Investigating Differences and Similarities between Betaxolol Polymorphs
by
 , , and
, , and
Patrizia Rossi
1 ,
,
Paola Paoli
1,*, 
Stella Milazzo
1,
Laura Chelazzi
2,
Andrea Ienco
3 and
Luca Conti
4
1
Department of Industrial Engineering, University of Florence, via Santa Marta 3, 50139 Florence, Italy
2
Centro di Cristallografia Strutturale, University of Florence, via della Lastruccia 3, Sesto F.no, 50019 Florence, Italy
3
Consiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti OrganoMetallici (CNR-ICCOM), via Madonna del Piano 10, Sesto F.no, 50019 Florence, Italy
4
Department of Chemistry “U. Schiff”, University of Florence, via della Lastruccia 3, Sesto F.no, 50019 Florence, Italy
*
Author to whom correspondence should be addressed.
Crystals 2019, 9(10), 509; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100509
Submission received: 29 August 2019
/
Revised: 13 September 2019
/
Accepted: 19 September 2019
/
Published: 29 September 2019
(This article belongs to the Special Issue Structure, Properties and Behavior of Solid Materials Explored by In-Silico and Experimental Methods)
Abstract
:Betaxolol belongs to the class of β1-adrenergic blocking agent. Several polymorphs of racemic betaxolol have been reported in the literature, but only one of them (BE_I) had the crystal structure determined from single-crystal X-ray diffraction. Here, we present a new crystalline phase of betaxolol (BE_IV). Its solid-state structure has been obtained from single-crystal X-ray diffraction data. The molecular and crystal arrangements of betaxolol in BE_IV have been further investigated by molecular modelling, by Cambridge Structural Database (CSD) surveys and by Hirshfeld surface analysis. A comparison with the solid-state structure of BE_I have been carried out. In the two polymorphs the 2-hydroxy-3-(isopropylamino)-propoxy chain, which is common to other β-blocker drugs, adopts a different conformation. In addition, the rotational isomer found in BE_IV is different with respect to the four already observed in the solid-state structure of analogous compounds. In both the polymorphs, the most significant interaction is due to the H-bonds involving the OH group as donor and the NH as acceptor, while the interaction where OH works as acceptor (NH acts as donor) is definitely less important. The resulting H-bond patterns are however different: Alternate R2,2(10) a > a (OH donors) and R2,2(10) b > b (OH acceptors) in BE_I vs. alternate R4,4 (8) a > b > a > b (OH donors) and R2,2 (10) b > b (OH acceptor) in BE_IV.
1. Introduction
If a substance exists in more than one crystal form, these different solid phases are designated as polymorphs and the phenomenon is known as polymorphism. For a given compound, only the form having the lowest Gibbs energy is thermodynamically stable. However, in some cases, due to the height of the energy barrier separating some polymorphs and the most stable one, the metastable forms remain stable for a period of time long enough to be used for practical purposes as if they were stable forms [1,2]. There are cases where obtaining a given polymorph is very difficult even if this solid form had previously been usually obtained for long time. This is the case of the so-called elusive polymorphs, which are quite often found in molecules having high conformational freedom [3].
The arrangement of the molecules of an Active Pharmaceutical Ingredient (API) in a solid determines its physical and chemical properties, which are strictly related with the performance. In addition, the API’s solid form can affect pharmaceutical operations such as: Milling, mixing, filtering, washing, drying, tableting and dissolution. Finally, polymorphism poses patent issues. Hence, the importance of a detailed solid-state characterization when dealing with APIs.
In the past, we have used X-ray diffraction, both from single crystal and microcrystalline powder, and computational methods to investigate solid-state features and correlate them to the chemical and physical properties of APIs as well as of metal complexes [4,5,6,7]. For example, recently we have reported on a new crystal form of dexketoprofen (β) [8]. Its crystal and molecular structure has been studied by experimental and in silico techniques and compared with the already known solid form (α); thermal data suggest that the two polymorphs are in a monotropic relationship. A similar methodological approach has been used to characterize the solid forms of metoprolol both as free base and as succinate, tartrate and fumarate salts [9,10,11]. In particular X-ray diffraction data and computational results have been used to try to correlate solid-state features to thermal behavior (e.g., isotropic vs. anisotropic thermal expansion).
Betaxolol, or (R,S)-1-{p-[2-(cyclopropylmethoxy) ethyl] phenoxy}-3-isopropylamino-2-propanol (see Scheme 1a), shares with metoprolol and a large number of β-blocker drugs the 2-hydroxy-3-isopropylaminopropoxy side arm and acts as a cardioselective β1 -adrenergic blocking agent. The main use of β1-blockers is the treatment of hypertension. Amongst β-blockers, betaxolol is a long acting highly selective β1 blocking agent (half-life ~ 19 h) and has several advantages compared to the others [12].
The drug is usually manufactured and marketed as a racemic mixture, notwithstanding the fact that the β1 blocking activity resides in the S enantiomer. Most of the racemic β blocker drugs are in fact effective because their S-enantiomer shows great structural similarities to the adrenergic hormone noradrenaline, whereas the R-enantiomer is responsible mostly for side effects [13].
The crystal structure of the first known phase of racemic betaxolol (BE_I in the following) was published in 2008 by Canotilho et al. [14]. Single crystals of BE_I were obtained from methanol:water (20:80 v/v) mixture and characterized by single crystal X-ray diffraction. BE_I crystallized in the triclinic system in the P-1 space group. In the asymmetric unit one disordered molecule of betaxolol is present; such disorder was modelled by using two positions for the atoms of the cyclopropylmethoxy fragment. In 2013 another metastable polymorph (polymorph II, BE_II) was found and characterized by Maria et al. [15], although neither its crystal parameters were reported nor its structure was solved, this new phase was characterized using thermal analysis (DSC and PLTM), spectroscopic methods (MAS and NMR) and X-ray powder diffraction (XRPD). The authors established a monotropic relationship between polymorph I and polymorph II, where the latter is the metastable form [15]. Finally, Maria et al. speculated on the existence of a third low ordered intermediate metastable phase (polymorph III, BE_III).
We present here the molecular and crystal structure of a new solid form of betaxolol (BE_IV, hereafter), which has also been investigated with the programs Mercury CSD [16] and Crystal- Explorer17 [17] to identify the most important intermolecular contacts in the crystal packing. The results are discussed and compared to those obtained for BE_I.
2. Materials and Methods
Betaxolol hydrochloride [(R,S)-1-{p-[2-(cyclopropylmethoxy)ethyl]phenoxy}-3-isopropylamlno-2-propanol hydrochloride (CAS 63659-19-8, product code B5683-50MG)] and all the solvents and resins were purchased from Sigma-Aldrich (Sigma-Adrich, Milano, Italy) and used without further purification.
2.1. Synthesis and Crystallization
Single crystals of form IV of (R,S)-betaxolol (BE_IV) were prepared from its hydrochloride salt. Betaxolol hydrochloride was dissolved in a minimal amount of mQ water (200 μL for 200 mg of betaxolol) and passed through an anion exchange resin (Dowex Marathon 11 chloride form - Sigma Aldrich CAS 69011-19-4) allowing to obtain the free base form of the drug directly in water.
The solvent was completely removed by distillation under reduced pressure and the resulting solid was dissolved in methanol: Water (20:80 percent (%), v/v). By means of a very slow evaporation of the organic phase at 3–4 °C, BE_IV single crystals were obtained after three weeks.
2.2. Single Crystal X-Ray Data Collection and Structure Solution
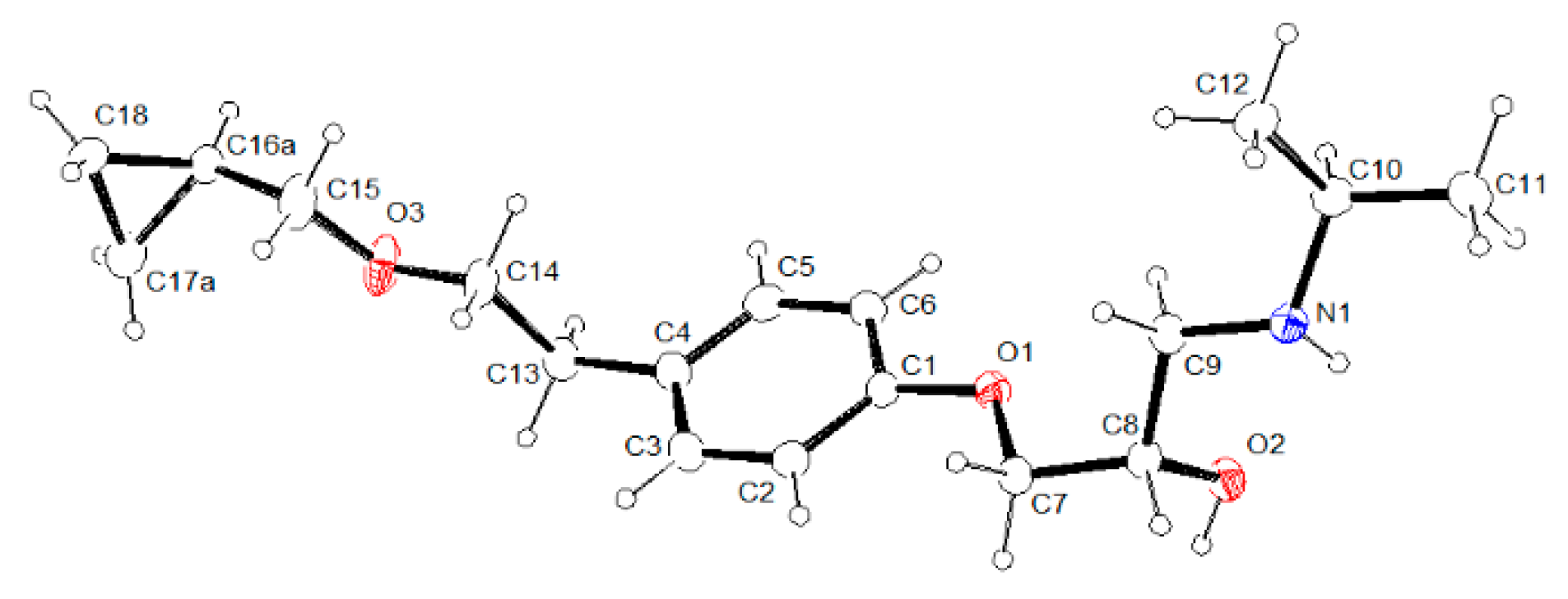
The crystal structure of betaxolol BE_IV was investigated by single-crystal X-ray diffraction. Measurements were carried out at 100, 200 and 300 K, with an Oxford Diffraction Excalibur diffractometer (Rigaku Oxford Diffraction, The Woodlands, Texas, USA) using a Mo-Kα radiation (λ = 0.71073 Å). In all cases, data collection was performed with the program CrysAlis CCD [18]. Data reduction was carried out with the program CrysAlis RED (CrysAlis RED, 2006) (Agilent Technologies UK Ltd., Yarnton, England) [18]. Absorption correction was performed with the program ABSPACK in CrysAlis RED. Betaxolol crystallizes in the monoclinic crystal system, space group I2/a. The structure was solved by using the SIR-97 package [19] and subsequently refined on the F2 values by the full-matrix least-squares program SHELXL-2017 (University of Gottingen, Gottingen, Germany) [20]. Geometrical calculations were performed by PARST97 (University of Parma, Parma, Italy) [21], and molecular plots were produced by the programs ORTEP-3 [22] (University of Glasgow, Glasgow, UK), Mercury (v4.1.3) (Cambridge Structural Data Centre, Cambridge, UK) [16] and Discovery Studio 2019 Client (Dassault Systèmes BIOVIA: San Diego, CA, USA) [23]. All the hydrogen atoms were found in the Fourier density map with the exclusion of the ones belonging to the disordered fragment (see below) and their coordinated were freely refined, while their thermal parameters were refined in agreement to the atom to which they are bonded. All the non-hydrogen atoms were refined anisotropically. The atoms C16, C17 of the cyclopropyl fragment are in disordered position. Such disorder was modelled using for each atom two positions having a final occupancy factor of 0.74 and 0.26, respectively. Double positions were used also for the hydrogen atoms bonded to C15 and C18. The molecular and crystal structures of BE_IV determined at 100, 200 and 300 K are not significantly different (see for example, Figure S1), as a consequence in the paper only the results from crystal data collected at 100 K will be reported and discussed. Table 1 lists crystal data and refinement parameters of BE_IV, at 100 K (see Table S1 for 200 and 300 K crystal data). In Figure 1 an ORTEP-3 view of the asymmetric unit of BE_IV is reported with the labelling scheme. Structural data can be obtained free of charge from The Cambridge Crystallographic Data Centre CCDC deposition number 1949001) [24].
2.3. Computational Methods
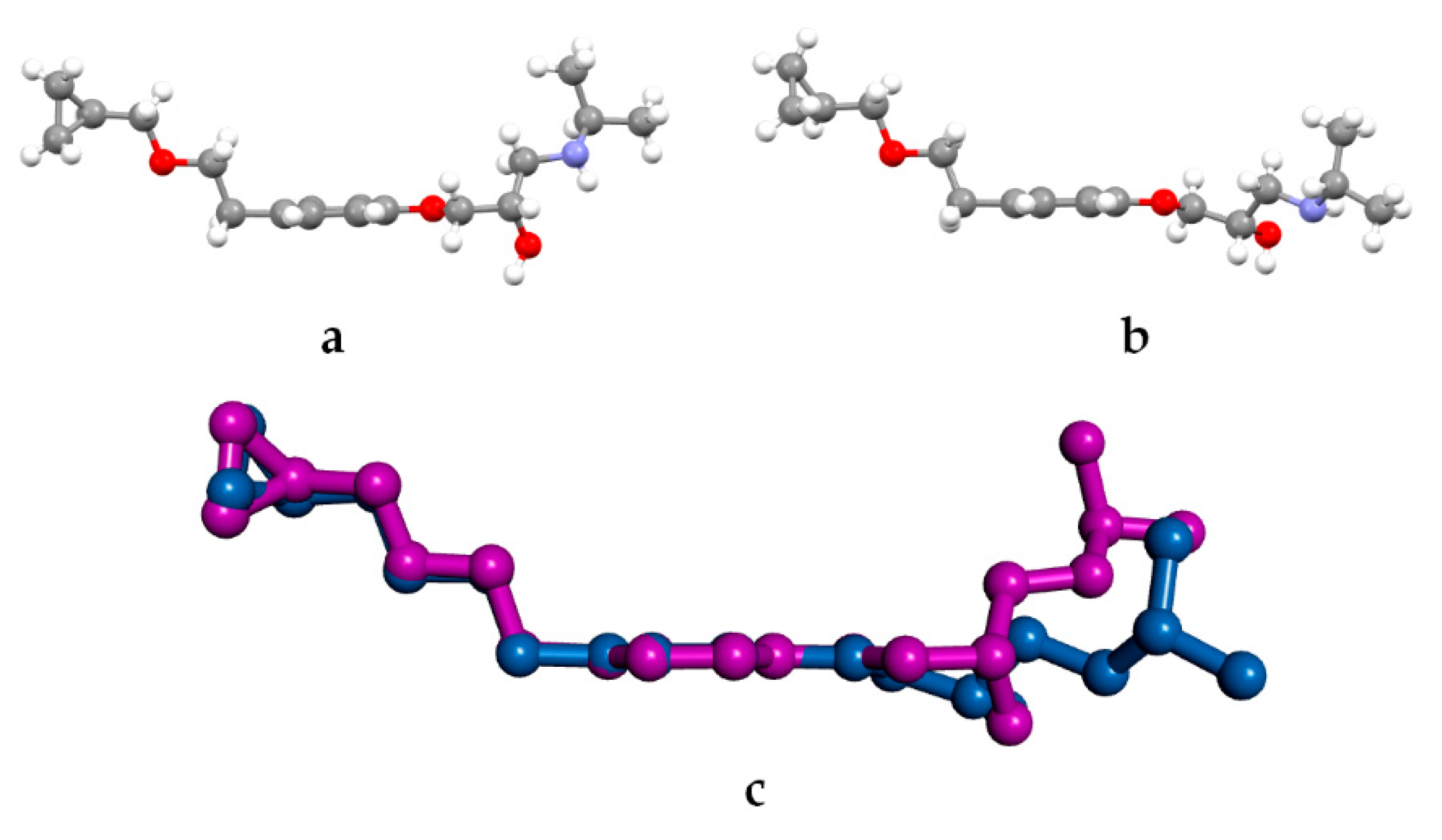
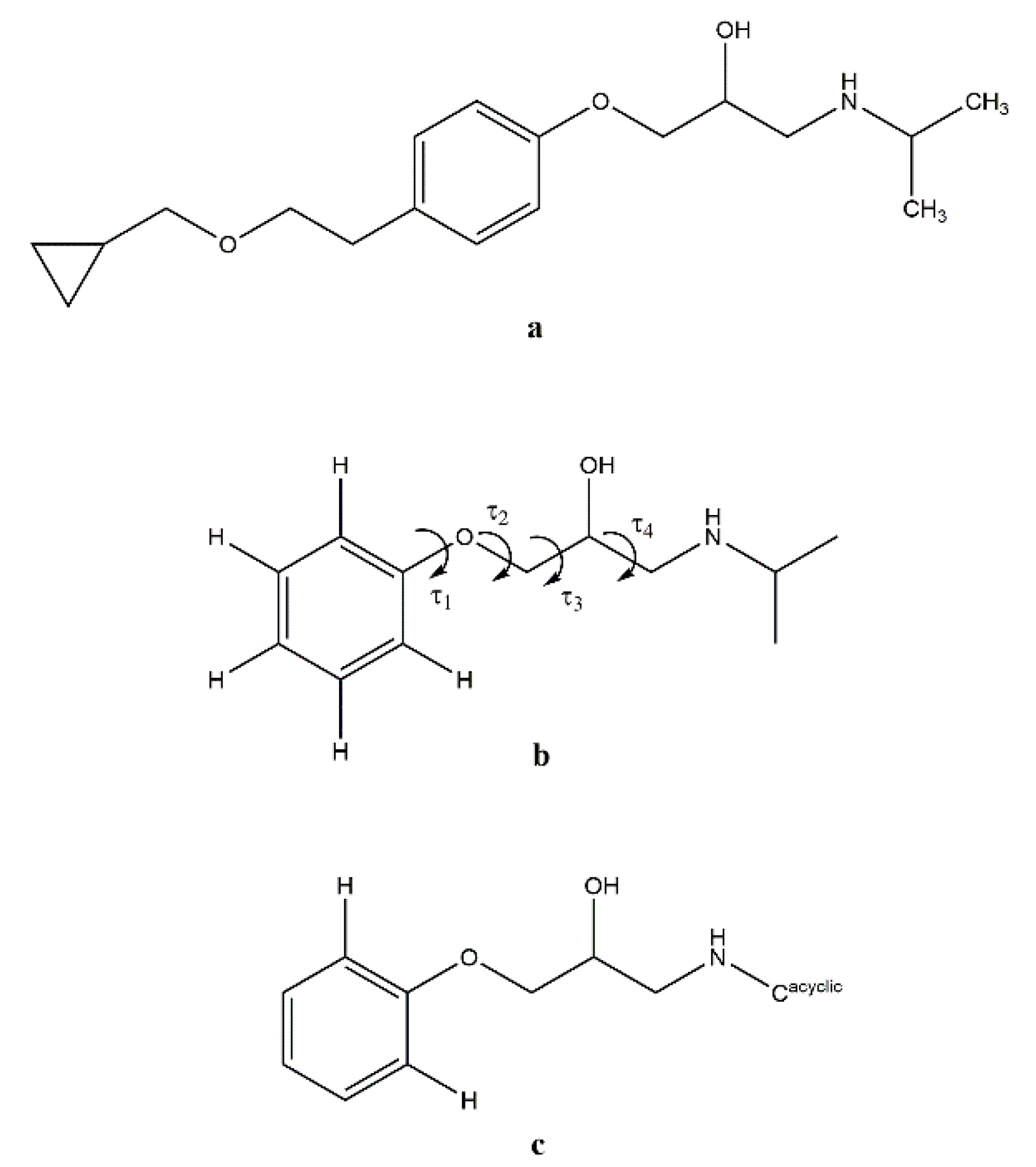
GAUSSIAN09 [25] was used for quantum chemical (QC) calculations using the following functionals: B3LYP [26,27] and B97-D [28]. The basis set was 6-311G (d,p) [29]. The Berny algorithm was used. [30]. The reliability of the stationary points was assessed by evaluation of the vibrational frequencies. The model molecule (BS) shown in Scheme 1b was used for the QC calculations. The starting conformation for the hydroxyamino side chain was that found in the crystal structure of BE_IV, i.e., ttg+t (BS_G+T, hereafter). A relaxed Potential Energy Surface (PES) scan was carried out about the τ3 dihedral angle (Scheme 1b; increment size 18°, steps 20, HF/6-311G (d,p) model chemistry).
The CSD Materials software [16] was used to analyze the crystal packing arrangement and calculating intermolecular energies using the UNI intermolecular potential [31,32] in order to identify the intermolecular interactions which are most significant from an energetic point of view.
CrystalExplorer17 [17] was used to compute Hirshfeld surfaces (HS) and their associated 2D (two-dimensional) fingerprint plots. Interaction energies between betaxolol molecules in BE_IV and BE_I at the HF/3-21G and B3LYP/6-31G** levels of theory were also calculated. In both the crystal structures the cyclopropylmethoxy chain is disordered over two positions and the set of coordinates having the highest occupancy factor was used to generate the HSs and for energy calculations.
3. Results and Discussion
Three polymorphs of betaxolol are known. The first one (BE_I) was fully characterized at the solid state by Canotilho et al. [14] in 2008. The structure of the second metastable form (BE_II) is not known but this polymorph has been characterized using thermal analysis (DSC and PLTM), spectroscopic methods (MAS and NMR) and X-ray powder diffraction (XRPD) by Maria et al. [15] in 2013. Finally, the existence of the third metastable form (BE_III) has only been hypothesized [15]. The betaxolol phase that we describe here (BE_IV) is then the fourth for this API. Considering that for BE_II and BE_III no structural characterization has been made, the difference between these two phases and BE_IV was established on the basis of the comparison of the experimental XRPD patterns of BE_II and BE_III reported in ref [15] with the theoretical one of BE_IV and on the different melting point observed for BE_II (Tm = 33 °C) and BE_IV. For this last polymorph melting point was estimated (see Section 3.1) on the basis of a hot stage microscopy analysis (crystals melt at about 53 °C, see Figure 2).
3.1. Synthesis
Preliminary to the crystal preparation, betaxolol hydrochloride has been converted to its free amine form. To this aim, the employment of an anion exchange resin (see experimental section for further details) was found to be a successful method to obtain the free amine form of the drug, thus representing a valid alternative to the solvent extraction method commonly reported in literature [33,34].
Of worth noting, in the crystal preparation procedure, the kinetic of the evaporation of the organic phase from samples revealed to strongly influence the crystal phase of the solid obtained at the end of the procedure. In particular, a fast removal of the organic phase at room temperature afforded to obtain the phase I of the drug after only 4 days, while cooling of the organic mixture to 3–4 °C afforded to obtain a few single crystals of the IV phase (after three weeks). Although numerous attempts were made to reproduce the crystallization process leading to the new polymorph BE_IV, the thermodynamically stable phase I (BE_I) was always obtained. In this respect BE_IV belongs to the class of elusive polymorphs.
3.2. Molecular Structure from Single-Crystal X-Ray Diffraction and Computational Methods
BE_IV crystallizes in the monoclinic system, I2/a space group, with one molecule of betaxolol in the asymmetric unit. As already observed in BE_I, also in BE_IV the cyclopropyl group is affected by disorder which has been modelled by using two positions (a and b, see Figure S2) for the atoms C16 and C17 (with an occupancy factor of 0.74 and 0.26, for a and b, respectively).
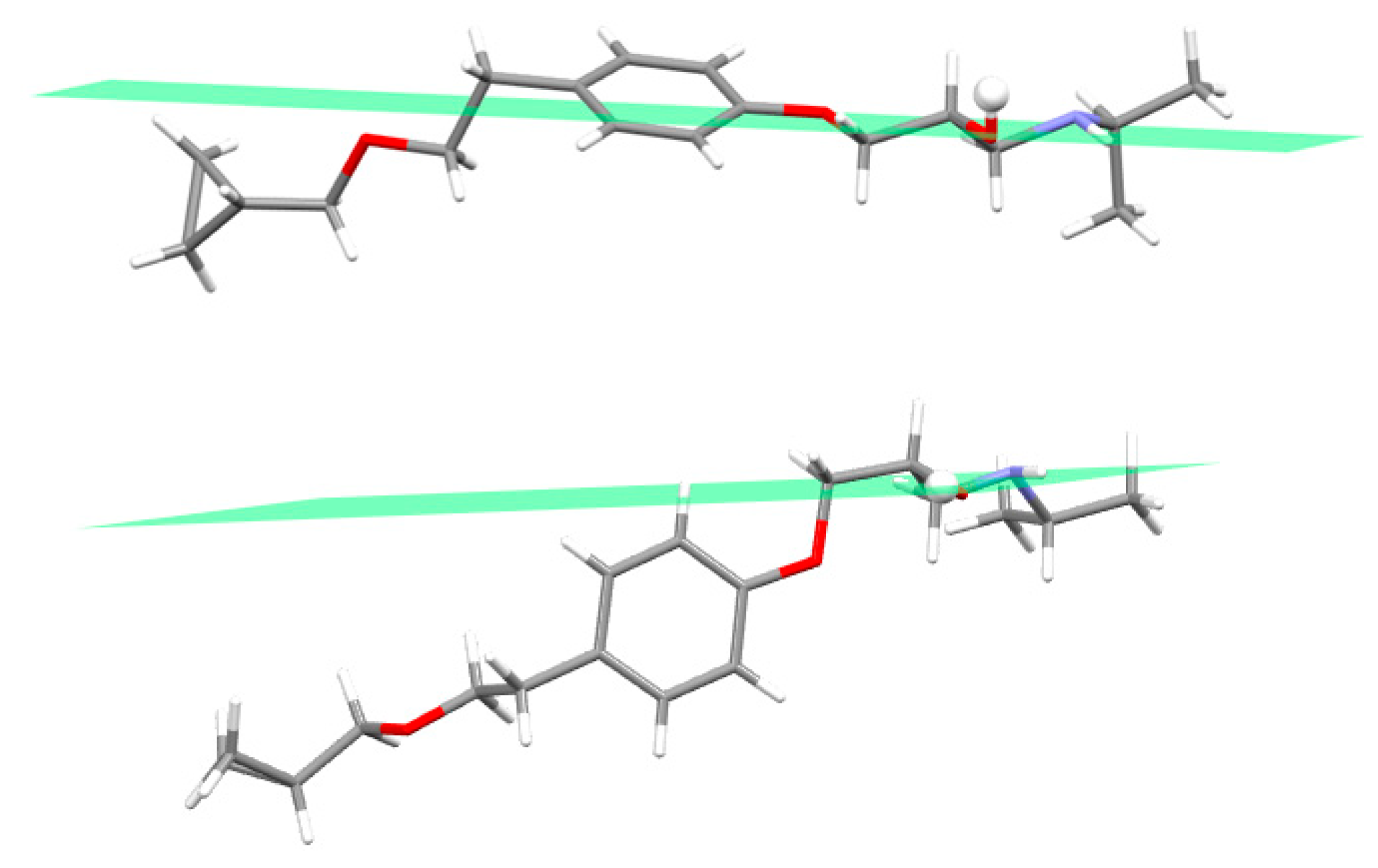
As a whole the betaxolol molecule in BE_IV is U-shaped, i.e., the two arms are on the same side of the phenoxy ring (see Figure 3a). In particular the molecule possesses a central planar fragment (the related mean plane will be referred as A in the following) containing the phenoxy group and the C7 and C13 atoms (see Figure 1 for the labelling), also C13, C14, O3 and C15 lie on a mean plane (B in the following) as well as C7, C8, C9 and N1 (C in the following). The angles formed by the B and C planes with the central one (A) are 79.8(2) and 58.6(2)°, respectively vs. 86 and 68°, in BE_I [14]. The betaxolol molecules in BE_IV and BE_I well superimpose as for the central phenoxy group and the cyclopropylmethoxyethyl side chain, while they differ for the arrangement of 2-hydroxy-3-isopropylamino- propoxy side arm (see Figure 3). As for the latter the sequence of the torsion angles τ1–τ4 (Scheme 1b, Table S2) is trans-trans-gauche (+)-trans (ttg+t) and trans-trans-gauche(-)-trans (ttg-t), in BE_IV and BE_I, respectively.
In a previous paper dealing with metoprolol [10] we have concluded that the hydroxyamino side arm is quite flexible, being able to change its 3D arrangement in response to the environment, as provided by X-ray (crystal environment), MD (in vacuum and simulated solvent) and QC (in vacuum) data. The fact that in BE_IV the 2-hydroxy-3-isopropylamino-propoxy side arm adopts a conformation (i.e., ttg+t) different with respect to the four already observed (aT, tttg+, ttg-t and ttg+g+) in the Cambridge Structural Database [35] (see Scheme 1c for the searched fragment), further strengthen that observation [10]. The rotational isomer found in BE_IV (ttg+t) is the highest in energy amongst the five, as provided by the geometry optimization of the model molecule BS_G+T (see experimental section). For example, the energy difference between BS_G+T (ttg+t conformation as in BE_IV) and BS_G-T (ttg-t conformation as in BE_I) is 5.4 kJ/mol. The energy barrier separating the BS_G+T and BS_G-T isomers should be about 12.5 kJ/mol, as roughly estimated by a relaxed Potential Energy Surface scans about τ3.
In addition, the relative arrangement of the OH/NH groups in BE_I and BE_IV is different: In BE_I the hydroxy hydrogen atom is definitely out of the OCCNH mean plane, while in BE_IV the HOCCNH atoms are well in a plane (Figure 4, Scheme S1 and Table S2).
In conclusion, in BE_I and BE_IV the betaxolol molecules differ for the overall conformation of the 2-hydroxy-3-isopropylamino-propoxy side arm as well as for the relative arrangement of OH/NH groups.
3.3. Crystal Structure from Single Crystal X-Ray Diffraction and Computational Methods
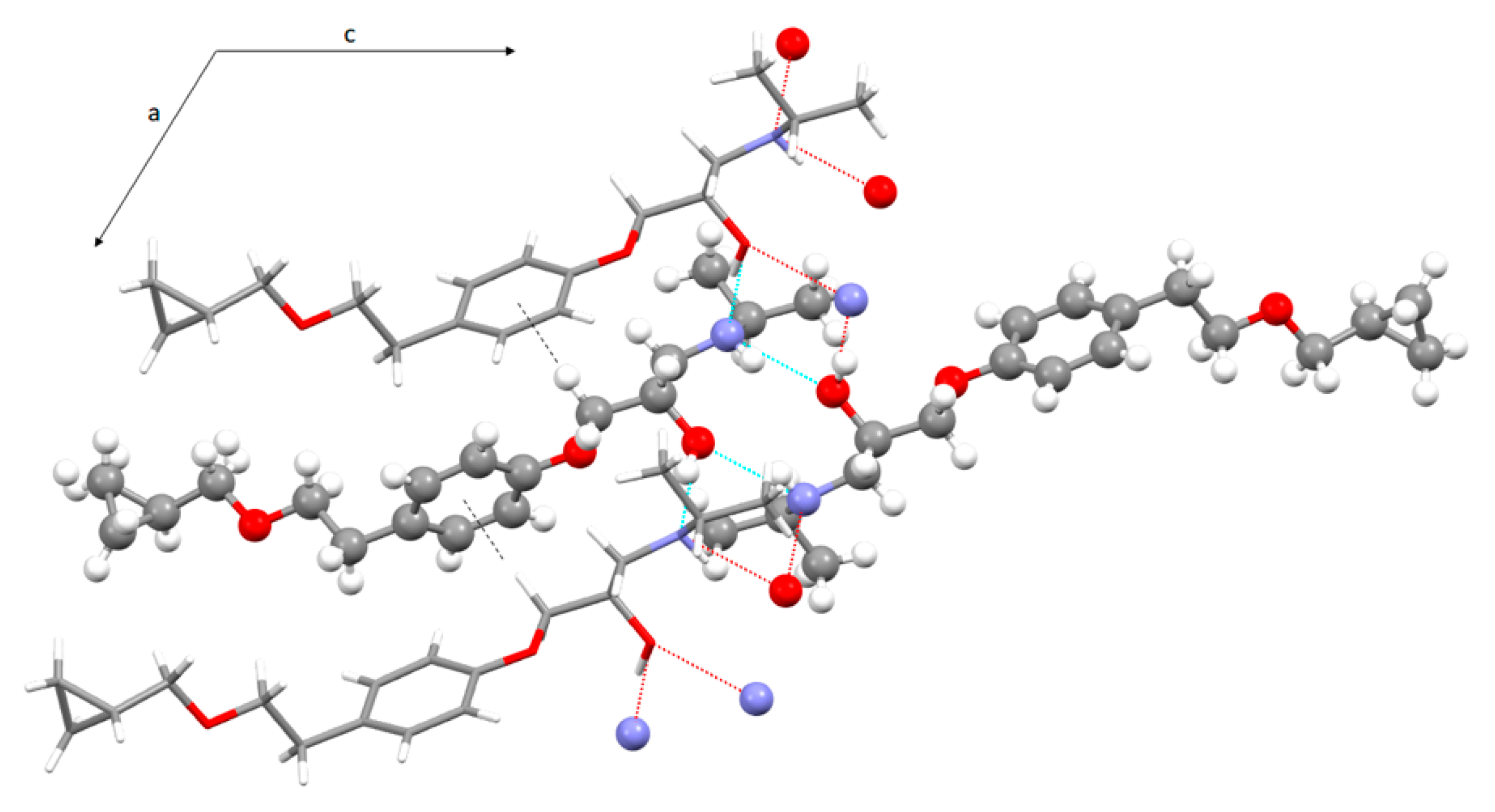
In the crystal lattice of BE_IV betaxolol molecules are linked via strong H-bonds [36] involving the hydroxy and amino groups which act both as H-bond donors and acceptors. In particular zig-zag chains of iso-oriented alternating R and S molecules extending along the a axis direction are formed through OH…N bonds (1.84(3) Å, 167(3)°, Table 2). In addition, pairs of homochiral molecules are H-bonded via NH…O interactions (2.22(3) Å, 157(2)°). As a result, the crystal lattice is made up of chains of alternating betaxolol enantiomers H-bonded through the OH donor developing along the a axis, these chains are facing each other via H-bonds through the hydroxy groups acting as acceptors (Figure 5).
As already found for analogous species [10], when the hydroxyl group acts as a donor toward the N atom of an inverted neighboring betaxolol molecule the resulting hydrogen bond is strong; by contrast the hydroxy group acts as a definitely weaker hydrogen-bond acceptor toward the NH group of an identical molecule. As a whole, these interactions give rise to two alternating hydrogen-bond patterns of R 4,4 (8) and R 2,2 (10) type (R 4,4 (8) a > b > a > b and R 2,2 (10) b > b) [37], which are responsible for the formation of infinite double chains of alternating R and S betaxolol molecules extending along the a-axis direction (Figures S3–S5). Finally, a relatively weak interaction of the CH…π type (C7-H7A…(C1-C6)centroid = 2.78(3) Å and 132(2)°), exists between adjacent molecules belonging to the same chain.
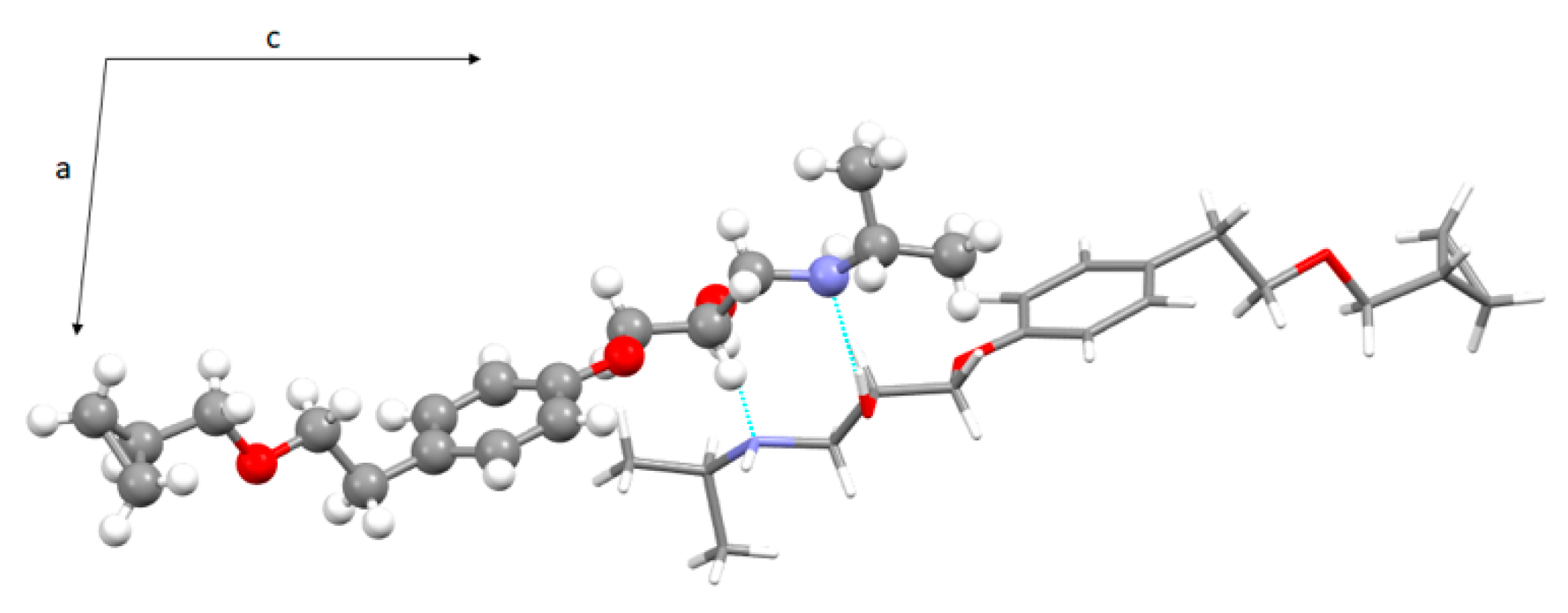
A comparison with the crystal structure of BE_I [15] shows several analogies: The hydroxy and amino groups are involved both as acceptors and donors of H-bonds; the OH group is definitely a stronger H bond donor than acceptor (Table S3 in the supporting information); chains of facing betaxolol molecules extend along the shortest axis direction (Figure 6).
However, in the two crystal packings R and S enantiomers pack differently: Omochiral stacks in BE_I vs. heterochiral stacks in BE_IV (Figure 7). More in details: In BE_I, the net of H-bonds give rise to two R2,2(10) motifs (R2,2(10) a > a (OH donors) and R2,2(10) b > b (OH acceptors)) which alternate along the doubled chain. In BE_IV, R4,4,(8) and R2,2 (10) patterns alternate along the coupled chains. The latter motif (OH acceptors) involves two homochiral betaxolol molecules, while R4,4(8) results from the arrangement of four molecules (the hydroxy groups act both as acceptors and donors of H-bonds).
As already evidenced by us [10], identical R2,2(10) motifs were found in the metoprolol (XITNIA refcode) [11], propranolol (PROPRA10 refcode) [38] and a reaction intermediate in an alternative route for the synthesis of atenolol (KAZPOQ refcode) [39].
A search in the CSD for the R4,4(8) pattern (see Scheme S2 for the searched fragment) gives a few crystal structures (20 hits), 4 of them [40,41,42,43] shows alternating R4,4(8)-R2,2(10) motifs as in BE_IV, in 3 of them [40,41,42] the R2,2(10) motif is made up of the H-bond donor hydroxy group as in BE_IV.
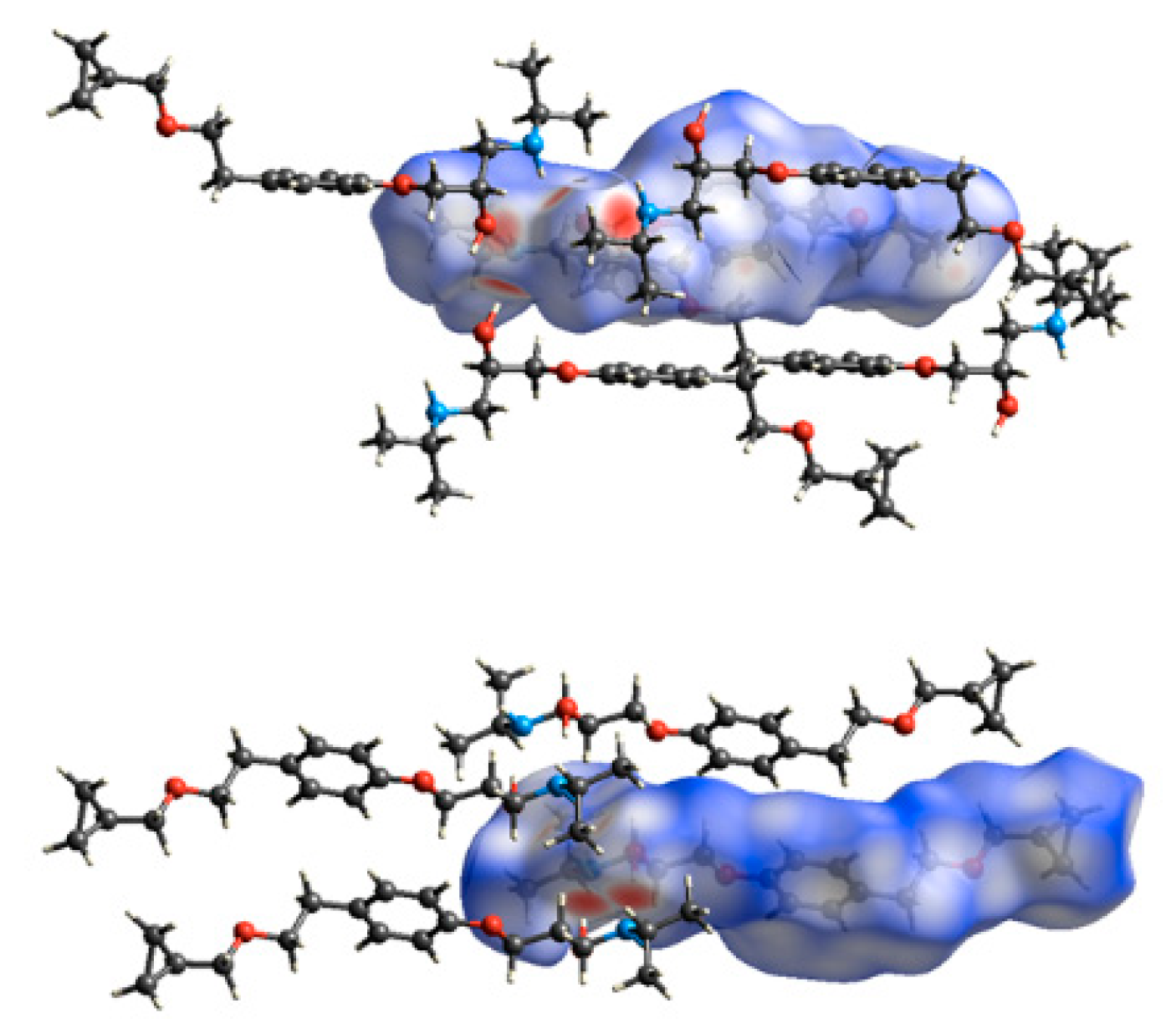
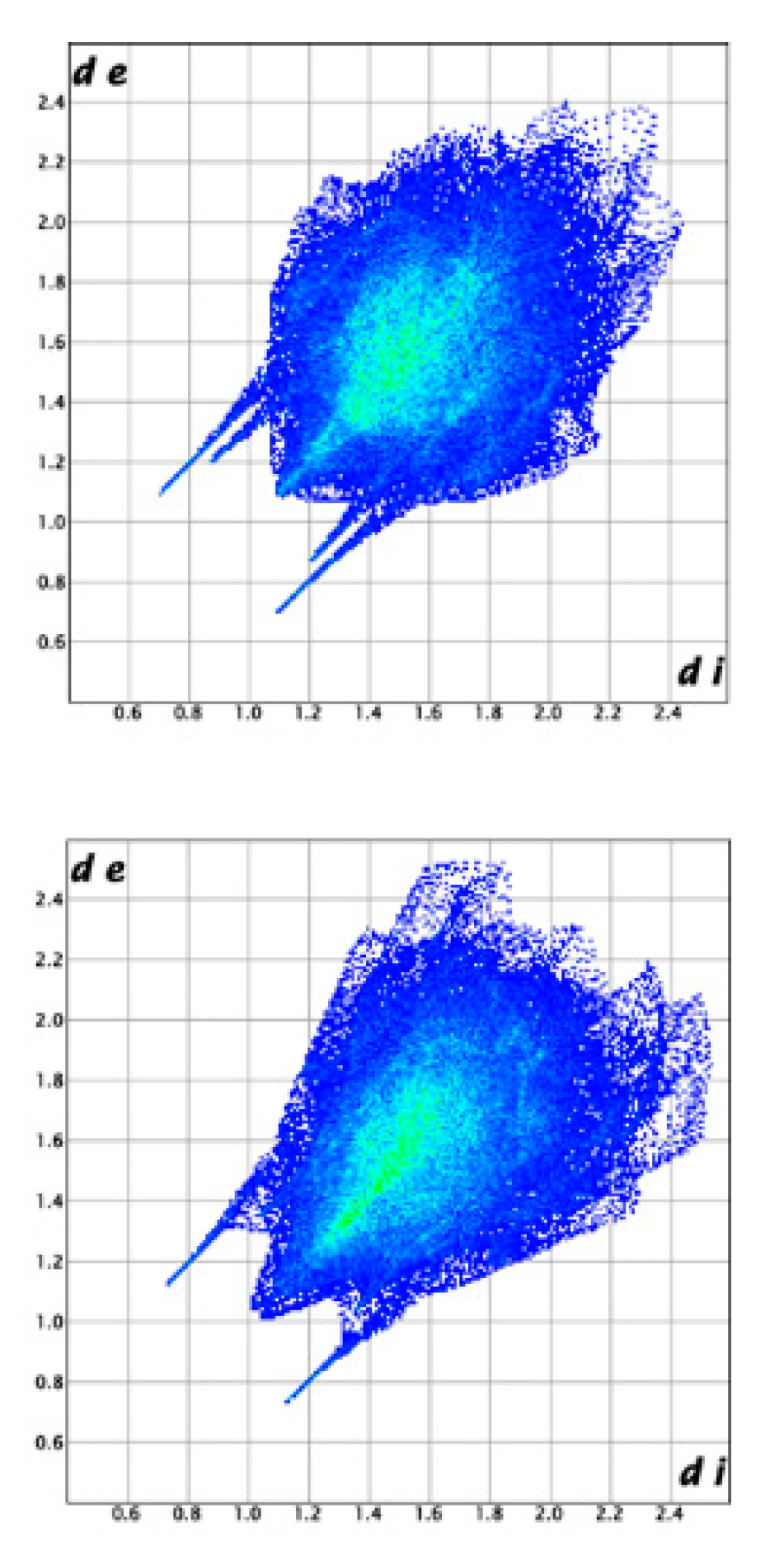
The intermolecular interactions that build up the crystal packings of BE_IV and BE_I were further studied using the Hirshfeld Surface (HS) analysis. Figure 8 shows the corresponding HSs mapped with dnorm. In both cases, the dominant interactions are the O-H…N hydrogen bonds (two large red spots); the weaker O…H-N interactions show up as pale-red regions. In the corresponding fingerprint plots, given in Figure 9, a pair of spikes account for the OH…N (upper left) and N…OH (bottom left) hydrogen bonds and two lateral wings relate to the C-H…π contacts. In BE_IV there is an additional pair of spikes due to the H-bonds involving the hydroxy group as acceptor which in BE_I do not show up as spikes because of the longer O…HN distance (2.562 Å vs. 2.22(3)Å). In both the crystal structures, the relative contributions to the Hirshfeld surface area due to N…H, O…H and C…H contacts are very similar (1.2, 10.2 and 9.6% for BE_I; 1.6, 9.8 and 11.4% for molecule BE_IV).
Intermolecular interaction energies (Table S4 in the supporting information) were then calculated to get a quantitative picture of the interaction strength between molecular pairs in BE_IV and BE_I. All the used models (UNI, HF and B3LYP) confirm that the most important interaction in energetic terms is that involving the hydroxy group as a donor (contact distances = 1.84(3) and 2.039 Å, in BE_IV and BE_I, respectively). The O…HN intermolecular hydrogen bond appear definitely less important, based on energetic grounds, being the second strongest interaction that between molecules interacting through CH…π contacts (contact distances = 2.78(3) and 3.18 Å).
In conclusion, in both the betaxolol solid forms the most significant intermolecular interaction involves the OH as donor and the NH as acceptor. The resulting H-bond patterns are however different: Alternate R2,2(10) a > a (OH donors) and R2,2(10) b > b (OH acceptors) in BE_I, alternate R4,4 (8) a > b > a> b (OH donors/acceptors) and R 2,2 (10) b > b (OH acceptor) in BE_IV.
4. Conclusions
The paper reports on the solid-state structure of a new elusive solid form of betaxolol (BE_IV). In BE_IV and BE_I the molecular conformation adopted by the hydroxyamino chain is different. This is not surprising given that the 2-hydroxy-3-isopropylamino-propoxy side arm can exist in different conformations and is quite mobile (as provided by a CSD survey and modelling data). The betaxolol molecules also differ for the relative orientation of the OH/NH groups, which give rise to different H-bonds patterns in the two polymorphs: Alternate R2,2(10) a > a (OH donors) and R2,2(10) b > b (OH acceptors) in BE_I vs. alternate R 4,4 (8) a > b > a > b (OH donors/acceptors) and R2,2 (10) b > b (OH acceptor) in BE_IV. In both the solid forms the dominant interactions, from an energetic point of view, are the O-H…N hydrogen bonds, followed by CH…π contacts, being the O…HN intermolecular hydrogen bonds less important. Finally, the higher melting point of the BE_IV form is (53 vs. 33 °C; density differences are definitely less significant: 1.15 vs. 1.13 mg/cm3) suggests a growing cohesive force in this solid form. Actually, in BE_IV each betaxolol molecule is H-bonded with 3 symmetry related molecule vs. 2 in BE_I.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4352/9/10/509/s1: Table S1: Cell parameters of BE_IV at 200 and 300 K; Table S2: Selected dihedral angles of BE_I and BE_IV (S enantiomer); Table S3: Selected intermolecular interactions in BE_I (ROKNUB refcode in the Cambridge Structural Database); Table S4. Intermolecular interaction energies; Scheme S1: Dihedral angles definition for the ethanolamine fragment; Scheme S2: Fragment searched in the CSD for the R4,4(8) pattern; Figure S1: Superimposition of the betaxolol molecule in BE_IV at 100 (blue), 200 (green) and 300 K (pink). Figure S2: Double configuration of the cyclopropylmethoxy fragment; Figures S3 and S4: Views of the crystal packing of BE_IV. Figure S5: View of the crystal packing of BE_IV along the c axis direction highlighting the zig-zag arrangement of the alternating R and S betaxolol molecules within each chain. For sake of clarity for cyclopropylmethoxy the model having the highest occupancy factor was shown.
Author Contributions
Conceptualization, P.P. and P.R.; methodology, L.C. (Luca Conti) and S.M.; software, A.I.; investigation, P.P., P.R. and L.C. (Laura Chelazzi); data curation, P.P. and P.R.; writing—original draft preparation, P.P., P.R. and S.M.; writing—review and editing, P.P., P.R. and S.M.; supervision, P.P.
Funding
This research received no external funding.
Acknowledgments
Authors thank the Centro di Cristallografia Strutturale (CRIST) of the University of Florence for the X-ray diffraction facilities and Samuele Ciattini for his valuable technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prasanthi, N.L.; Sudhir, M.; Jyothi, N.; Sri vajrapriya, V. A review on polymorphism perpetuates pharmaceuticals. Am. J. Adv. Drug Deliv. 2016, 4, 58–63. [Google Scholar]
- Redinha, J.S.; Lopes Jesus, A.J.; Crystal. Crystal Growth of Pharmaceuticals from Melt. In Crystallization and Materials Science of Modern Artificial and Natural Crystals, 1st ed.; Borisenko, E., Kolesnikov, N., Eds.; InTech: Rijeka, Croatia, 2012; Volume 10, pp. 225–248. [Google Scholar]
- Dunitz, J.D.; Bernstein, J. Disappearing polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Chelazzi, L.; Altamura, M.; Fedi, V.; Giannotti, D. Solid State Investigation and Characterization of a Nepadutant Precursor: Polymorphic and Pseudopolymorphic Forms of MEN11282. Cryst. Growth Des. 2016, 16, 5294–5304. [Google Scholar] [CrossRef]
- Rossi, P.; Macedi, E.; Paoli, P.; Bernazzani, L.; Carignani, E.; Borsacchi, S.; Geppi, M. Solid-Solid Transition between Hydrated Racemic Compound and Anhydrous Conglomerate in Na-Ibuprofen: A Combined X-ray Diffraction, Solid-State NMR, Calorimetric, and Computational Study. Cryst. Growth Des. 2014, 14, 2441–2452. [Google Scholar] [CrossRef]
- Amatori, S.; Ambrosi, G.; Borgogelli, E.; Fanelli, M.; Formica, M.; Fusi, V.; Giorgi, L.; Macedi, E.; Micheloni, M.; Paoli, P.; et al. Modulating the Sensor Response to Halide Using NBD-Based Azamacrocycles. Inorg. Chem. 2014, 53, 4560–4569. [Google Scholar] [CrossRef]
- Crociani, B.; Antonaroli, S.; Burattini, M.; Paoli, P.; Rossi, P. Palladium complexes with a tridentate PNO ligand. Synthesis of η1-allyl complexes and cross-coupling reactions promoted by boron compounds. Dalton Trans. 2010, 39, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Paoli, P.; Ienco, A.; Biagi, D.; Valleri, M.; Conti, L. A new crystal form of the NSAID dexketoprofen. Acta Cryst. 2019, 75, 783–792. [Google Scholar] [CrossRef]
- Paoli, P.; Rossi, P.; Macedi, E.; Ienco, A.; Chelazzi, L.; Bartolucci, G.L.; Bruni, B. Similar but Different: The Case of Metoprolol Tartrate and Succinate Salts. Cryst. Growth Des. 2016, 16, 789–799. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. Metroprolol Fumarate: Crystal Structure from Powder X-ray Diffraction Data and Comparison with the Tartrate and Succinate Salts. Cryst. Growth Des. 2018, 18, 7015–7026. [Google Scholar] [CrossRef]
- Rossi, P.; Paoli, P.; Chelazzi, L.; Conti, L.; Bencini, A. The solid-state structure of the β-blocker metoprolol: A combined experimental and in silico investigation. Acta Cryst. 2019, C75, 87–96. [Google Scholar]
- Cierpka-Kmieć, K.; Hering, D. Tachycardia: The hidden cardiovascular risk factor in uncomplicated arterial hypertension. Cardiol. J. 2019, in press. [Google Scholar] [CrossRef]
- Mehvar, R.; Brocks, D.R. Stereospecific Pharmacokinetics and Pharmacodynamics of Beta-Adrenergic Blockers in humans. J. Pharm. Pharmaceut. Sci. 2001, 4, 185–200. [Google Scholar]
- Canotilho, J.; Castro, R.A.E.; Rosado, M.T.S.; Silva, M.R.; Beja, A.M.; Paixão, J.A.; Redinha, J.S. The structure of betaxolol from single crystal X-ray diffraction and natural bond orbital analysis. J. Mol. Struct. 2008, 891, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Maria, T.M.R.; Castro, R.A.E.; Silva, M.R.; Ramos, M.L.; Justino, L.L.G.; Burrows, H.D.; Canolitho, J.; Eusébio, M.E.S. Polymorphism and melt crystallisation of racemic betaxolol, a β-adrenergic antagonist drug. J. Therm. Anal. Calorim. 2013, 111, 2171–2178. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17, version 17; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- CrysAlisPro, Oxford Diffraction, version 2011; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Da Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT- Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Nardelli, M. PARST95—An update to PARST: A system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Cryst. 1995, 28, 659. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Discovery Studio Visualizer, version 19.1.0.18287; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019.
- Access Structures. Available online: https://www.ccdc.cam.ac.uk/structures (accessed on 24 September 2019).
- GAUSSIAN09; revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Gavezzotti, A. Are Crystal Structures Predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A. The Crystal Packing of Organic Molecules: Challenge and Fascination Below 1000 Da. Crystallogr. Rev. 1998, 7, 5–121. [Google Scholar] [CrossRef]
- Canotilho, J.; Castro, R.A.E. The structure of betaxolol studied by infrared spectroscopy and natural bond orbital theory. Spectrochimica Acta Part A. 2010, 76, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, C.; Caira, M.R.; Bojita, M.T.; Nassimbeni, L.R.; Mhlongo, W.T. Solid state characterization of metoprolol free base and metoprolol tartrate. FARMACIA 2006, 54, 9–17. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology, 1st ed.; Oxford Science Publications: Great Britain, UK, 1999. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Ammon, H.L.; Howe, D.-B.; Erhardt, W.D.; Balsamo, A.; Macchia, B.; Macchia, F.; Keefe, W.E. The crystal structures of dichloroisoproterenol, propranolol and propranolol hydrochloride. Acta Cryst. 1977, B33, 21–29. [Google Scholar] [CrossRef]
- Akisanya, J.; Parkins, A.W.; Steed, J.W. A Synthesis of Atenolol Using a Nitrile Hydration Catalyst. Org. Process Res. Dev. 1998, 2, 274–276. [Google Scholar] [CrossRef]
- Ning, F.; Anderson, R.J.; Hibbs, D.E.; Groundwater, P.W. A new, one-step synthesis of 1-heteroaryl-2-alkylaminoethanols. Tetrahedron Lett. 2010, 51, 843–845. [Google Scholar] [CrossRef]
- Mereiter, K.; Frohlich, J.; Ilk, R. CCDC 164293: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2001. [Google Scholar] [CrossRef]
- Batey, R.A.; MacKay, D.B.; Santhakumar, V. Alkenyl and Aryl Boronates-Mild Nucleophiles for the Stereoselective Formation of Functionalized N-Heterocycles. J. Am. Chem. Soc. 1999, 121, 5075–5076. [Google Scholar] [CrossRef]
- Palchykov, V.A.; Zarovnaya, I.S.; Tretiakov, S.V.; Reshetnyak, A.V.; Omelchenko, I.V.; Shishkin, O.V.; Okovytyy, S.I. Synthesis and characterization of sulfolane-based amino alcohols: A combined experimental and computational study. J. Mol. Struct. 2018, 1157, 149–158. [Google Scholar] [CrossRef]
Scheme 1.
(a) Betaxolol molecule; (b) model molecule (BS) used for the QC calculations; (c) fragment searched in the Cambridge Structural Database with evidenced the four analyzed torsion angles.
Scheme 1.
(a) Betaxolol molecule; (b) model molecule (BS) used for the QC calculations; (c) fragment searched in the Cambridge Structural Database with evidenced the four analyzed torsion angles.

Figure 1.
ORTEP-3 view of the betaxolol molecule BE_IV. For sake of clarity only the set of coordinates having the highest occupancy factor has been reported (occupancy factor 74%).
Figure 1.
ORTEP-3 view of the betaxolol molecule BE_IV. For sake of clarity only the set of coordinates having the highest occupancy factor has been reported (occupancy factor 74%).

Figure 2.
Hot stage microscopy images collected at 52 and 53 °C of a crystal of BE_IV.

Figure 3.
(a) Ball and stick view of the conformation of betaxolol in BE_IV; (b) ball and stick view of the conformation of betaxolol in BE_I; (c) superimposition of betaxolol in BE_I (blue) and BE_IV (pink). For sake of clarity only the set of coordinates having the highest occupancy factor has been reported.
Figure 3.
(a) Ball and stick view of the conformation of betaxolol in BE_IV; (b) ball and stick view of the conformation of betaxolol in BE_I; (c) superimposition of betaxolol in BE_I (blue) and BE_IV (pink). For sake of clarity only the set of coordinates having the highest occupancy factor has been reported.

Figure 4.
Betaxolol molecules in BE_I (top) and BE_IV (bottom) highlighting the relative position of the mean plane (in green) passing through the OCCNH atoms and the OH hydrogen atom. For sake of clarity only the set of coordinates having the highest occupancy factor has been reported.
Figure 4.
Betaxolol molecules in BE_I (top) and BE_IV (bottom) highlighting the relative position of the mean plane (in green) passing through the OCCNH atoms and the OH hydrogen atom. For sake of clarity only the set of coordinates having the highest occupancy factor has been reported.

Figure 5.
View of the H-bond and CH…π network in the crystal packing of BE_IV along the b axis direction (S enantiomer in ball and stick, R enantiomer in stick). For sake of clarity only the set of coordinates having the highest occupancy factor was shown.
Figure 5.
View of the H-bond and CH…π network in the crystal packing of BE_IV along the b axis direction (S enantiomer in ball and stick, R enantiomer in stick). For sake of clarity only the set of coordinates having the highest occupancy factor was shown.

Figure 6.
View of the H-bond network in the crystal packing of BE_I along the b axis direction (S enantiomer in ball and stick, R enantiomer in stick). For sake of clarity only the set of coordinates having the highest occupancy factor was shown.
Figure 6.
View of the H-bond network in the crystal packing of BE_I along the b axis direction (S enantiomer in ball and stick, R enantiomer in stick). For sake of clarity only the set of coordinates having the highest occupancy factor was shown.

Figure 7.
View of the crystal packing of: (A) BE_I and (B) BE_IV (S enantiomer in green, R enantiomer in pink). For sake of clarity, only the set of coordinates having the highest occupancy factor was shown).
Figure 7.
View of the crystal packing of: (A) BE_I and (B) BE_IV (S enantiomer in green, R enantiomer in pink). For sake of clarity, only the set of coordinates having the highest occupancy factor was shown).

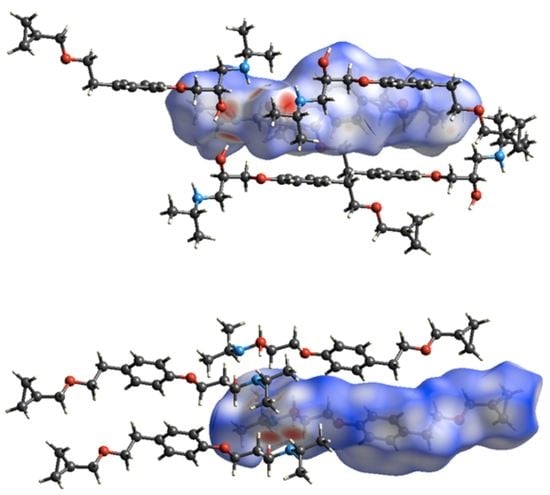
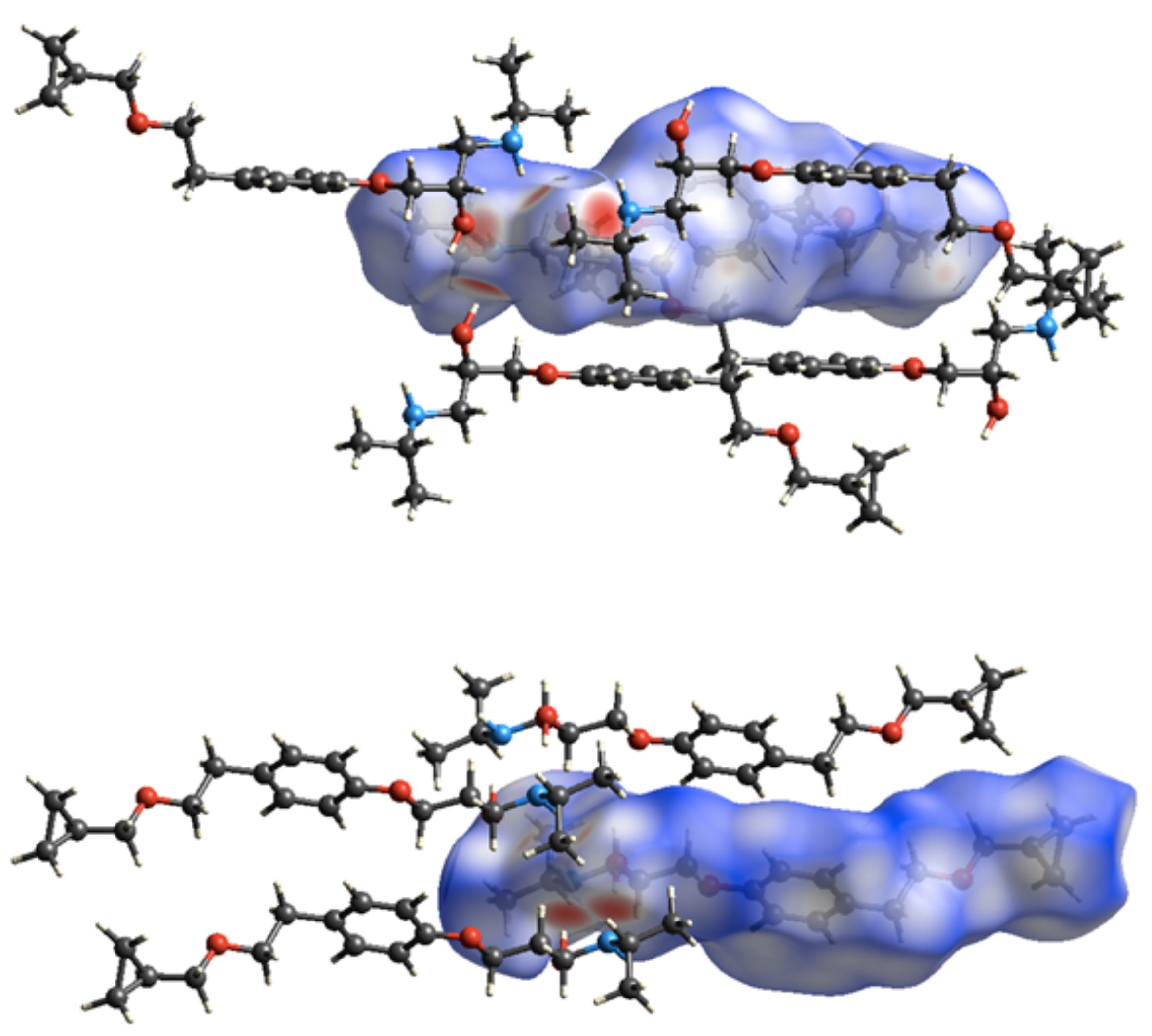
Figure 8.
Hirshfeld surfaces of the BE_IV (top) and BE_I (bottom) together with the neighboring interacting molecules via hydrogen bonds and C-H…π contacts.
Figure 8.
Hirshfeld surfaces of the BE_IV (top) and BE_I (bottom) together with the neighboring interacting molecules via hydrogen bonds and C-H…π contacts.

Figure 9.
Fingerprint plots of the BE_IV (top) and BE_I (bottom).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic data and refinement parameters for BE_IV.
| Formula | C18H29NO3 |
|---|---|
| MW | 307.42 |
| T (K) | 100 |
| λ (Å) | 0.71073 |
| Crystal system, space group | Monoclinic, I2/a |
| Unit cell dimensions (Å, °) | a = 9.3146(7) b = 19.619(1); β = 103.281(8) c = 19.898(1) |
| Volume (Å3) | 3539.0(4) |
| Z, Dc (mg/cm3) | 8, 1.154 |
| μ (mm−1) | 0.077 |
| Rwp (%) | 6.87 |
| GOF’s | 1.056 |
Table 2.
Selected intermolecular interactions in BE_IV 1.
| H-Bonds & CH-π Interactions | Distance (Å) | Distance (Å) | Angle (°) |
|---|---|---|---|
| X-H…Y | X…Y | H…Y | X-H…Y |
| O2-H2o...N1 2 | 2.786(3) | 1.84(3) | 167(3) |
| N1-H1n…O2 3 | 3.038(3) | 2.22(3) | 157(2) |
| C-H…π | H…(C6)centroid (Å) | CH…(C6)centroid (°) | |
| C7-H7a…(C1-C6) 4centroid | 2.78(3) | 132(2) |
1 geometrical data refer to X-ray diffraction data collected at 100K; 2 = x + 1/2, −y,+z; 3 = −x + 3/2, + y,−z + 1; 4 = x−1/2, −y, z.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rossi, P.; Paoli, P.; Milazzo, S.; Chelazzi, L.; Ienco, A.; Conti, L. Investigating Differences and Similarities between Betaxolol Polymorphs. Crystals 2019, 9, 509. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100509
AMA Style
Rossi P, Paoli P, Milazzo S, Chelazzi L, Ienco A, Conti L. Investigating Differences and Similarities between Betaxolol Polymorphs. Crystals. 2019; 9(10):509. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100509
Chicago/Turabian StyleRossi, Patrizia, Paola Paoli, Stella Milazzo, Laura Chelazzi, Andrea Ienco, and Luca Conti. 2019. "Investigating Differences and Similarities between Betaxolol Polymorphs" Crystals 9, no. 10: 509. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100509
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.