A Preview of a Construction of a Crystal Lattice Based on Intermolecular Interactions


1
Institute of Chemical Process Fundamentals, Czech Academy of Sciences, Rozvojová 135, 165 02 Prague, Czech Republic
2
Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Crystals 2019, 9(3), 159; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9030159
Submission received: 25 February 2019
/
Revised: 13 March 2019
/
Accepted: 14 March 2019
/
Published: 19 March 2019
(This article belongs to the Special Issue NMR Crystallography)
Abstract
:A general procedure of crystal packing reconstruction using a certain number of intermolecular interactions is introduced and demonstrated on the crystal structure of l-histidine·HCl·H2O. Geometric restrictions based on intermolecular interactions are used for formation of a molecular pair as a basic repetitive motif of the crystal packing. The geometric restrictions were applied gradually within a supervised procedure, narrowing the scope of possible arrangement of two adjacent molecules. Subsequently, a pair of histidine molecules was used for construction of a molecular chain. The chain formed contained translation information on histidine molecules in one dimension, which coincided with one of the cell parameters. Furthermore, the periodicity in the second and third dimensions can be accomplished by chain assembly into sheets (2D), and sheets can be arranged into the final 3D structure. For this purpose, the rest of the available intermolecular interactions could be used to control the mutual assembly of molecular chains and sheets. Complete molecular packing would enable derivation of standard crystallographic parameters that can be used for verification of the structural model obtained. However, the procedure described for construction of the whole 3D structure from molecular chains was not attempted, and is only briefly outlined here. The procedure described can be employed especially when standard crystallographic parameters are not available and traditional methods based on X-ray diffraction fail.
1. Introduction
X-ray structure analysis has an irreplaceable role in the determination of crystal structures of organic compounds, as supported by almost a million entries in the Cambridge Structure Database (CSD) [1]. Regarding single crystal X-ray analysis, the final structure is obtained in several steps performed in a specific order [2]. The chosen single crystal is mounted in a diffractometer, and within a few minutes, several diffraction frames are acquired. Based on the reflection positions, unit cell parameters are calculated, revealing the crystal system. The accuracy of the cell parameters rises with the number of acquired reflections. Subsequently, a particular space group of the system is proposed. With a sufficient number of reflections, the unit cell parameters and the appropriate space group, the crystal structure can be solved. Nowadays, there are plenty of methods enabling structure solution. The original heavy atom methods [3] have been progressively substituted by direct methods [4]. The X-ray beam interacts with the electrons and diffracts on periodic electron density in a given direction. The initial structure solution always consists of a coarse skeleton of heavy atoms, as lighter atoms scatter weakly due to the low number of electrons. Light atoms have to be localized and refined at the very end of structure refinement. It has to be pointed out that even the positions of the lightest atoms, the hydrogen atoms, can be localized from residual electron density maps using good-quality diffraction data. The position of the hydrogen atom indicates the location of its electron, which is pulled towards the bonding partner; therefore, it does not coincide with the position of the hydrogen nucleus. Having just one electron, the contribution of hydrogen atoms to the diffraction pattern is rather small.
An alternative experimental technique for the characterization of solids is solid-state NMR spectroscopy (SS-NMR). NMR-based crystallography is a rapidly developing field generating around one thousand research articles per year, according to Web of Science (WOS) [5]. In contrast to X-ray diffraction, hydrogen atoms are the most sensitive for NMR spectroscopy. Furthermore, SS-NMR does not require a long-range order in the studied materials, and is therefore suitable for characterization of disordered and amorphous samples [6,7,8,9,10]. X-ray diffraction and SS-NMR are thus complementary techniques in many respects. The progress of NMR crystallography in recent years can mostly be attributed to the development of fast and reliable computational techniques (mostly based on density functional theory) that enable the linking of experimentally observable parameters with structural models [11,12,13].
However, the main challenge of NMR crystallography is represented by direct structure elucidation. Although it was demonstrated that de novo crystal structure predictions are possible for NMR crystallography, all current procedures place high demands on computer time, because the NMR parameters of a large set of initially predicted crystal structures have to be calculated and compared with the experimental data [14,15]. Therefore, the majority of current NMR crystallography procedures use crystallographic parameters as an input, namely, cell lattice parameters and a space group, which are generally inaccessible by NMR methods.
In this paper, the hypothetical possibility of reconstructing the molecular packing network and determining the crystallographic parameters using just NMR data is explored. Structural data of l-histidine·HCl·H2O are used for a case simulation, considering that cell parameters could not be determined from X-ray powder diffraction for some reason, which is quite common for monoclinic and triclinic systems. This particular system was chosen because the recent pioneering work of Nishiyama and coworkers has demonstrated that intermolecular distances can be obtained with SS-NMR experiments [16,17]. The newly developed approaches were demonstrated on a crystalline sample of l-histidine·HCl·H2O. In addition to 10 intramolecular hydrogen interactions, the 1H DQ-SQ correlation spectrum collected in ref. [16] also revealed 7 intermolecular hydrogen-hydrogen interactions. Moreover, two intermolecular contacts between nitrogen and hydrogen atoms were observed in the 2D 14N(SQ)-1H(SQ) spectrum. Altogether, 9 intermolecular distances were described in ref. [16].
Considering that quantification of intermolecular distances will be feasible experimentally in the future, the procedure for the derivation of standard crystallographic parameters from such data was attempted here. Solid state NMR can easily determine the number of non-equivalent molecules in the asymmetric unit (Z’) [18]. In accordance with the experiments, Z’ = 1 was used in this simulation. To focus on the intermolecular interactions and to simplify the structure prediction procedure, we did not intend to determine the conformation of the histidine molecule, and used its known conformation found in solid state.
2. Results and Discussion
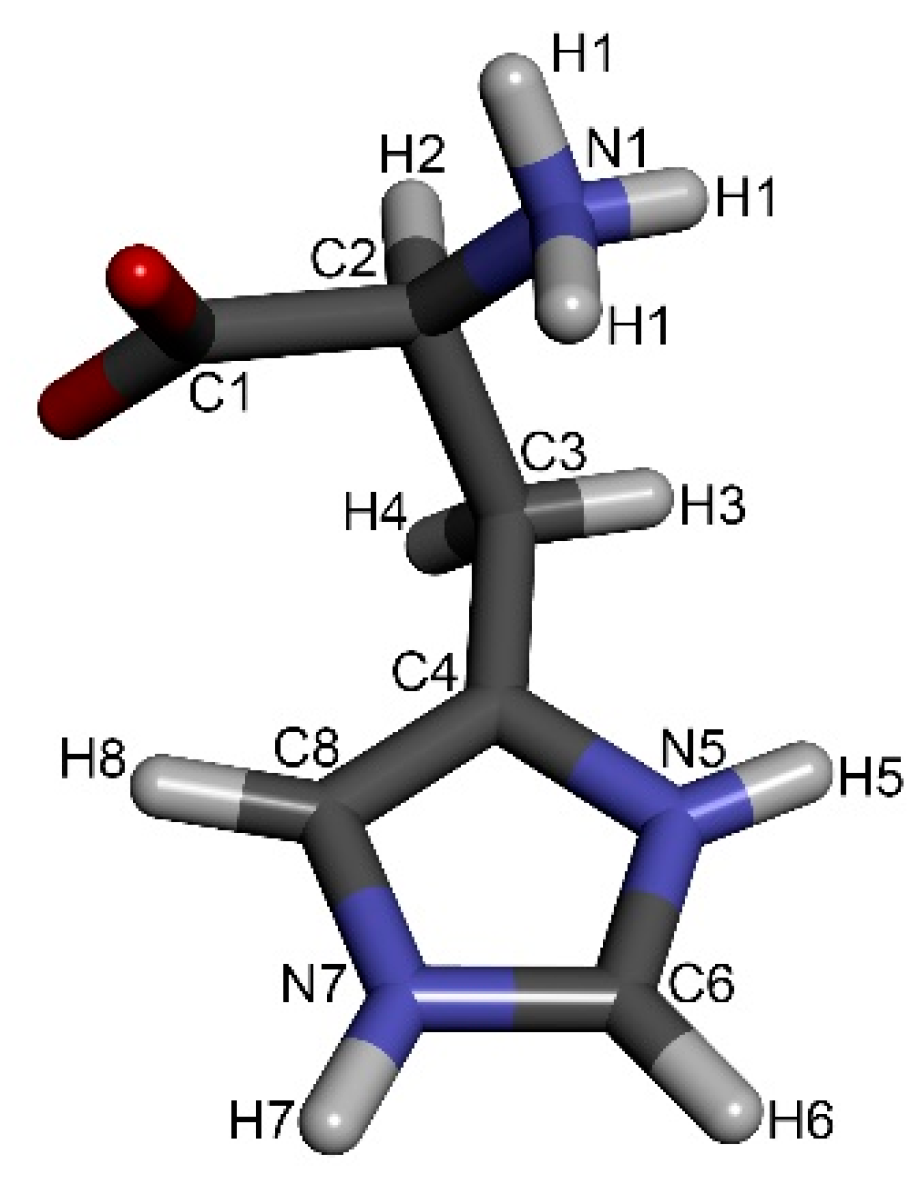
When examining the data found in ref. [16] closely, the most information on involvement in intermolecular interactions was obtained for the imidazole moiety of the histidine molecule (Table 1). Three intermolecular hydrogen-hydrogen interactions were detected; namely, H5···H8’, H5···H7’ and H6···H8’ (for numbering see Figure 1). Additionally, there were the two above-mentioned nitrogen-hydrogen interactions, H5···N7’ and N5···H7’. The rest of the intermolecular interactions described belong to the interaction between the imidazole moiety and the CH2 group (H8···H3) and 3 interactions of histidine with water molecules trapped in the crystal lattice (H1···w, H5···w, H8···w).
Generally, for the construction of a 3D network of molecular packing, it is necessary to start with a single molecule of proper geometry. To establish an initial molecule of proper geometry, intramolecular distances can be used. In this work, the original coordinates found in l-histidine·HCl·H2O from neutron diffraction are used for simplification [19]. Subsequently, the geometry optimization of the crystal structure was performed with the CASTEP program [20].
The obtained molecular geometry was used for the creation of a molecular pair in the mutual arrangement meeting the geometric restrictions based on available intermolecular distances. The initial molecule, labeled as molecule A in the following text, is fixed in space. The second molecule (molecule B) is then moved into proximity with molecule A and arranged in 3D space, meeting the requirements given by the intermolecular interactions listed above. To define the position of a single atom of molecule B with respect to molecule A, three independent distances between this atom and three atoms in molecule A are necessary. However, a set of three distances leads to two possible solutions related by a plane of symmetry defined by the three atoms in molecule A. When defining the position of a whole molecule B, a second set of three distances is required. For the case of l-histidine·HCl·H2O, three different distances were found, e.g., for hydrogen H5, namely d(H5···H7’) = 3.15 Å, d(H5···H8’) = 3.25 Å and d(H5···N7’) = 3.44 Å (Table 1). Unfortunately, the three spheres defined by this distance do not intersect in a single point. A similar situation was found for atom H8, for which three interactions were also given, namely d(H3···H8’) = 2.90 Å, d(H5···H8’) = 3.25 Å and d(H6···H8’) = 3.03 Å (Table 1). Obviously, the three distances originate from interactions with at least two different molecules in the crystal lattice. Such a possibility always has to be taken into account. Overall, a specific position of H5 or H8 toward molecule A cannot be defined with certainty using the available data.
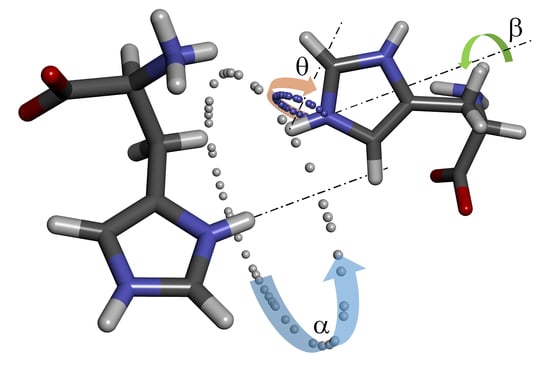
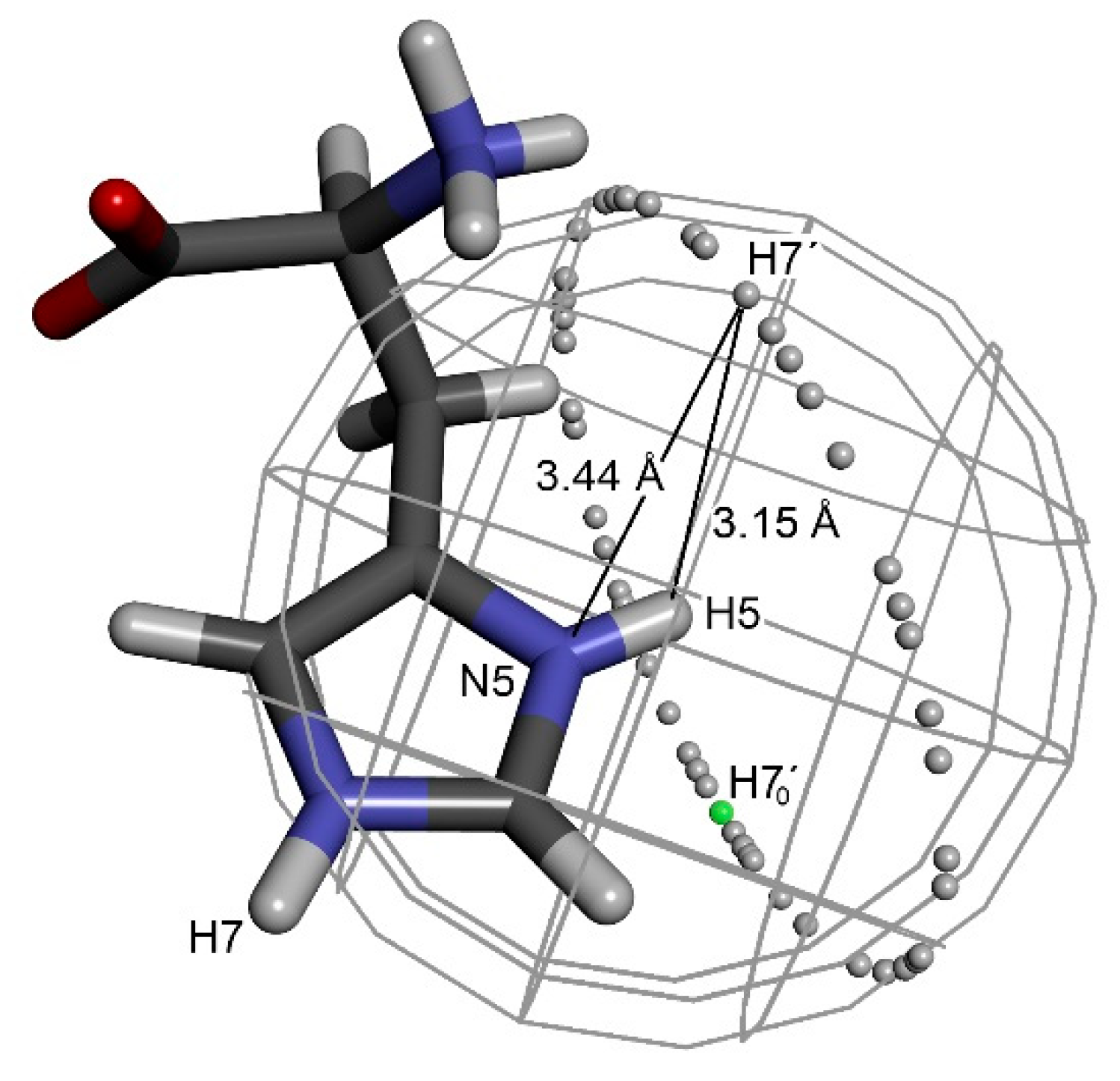
To narrow the space where molecule B can be located and to define its proper orientation, it was necessary to find other interactions that could be attributed to a single molecule. Useful geometric restrictions were found in the imidazole moiety, namely a combination of three interatomic distances between four nuclei N5–H5···H7’–N7’. The imidazole moiety is very convenient, as its geometry can be easily optimized, and the following steps can be also performed just with the imidazole fragment of the molecule. The hydrogen atom H7’ of molecule B has to be placed at a distance of 3.15 Å from hydrogen H5. This constraint reduced the possible space to a sphere with a radius of 3.15 Å centered on H5. Simultaneously, the second constraint keeps hydrogen H7’ at a distance of 3.44 Å from nitrogen N5, reducing the available space from a sphere to a circle (Figure 2). Each location on such a ring can be characterized by angle α defined as H70’–H5–H7’ angle, where H70’ is an arbitrarily chosen initial position of H7’.
Having H7’ fixed at the circle around molecule A, nitrogen N7’ has to be placed at a distance of 3.84 Å from hydrogen H5. This constraint locates N7’ on a smaller circle around any chosen position of H7’. The graphical presentation of the possible location of N7’ resembles precession around the imaginary axis H5···H7’. The chosen orientation of molecule B can then be described by precession angle θ (N5–H5–H7’–N7’). Obviously, molecule B can also freely rotate around the H7’–N7’ bond. This rotation defines the last torsion angle (β, H5–H7’–N7’–C6), which is necessary for a complete description of the position and orientation of molecule B towards original molecule A under the given geometric constraints (Figure 3).
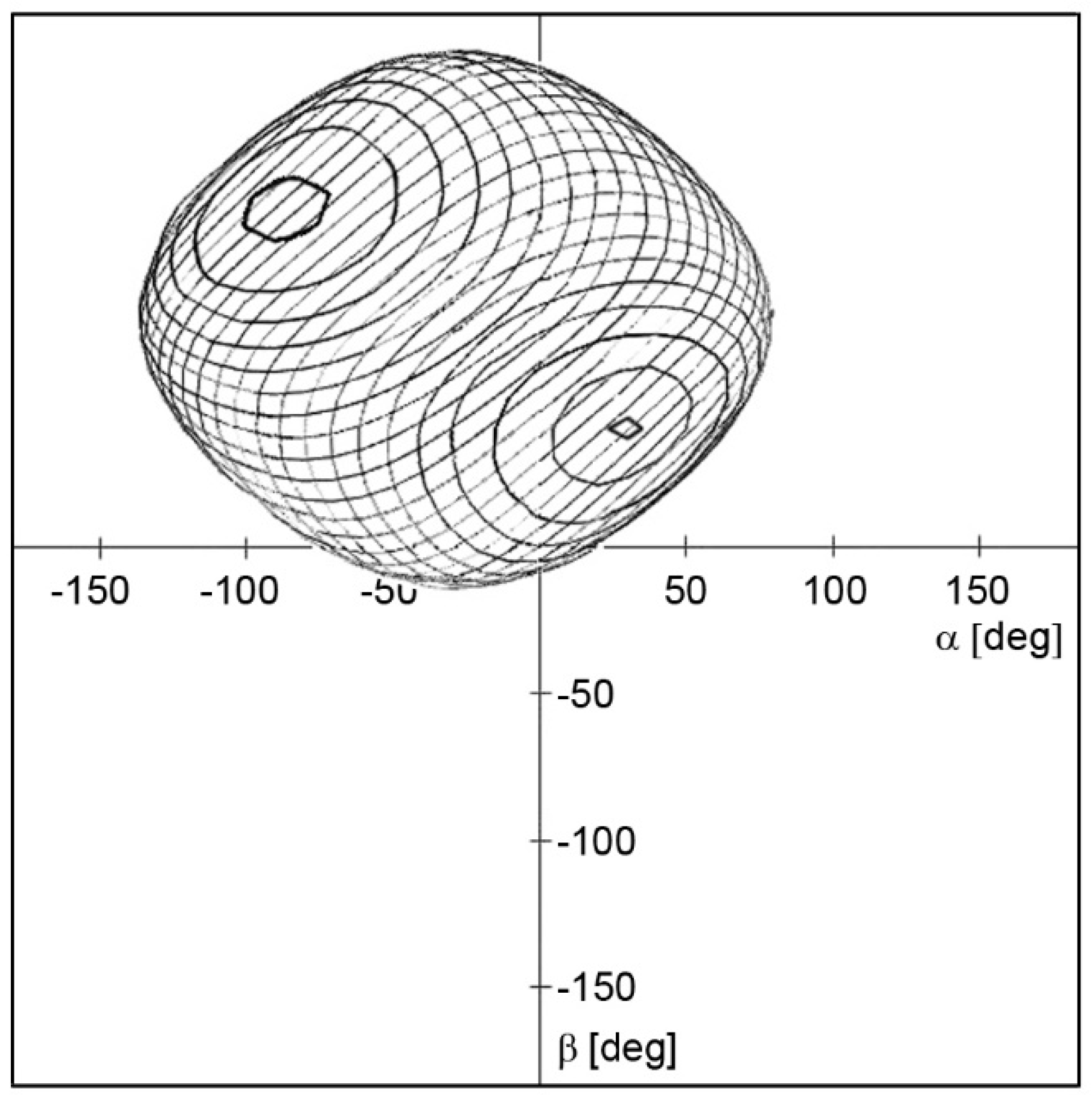
Inspection of the 3D space defined by the three angles defined using a 5-degree step for each angle provides over 370,000 possible arrangements of two histidine molecules. Naturally, a significant part of these solutions can be immediately omitted due to mutual intersection of van der Waals radii of some atoms. On the other hand, some of the solutions evince the approaching of hydrogen H8’ to hydrogen H6 to a distance of 3.03 Å, which was reported as another observable intermolecular interaction, vide supra. The additional condition of d(H6···H8’) = 3.03 Å reduces the 3D space significantly to the surface of the spherical shape depicted in Figure 4. The number of possible arrangements using the 5° grid drops to several thousand.
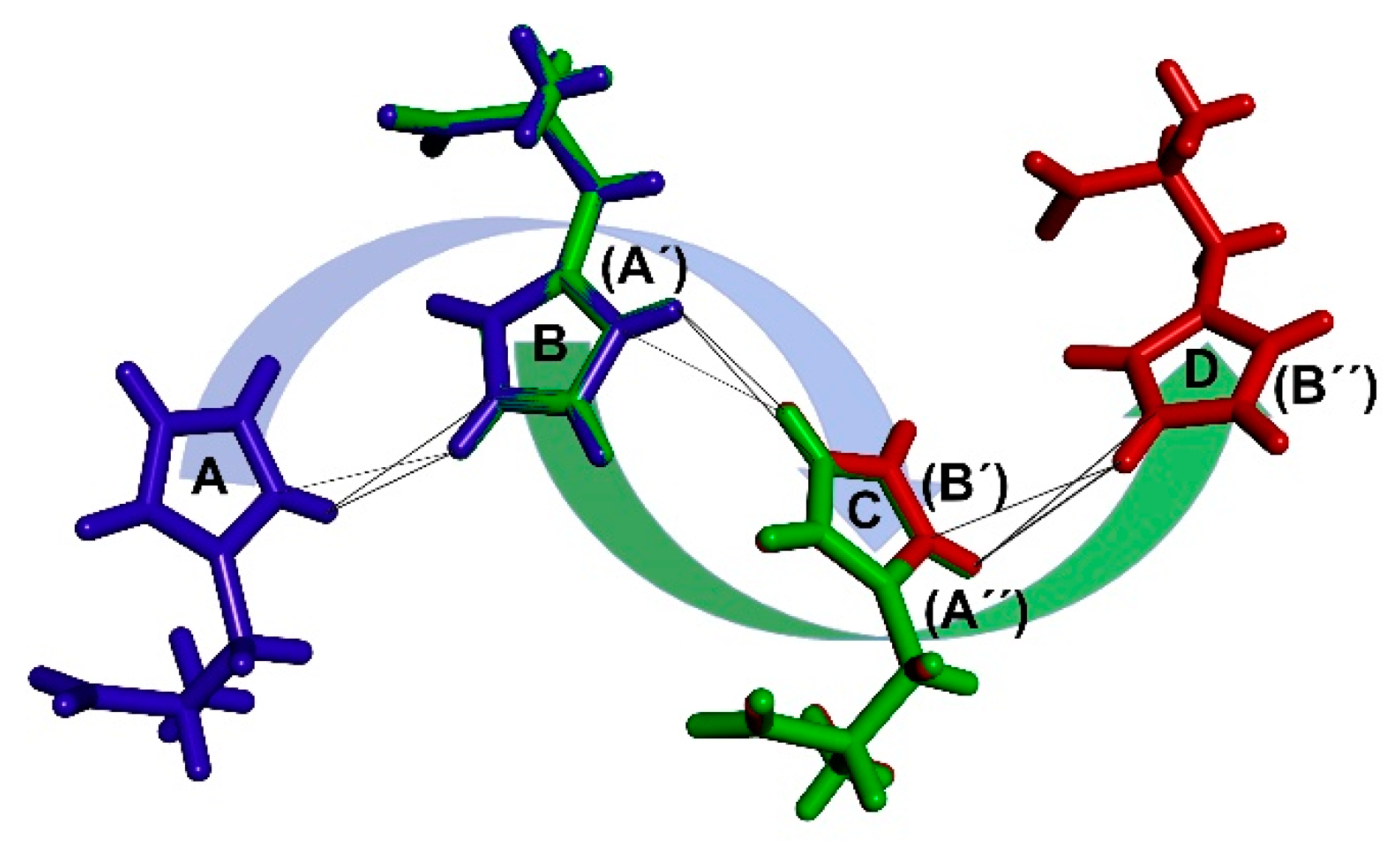
By systematically selecting one particular arrangement of the two histidine molecules after another and using it as a basic repetitive motif, it is possible to start the formation of larger conglomerates. One of the consequences of the symmetrical arrangement of molecules in the crystal lattice is that every molecule must have the same surroundings as the others, considering one independent molecule in the asymmetric unit. Therefore, multiplying a pair of histidine molecules and placing molecule A into the position of molecule B generates another molecule (molecule C). This step can be repeated to generate a molecular chain of an arbitrary length. The mutual arrangement of molecule B to molecule A is in this way transposed to the spacing of molecule C to molecule B and so on (Figure 5).
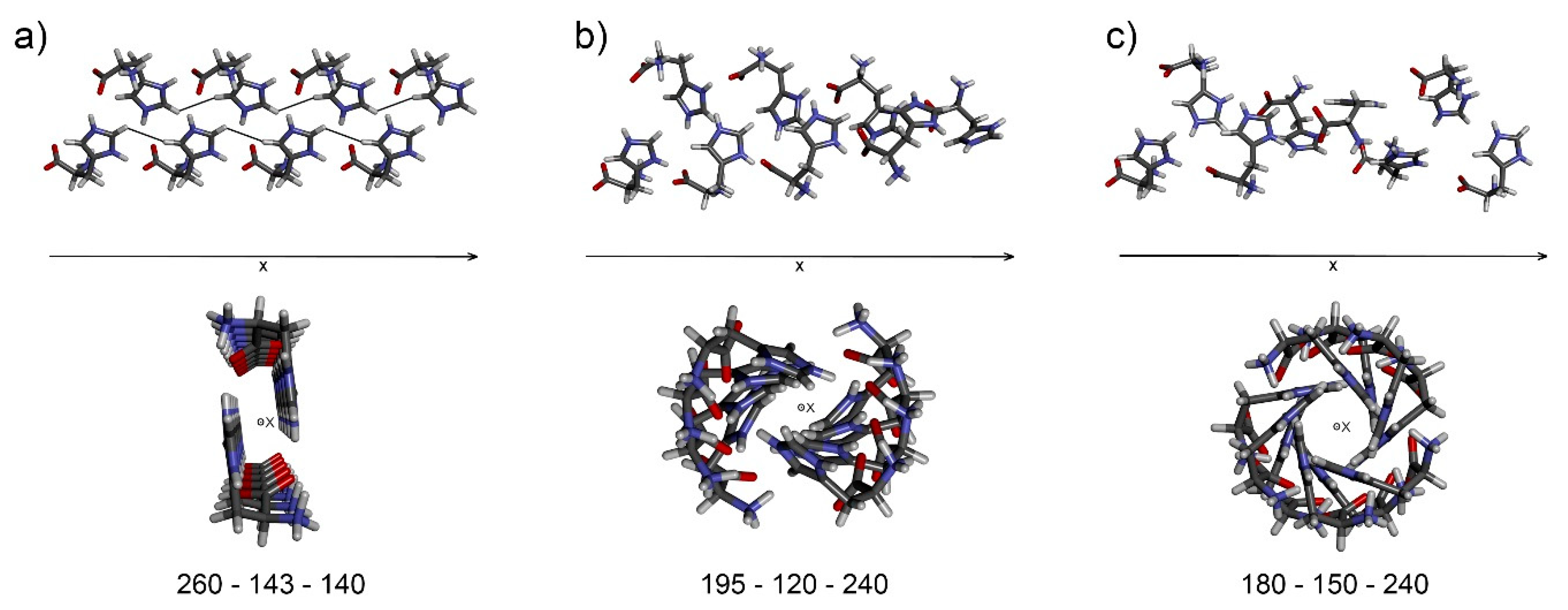
As a consequence of crystal lattice periodicity, certain arrangements provide linear chains with identical distances between equivalent atoms in molecules A and C, while the other solutions provide helical structures. The helical arrangement can be acceptable but only for an exact 2-, 3-, 4- or 6-fold axis, as only such axes can be found in real crystal systems. The additional condition of chain linearity or defined helicity further reduces the scope of possible solutions. The number of possible solutions dropped to six using 1-degree incrementation of the three rotation angles. One particular solution (Figure 6a) places hydrogen H8’’ (molecule C) at a distance of 3.25 Å from hydrogen H5 (molecule A), in accordance with the list of found intermolecular interactions (Table 1). For examples of molecular chains obtained, see Figure 6b,c.
In summary, the arrangement of histidine molecules within a final chain was accomplished using six constraints. Three distances in the N5–H5···H7’–N7’ system, interactions H5···H8’ and H6···H8’, and the condition of chain linearity provided a unique solution. The definition of the basic repetitive motif is an essential step for the subsequent construction of the 3D molecular packing. Such a process was not attempted here; therefore, the following paragraphs serve just as a tentative indication of how it could be done.
The chain formed contains translation information on histidine molecules in one dimension. This translation might coincide with one of the cell parameters. To reveal the periodicity in the second and third dimension, the chains have to be assembled into sheets (2D) and the sheets into the final 3D structure. For this purpose, the rest of the available intermolecular interactions can be used in order to control the mutual assembly of molecular chains. The so-far-unused intermolecular interaction between H3···H8’ can be utilized for arrangement of two adjacent chains. A sphere with a radius of 2.90 Å made around each H8 atom in the chain defines the possible location of H3 atoms from the second chain (Figure 7a). Van der Waals presentation reveals a relatively narrow range in the space where the second chain can be placed (Figure 7b).
The axes of both chains have to be parallel in order to fulfill d(H3···H8’) = 2.90 Å for each pair of atoms. However, there are two possible arrangements generated by rotation of the second chain axis by 180 degrees (Figure 8). Furthermore, when the second chain is located at d(H3···H8’) = 2.90 Å, it can be tilted around the axis defined by H3 atoms in the chain arbitrarily until the violation of van der Waals radii is reached. The position of the second chain is defined by d(H3···H8’) distance and angle γ of the inclination. Furthermore, some space has to be left for the positioning of the water molecule. Unfortunately, its location in the crystal lattice can be determined only approximately, as signals of both its hydrogen atoms coincide in the published spectra. However, two of three intermolecular interactions attributed to the water molecule indicate its probable location. The intersection of spheres defined by d(H5···Hw) = 2.47 Å and d(H8···Hw) = 3.09 Å localizes possible placement of the water molecule among histidine molecules.
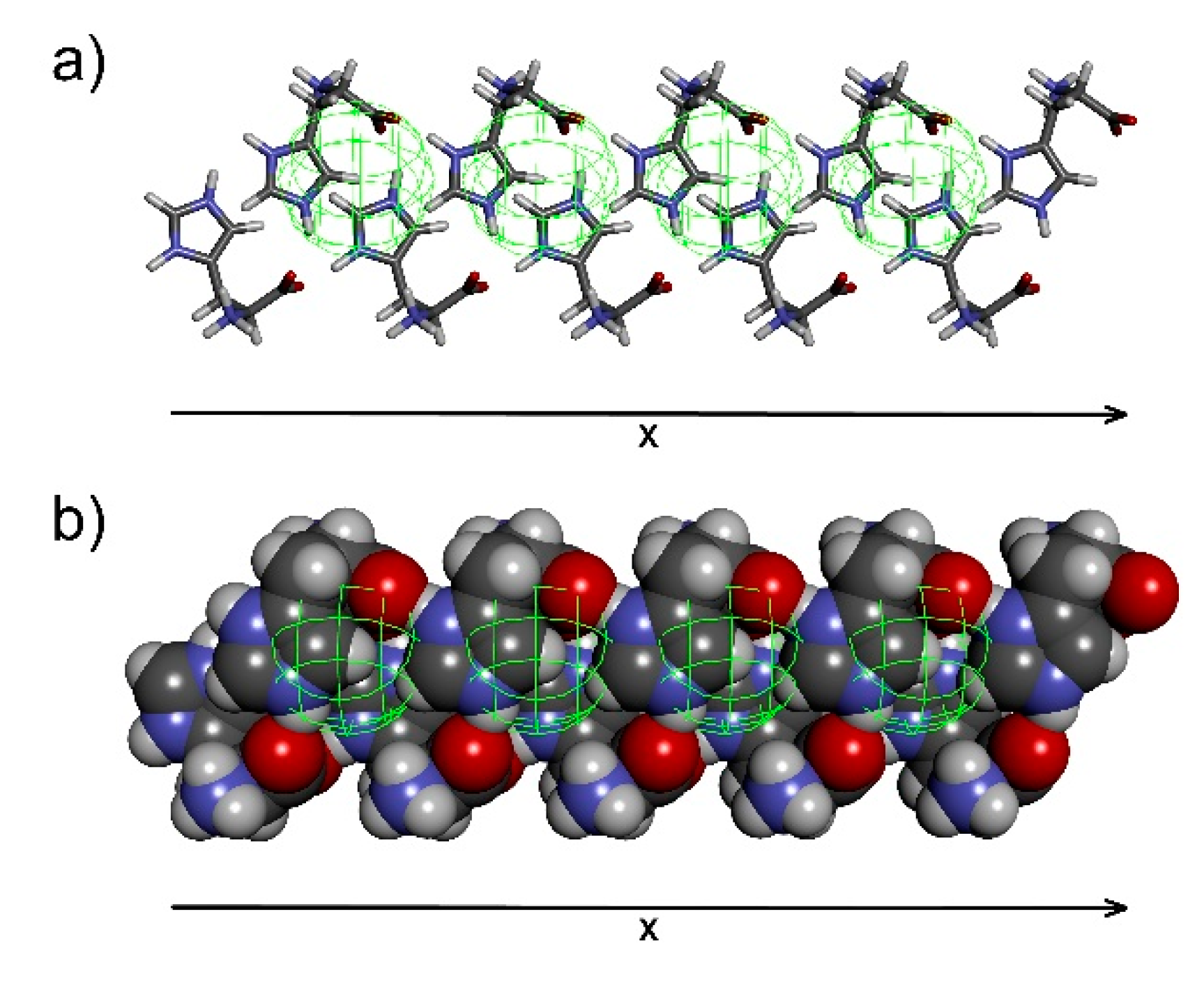
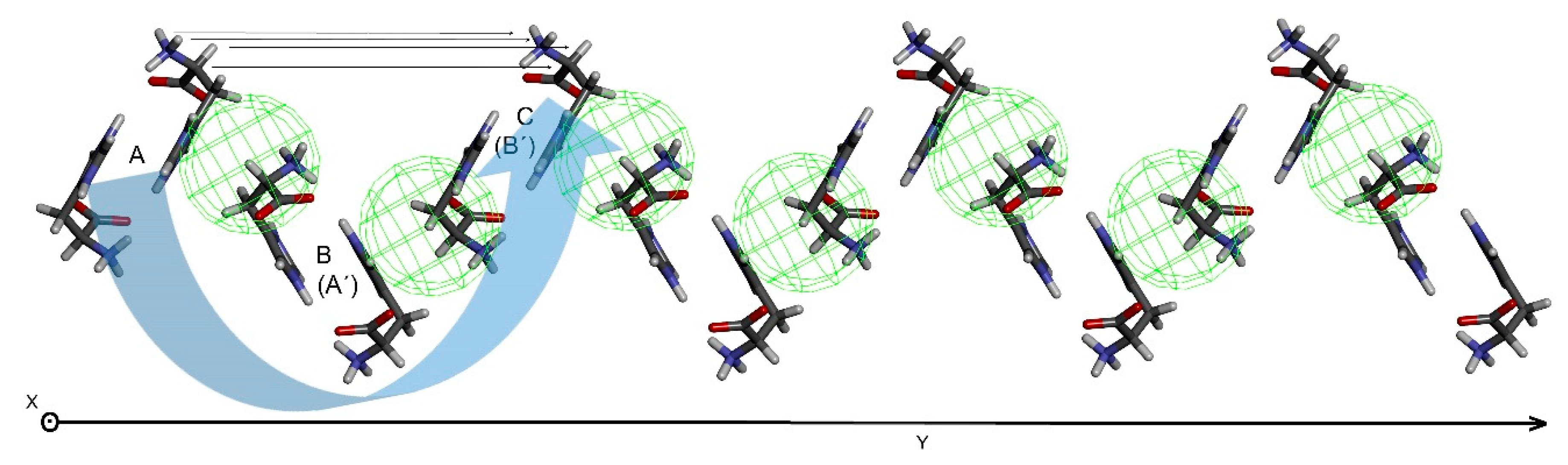
For the 2D-sheet generation, the chain pair is multiplied, and chain A is placed into the position of chain B, generating another chain, similar to the manner described for chain generation from a pair of histidine molecules. Only chains of certain mutual orientation can provide a linear 2D-sheet in which corresponding atoms in chain A and C are equidistant. The 2D-sheet formed contains translation information in the second dimension (Figure 9) which might coincide with one of the cell parameters. Perpendicular arrangements of histidine molecules in the sheet (chains A and C, see horizontal lines in Figure 9) to histidine molecules within a single chain (molecules A and C) should be tested preferentially, as perpendicular arrangements can provide perpendicular cell parameters in the final step.
An assembly of 2D-sheets into a 3D structure represents the final step of crystal lattice reconstruction. The multiplied sheets are brought closer. Individual sheets mutually interlock well and minimize voids in between. The only criterion is the possible collision of van der Waals radii. There are no distance restraints left, which might be used for the mutual positioning of neighboring 2D-sheets. However, the sheets should be positioned so that the volume of empty space in the crystal is minimized (Figure 10).
Finally, the approximate position of the water molecule in the crystal packing can be determined. However, it cannot be located precisely, as its hydrogen atoms are not spatially resolved in the available NMR data. The accurate location and orientation of the water molecule has to be examined ex post, taking into account possible involvement in hydrogen-bond networking. On the other hand, the location of the chloride anion cannot be determined at all, as there is no information of chloride atom involvement in the molecular packing. Therefore, the remaining voids in the crystal structure and possible hydrogen-bonding interactions have to be examined for its location.
Once the molecular packing is established, it is possible to derive crystal cell parameters. This information is hidden in the periodicity of a single histidine molecule, of each of its atoms. Selecting one particular atom in the histidine molecule and deleting the rest of them, the information on translation becomes obvious (Figure 11). Then cell parameters are easily determined, and the structure can be classified into the corresponding system, in this case an orthogonal crystal system identical with the system found in ref. [16].
For this particular solution, parameter a is found in the 2D-sheet between chain A and C (see Figure 9). Parameter b is identified as the distance between 2D-sheet A and B (see Figure 10). Finally, parameter c emerges as the distance between histidine molecules A and C within a single molecular chain (see Figure 5). Four histidine molecules were found in the crystal unit cell. One molecule is situated at the origin of the orthogonal system while the remaining positions correspond to the symmetry operations characteristic for the given space group. The deduction of possible space groups is then rather straightforward. There are 59 space groups available for the orthorhombic crystal system. The requirements for the space group are two: a primitive cell based on the resulting molecular packing (Figure 11b) and no mirror planes, as histidine is a chiral compound and only its l-form is present. Therefore, the number of possible space groups is reduced to 4, namely P222, P2221, P21212 and P212121. Based on symmetry operations, each space group generates different positions for four histidine molecules in the unit cell. The only match of positions generated by symmetry operations with the positions found in the reconstructed crystal structure was found for space group P212121 providing x, y, z, −x + 1/2, −y, z + 1/2, x + 1/2, −y + 1/2, −z and –x + 1/1, y + 1/2, −z + 1/2 sites.
The found crystallographic parameters can then be confronted with X-ray powder diffraction data for verification of the crystal model proposed. When a significant match is found, the crystal structure can be further refined by Rietveld refinement. Alternatively, the crystal’s structure can be optimized by quantum-chemical methods and NMR parameters can be calculated, which may serve for comparison [21] with experimental data.
3. Conclusions
A general procedure of crystal packing reconstruction using a certain number of intermolecular interactions was introduced and demonstrated on the crystal structure of l-histidine·HCl·H2O. The geometric restrictions are applied gradually within a supervised procedure, narrowing the scope of possible arrangement of two adjacent molecules. Using a list of available intermolecular interactions and implementing the additional condition that every molecule has the same surroundings in the crystal lattice, a basic repetitive motif of histidine molecules was derived. This motif was subsequently used for the construction of molecular chains containing information on translation in one dimension. The other two dimensions can then be obtained by the construction of 2D-sheets and their mutual assembly into the final 3D structure. However, the procedure for the whole 3D structure from molecular chains was not attempted; it was only briefly outlined here. Complete molecular packing would enable the derivation of standard crystallographic parameters that can be used for verification of the structural model. The procedure described can be employed especially when standard crystallographic parameters are not available and traditional methods based on X-ray diffraction fail. Similarly to other fields of NMR spectroscopy, hydrogen atoms show high relevance for direct structure solution using solid-state NMR data.
Generally speaking, further development of methods for detection and quantification of intra- and intermolecular interactions is in high demand. Intramolecular distances can serve for construction of the initial molecule with a proper conformation, while the intermolecular distances are used for a molecular assembly within crystal packing and derivation of standard crystallographic parameters. More interactions mean lower degrees of freedom and a narrower scope of possible solutions. Regarding intermolecular interactions, only the shortest ones ranging from 2.5–4.0 Å are meaningful. The number of required distances can vary for different crystallographic systems. It seems that the construction of molecular frameworks of systems of low symmetry will be easier due to the low number of molecules in the unit cell, and therefore also in the basic repetitive motif. In some cases, a coarse crystal packing could be obtained using just a fragment of the molecule. Furthermore, the construction of crystal packing of achiral molecules crystallizing in centrosymmetric groups will probably be more difficult, as a mirror image has to be considered in every single step.
4. Materials and Methods
4.1. DFT Calculations
The geometry of histidine hydrochloride monohydrate crystal structure was taken from the Cambridge Crystallographic Database (Refcode HISTCM12) [1,19]. The geometry optimization of the crystal structure was performed with the CASTEP program [20], which is a DFT-based code, using pseudopotentials to model the effects of core electrons and plane waves to describe the valence electrons. Electronic exchange and correlation effects were modelled using the PBE functional [22]. A plane wave basis set energy cutoff of 600 eV, default ‘on the fly generation’ pseudopotentials, and a k-point spacing of 0.05 Å−1 over the Brillouin zone via a Monkhorst-Pack grid [23] was used.
4.2. Construction of Molecular Framework
The search for the arrangement of two histidine molecules and the building of a molecular pair was done in MATLAB R2016b (MathWorks, USA). The inspection of the 3D space defined by angles α, β and θ (Figure 3) was done using a 5-degree step for each angle. The final optimization of the mutual arrangement within a molecular chain in order to obtain a linear object was done using a 1-degree step for each angle. In this step, a dispersion of distances between equivalent atoms in molecules A and C was minimized.
4.3. Visualization of Generated Structures
Individual steps of the creation of a basic repetitive motif and subsequent proposal of the construction of the complete molecular packing were visualized in Biovia Discovery Studio Visualizer (Accelrys Inc., USA).
Author Contributions
Conceptualization, J.S.; methodology, J.S. and V.H.; formal analysis, J.S. and M.D.; investigation, V.H., J.S. and M.D.; resources, J.S. and M.D.; writing—original draft preparation, J.S.; writing—review and editing, J.S. and M.D.; visualization, J.S.
Funding
This work was supported by The Czech Science Foundation (grant No. 15-12719S and 18-11851S).
Acknowledgments
The authors are grateful to Andrew Christensen for proofreading.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Blake, A.J.; Gould, R.O.; Main, P. Crystal Structure Analysis: Principles and Practice, 2nd ed.; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Patterson, A.L. A direct method for the determination of the components of interatomic distances in crystals. Z. Kristallogr. 1935, 90, 517–542. [Google Scholar] [CrossRef]
- Viterbo, D. Solution and Refinement of Crystal Structures. In Fundamentals of Crystallography, 2nd ed.; Giacovazzo, C., Ed.; Oxford University Press: New York, NY, USA, 2002; pp. 413–501. [Google Scholar]
- Web of Science. Available online: https://0-apps-webofknowledge-com.brum.beds.ac.uk/ (accessed on 31 January 2019).
- Dračínský, M.; Šála, M.; Hodgkinson, P. Dynamics of water molecules and sodium ions in solid hydrates of nucleotides. CrystEngComm 2014, 16, 6756–6764. [Google Scholar] [CrossRef] [Green Version]
- Kaleta, J.; Chen, J.W.; Bastien, G.; Dračínský, M.; Mašat, M.; Rogers, C.T.; Feringa, B.L.; Michl, J. Surface Inclusion of Unidirectional Molecular Motors in Hexagonal Tris(o-phenylene)cyclotriphosphazene. J. Am. Chem. Soc. 2017, 139, 10486–10498. [Google Scholar] [CrossRef] [PubMed]
- Dračínský, M.; Hodgkinson, P. Solid-state NMR studies of nucleic acid components. RSC Adv. 2015, 5, 12300–12310. [Google Scholar] [CrossRef] [Green Version]
- Florian, P.; Massiot, D. Beyond periodicity: Probing disorder in crystalline materials by solid-state nuclear magnetic resonance spectroscopy. CrystEngComm 2013, 15, 8623–8626. [Google Scholar] [CrossRef]
- Kerr, H.E.; Mason, H.E.; Sparkes, H.A.; Hodgkinson, P. Testing the limits of NMR crystallography: The case of caffeine-citric acid hydrate. CrystEngComm 2016, 18, 6700–6707. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemist’s Point of View. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.D.; Kudla, R.A.; Day, G.M.; Mueller, L.J.; Beran, G.J.O. Benchmark fragment-based H-1, C-13, N-15 and O-17 chemical shift predictions in molecular crystals. Phys. Chem. Chem. Phys. 2016, 18, 21686–21709. [Google Scholar] [CrossRef] [PubMed]
- Dračínský, M.; Bouř, P.; Hodgkinson, P. Temperature Dependence of NMR Parameters Calculated from Path Integral Molecular Dynamics Simulations. J. Chem. Theory Comput. 2016, 12, 968–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salager, E.; Day, G.M.; Stein, R.S.; Pickard, C.J.; Elena, B.; Emsley, L. Powder Crystallography by Combined Crystal Structure Prediction and High-Resolution H-1 Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 2564. [Google Scholar] [CrossRef] [PubMed]
- Salager, E.; Stein, R.S.; Pickard, C.J.; Elena, B.; Emsley, L. Powder NMR crystallography of thymol. Phys. Chem. Chem. Phys. 2009, 11, 2610–2621. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.N.M.; Malon, M.; Marsh, A.; Nishiyama, Y.; Brown, S.P. Fast Magic-Angle Spinning Three-Dimensional NMR Experiment for Simultaneously Probing H-H and N-H Proximities in Solids. Anal. Chem. 2016, 88, 11412–11419. [Google Scholar] [CrossRef] [PubMed]
- Duong, N.T.; Raran-Kurussi, S.; Nishiyama, Y.; Agarwal, V. Quantitative H-1-H-1 Distances in Protonated Solids by Frequency-Selective Recoupling at Fast Magic Angle Spinning NMR. J. Phys. Chem. Lett. 2018, 9, 5948–5954. [Google Scholar] [CrossRef] [PubMed]
- Dračínský, M.; Buděšínský, M.; Warzajtis, B.; Rychlewska, U. Solution and Solid-State Effects on NMR Chemical Shifts in Sesquiterpene Lactones: NMR, X-ray, and Theoretical Methods. J. Phys. Chem. A 2012, 116, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Fuess, H.; Hohlwein, D.; Mason, S.A. Neutron diffraction study of L-histidine hydrochloride monohydrate. Acta Cryst. 1977, B33, 654. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Belton, P.S. NMR studies of hydration in low water content biopolymer systems. Magn. Reson. Chem. 2011, 49, S127–S132. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
Figure 1.
Molecule of l-histidine with atom numbering according to Ref. [16].
Figure 1.
Molecule of l-histidine with atom numbering according to Ref. [16].

Figure 2.
Definition of a sphere around H5 and its reduction to a circle.

Figure 3.
Definition of the three rotational angles resulting from geometric constraints; d(H5···H7’) = 3.15 Å, d(H5···N7’) = 3.44 Å and d(N5···H7’) = 3.84 Å.
Figure 3.
Definition of the three rotational angles resulting from geometric constraints; d(H5···H7’) = 3.15 Å, d(H5···N7’) = 3.44 Å and d(N5···H7’) = 3.84 Å.

Figure 4.
2D projection of a spherical shape showing possible combinations of angles α, β and θ meeting additional condition d(H6···H8’) = 3.03 Å. The depicted contour plot was obtained for angle θ ranging from 130–360, 0–25 deg; other values of angle θ lead to forbidden interatomic contacts.
Figure 4.
2D projection of a spherical shape showing possible combinations of angles α, β and θ meeting additional condition d(H6···H8’) = 3.03 Å. The depicted contour plot was obtained for angle θ ranging from 130–360, 0–25 deg; other values of angle θ lead to forbidden interatomic contacts.

Figure 5.
Chain generation principle described using 0–0–0 combination of angles; blue—original pair, green—pair copy placing molecule A’ into B position generating molecule C (B’); red—2nd pair copy placing molecule A’’ into B’ position generating molecule D (B’’).
Figure 5.
Chain generation principle described using 0–0–0 combination of angles; blue—original pair, green—pair copy placing molecule A’ into B position generating molecule C (B’); red—2nd pair copy placing molecule A’’ into B’ position generating molecule D (B’’).

Figure 6.
Molecular chains generated for different sets of angles α, β, and θ; (a) 260–143–140, (b) 195–120–240, (c) 180–150–240.
Figure 6.
Molecular chains generated for different sets of angles α, β, and θ; (a) 260–143–140, (b) 195–120–240, (c) 180–150–240.

Figure 7.
Visualization of 2.90 Å spheres around hydrogen H8 in the molecular chain; (a) stick presentation, (b) space fill presentation.
Figure 7.
Visualization of 2.90 Å spheres around hydrogen H8 in the molecular chain; (a) stick presentation, (b) space fill presentation.

Figure 8.
Possible arrangements of two adjacent molecular chains generated by 180-degree rotation of the second chain (a) and (b). The chains are viewed end-on indicating possible location of a water molecule.
Figure 8.
Possible arrangements of two adjacent molecular chains generated by 180-degree rotation of the second chain (a) and (b). The chains are viewed end-on indicating possible location of a water molecule.

Figure 9.
2D-sheet generated by multiplication of a chosen pair of chains. The chains are viewed end-on. Green spheres indicate d(H3···H8’) = 2.90 Å through the sheet.
Figure 9.
2D-sheet generated by multiplication of a chosen pair of chains. The chains are viewed end-on. Green spheres indicate d(H3···H8’) = 2.90 Å through the sheet.

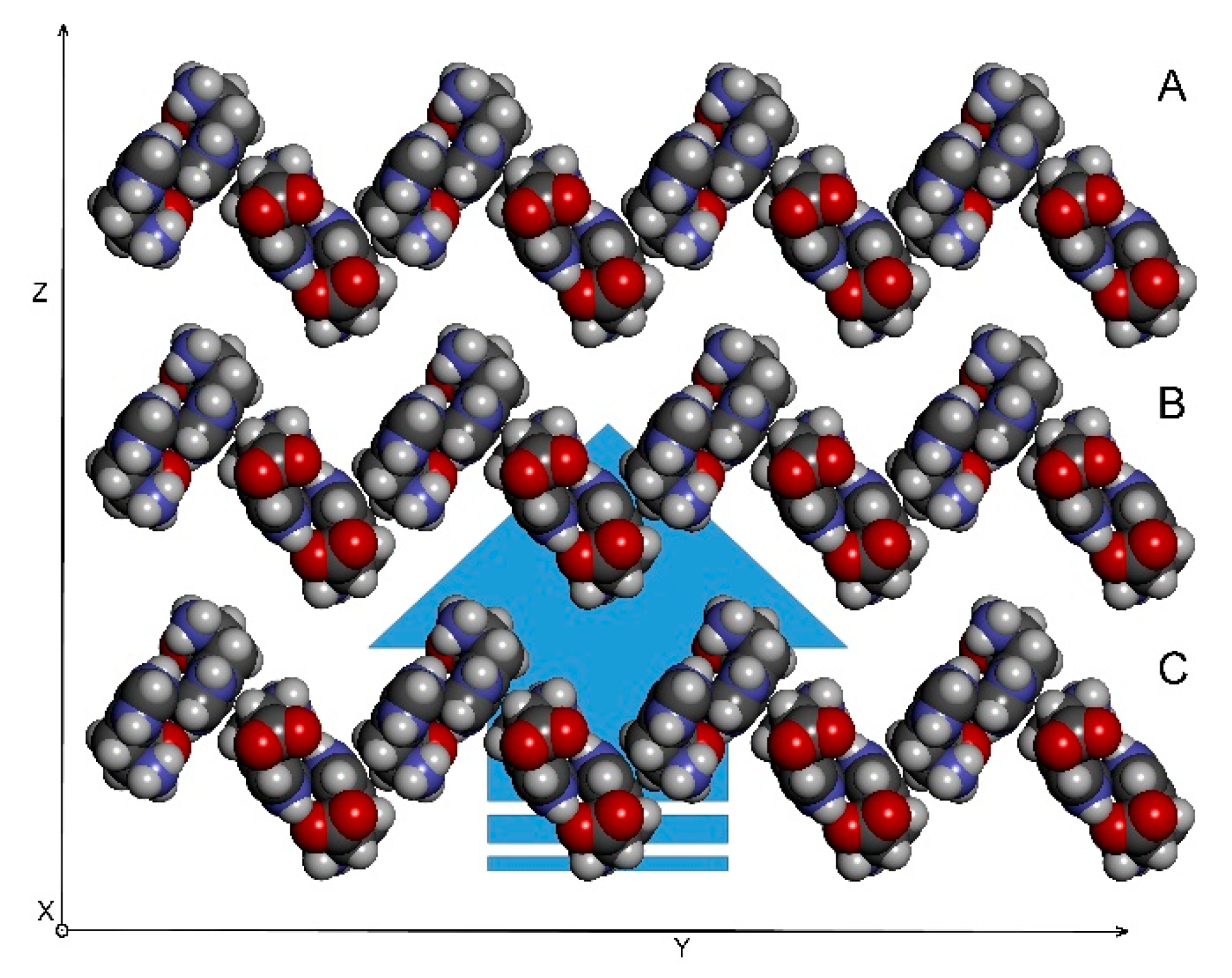
Figure 10.
An assembly of 2D-sheets into the final 3D structure. The coordinate system depicted was chosen arbitrarily and has only illustrative function.
Figure 10.
An assembly of 2D-sheets into the final 3D structure. The coordinate system depicted was chosen arbitrarily and has only illustrative function.

Figure 11.
A derivation of the cell lattice parameters from the molecular framework; (a) molecular packing in stick presentation, (b) projection of molecular packing using only one atom of each molecule.
Figure 11.
A derivation of the cell lattice parameters from the molecular framework; (a) molecular packing in stick presentation, (b) projection of molecular packing using only one atom of each molecule.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Intermolecular interactions detected in the crystal structure of l-histidine·HCl·H2O.
| Atoms | Distance [Å] |
|---|---|
| H3···H8’ | 2.90 |
| H5···H7’ | 3.15 |
| H5···H8’ | 3.25 |
| H6···H8’ | 3.03 |
| H1···Hw | 2.35 |
| H5···Hw | 2.47 |
| H8···Hw | 3.09 |
| H5···N7’ | 3.44 |
| N5···H7’ | 3.84 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hejtmánek, V.; Dračínský, M.; Sýkora, J. A Preview of a Construction of a Crystal Lattice Based on Intermolecular Interactions. Crystals 2019, 9, 159. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9030159
AMA Style
Hejtmánek V, Dračínský M, Sýkora J. A Preview of a Construction of a Crystal Lattice Based on Intermolecular Interactions. Crystals. 2019; 9(3):159. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9030159
Chicago/Turabian StyleHejtmánek, Vladimír, Martin Dračínský, and Jan Sýkora. 2019. "A Preview of a Construction of a Crystal Lattice Based on Intermolecular Interactions" Crystals 9, no. 3: 159. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9030159
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.