomers. J. Org. Chem. 2009, 74, 8924–8934. [Google Scholar] [CrossRef] [PubMed]
- Mendy, J.S.; Pilate, M.L.; Horne, T.; Dey, V.W.; Hossain, M.A. Encapsulation and selective recognition of sulfate anion in an azamacrocycle in water. Chem. Commun. 2010, 46, 6084–6086. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Saeed, M.A.; Fronczek, F.R.; Wong, B.M.; Deay, K.R.; Mendy, J.S.; Gibson, D. Charge-assisted encapsulation of two chlorides by a hexaprotonated azamacrocycle. Cryst. Growth Des. 2010, 10, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.; Rhaman, M.M.; Mendy, J.S.; Wang, J.; Fronczek, F.R.; Powell, D.R.; Leszczynski, J.; Hossain, M. Experimental and theoretical studies on halide binding with a p-Xylyl-based azamacrocycle. J. Phys. Chem. A 2015, 119, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab inition calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the minnesota density functional. Chem. Phys. Lett. 2011, 502, 1–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functional for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functional and systematic testing of four M06-class functional and 12 other functional. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functional with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Radom, L.; Schleyer, P.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000—A program to analyze and visualize atoms in molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Vanquickenborne, L.G. Quantum chemistry of hydrogen bonds. In Intermolecular Forces; Huyskens, P.L., Luck, W.A.P., ZeegersHuyskens, T., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; p. 41. [Google Scholar]
- Grabowski, S.J.; Leszczynski, J. Unrevealing the nature of hydrogen bonds: PI-electron delocalization shapes H-Bond features. Intramolecular and intermolecular resonance-assisted hydrogen bonds in hydrogen bonding new insights. In Challenges and Advances in Computational Chemistry and Physics; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2006; Volume 3. [Google Scholar]
- Lipkowski, P.; Grabowski, S.J.; Leszczynski, J. Properties of the halogen-hydride interaction: An ab initio and “atoms in molecules” analysis. J. Phys. Chem. A 2006, 110, 10296–10302. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen Bonds | NH…X (Ǻ) | ρ(BCPs) (au) | ∇2ρ (au) | X…X (Å) | N2…N2’ (Ǻ) | H (Ǻ) | ΔE (kcal/mol) | logK2 (Reference [19]) | |
|---|---|---|---|---|---|---|---|---|---|
| [H6L]F2 | HB1 | 1.612 (1.627) | 0.0514 | 0.0414 | 6.563 (6.502) | 11.314 (11.106) | 7.119 (7.172) | −95.9 (−108.9) | |
| HB2 | 1.510 (1.413) | 0.0691 | 0.0485 | 2.82 | |||||
| HB3 | 1.612 (1.627) | 0.0514 | 0.0414 | ||||||
| [H6L]Cl2 | HB1 | 2.147 (2.130) | 0.0296 | 0.0171 | 5.628 (5.628) | 10.768 (10.678) | 7.927 (7.736) | −52.2 (−66.5) | |
| HB2 | 2.068 (2.049) | 0.0368 | 0.0181 | 2.70 | |||||
| HB3 | 2.147 (2.130) | 0.0296 | 0.0171 | ||||||
| [H6L]Br2 | HB1 | 2.311 (2.312) | 0.0261 | 0.0135 | 5.683 (4.831) | 10.705 (10.344) | 8.054 (7.952) | −45.5 (−60.4) | |
| HB2 | 2.233 (2.233) | 0.0319 | 0.0143 | 2.28 | |||||
| HB3 | 2.311 (2.312) | 0.0261 | 0.0135 | ||||||
| [H6L]I2 | HB1 | 2.571 (2.548) | 0.0209 | 0.0128 | 5.435 (4.821) | 10.502 (10.252) | 8.347 (8.102) | −45.7 (−63.8) | 2.20 |
| HB2 | 2.509 (2.493) | 0.0245 | 0.0134 | ||||||
| HB3 | 2.571 (2.548) | 0.0209 | 0.0128 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Gu, J.; Hossain, M.A.; Leszczynski, J. Theoretical Studies on Hydrogen Bonds in Anions Encapsulated by an Azamacrocyclic Receptor. Crystals 2016, 6, 31. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6030031
Wang J, Gu J, Hossain MA, Leszczynski J. Theoretical Studies on Hydrogen Bonds in Anions Encapsulated by an Azamacrocyclic Receptor. Crystals. 2016; 6(3):31. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6030031
Chicago/Turabian StyleWang, Jing, Jiande Gu, Md. Alamgir Hossain, and Jerzy Leszczynski. 2016. "Theoretical Studies on Hydrogen Bonds in Anions Encapsulated by an Azamacrocyclic Receptor" Crystals 6, no. 3: 31. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst6030031